 Li Zhang
Li Zhang Yanling Teng
Yanling Teng Haoran Hu
Haoran Hu Huimin Zhu1
Huimin Zhu1 Desheng Liang
Desheng Liang Zhuo Li
Zhuo Li Lingqian Wu
Lingqian Wu- 1Center for Medical Genetics, Hunan Key Laboratory of Medical Genetics, Hunan Key Laboratory of Animal Models for Human Diseases, School of Life Sciences, Central South University, Changsha, Hunan, China
- 2Hunan Jiahui Genetics Hospital, Changsha, Hunan, China
Background: Seckel syndrome (SCKL) is a rare autosomal recessive inherited disorder, which is mainly characterized by intrauterine and postnatal growth restrictions, microcephaly, intellectual disability, and a typical “bird-head” facial appearance. Here, we aimed to identify the genetic etiology of a family with suspected SCKL.
Methods: This study enrolled a Chinese family suspected of SCKL with their detailed family history and clinical data. We performed karyotype analysis, copy number variation sequencing (CNV-seq), and trio whole-exome sequencing (WES) to explore the genetic etiology in the proband. Furthermore, the quantitative real-time polymerase chain reaction (PCR) and reverse transcription-PCR (RT-PCR) were conducted to confirm the pathogenicity of novel variants.
Results: The karyotype analysis and CNV-seq were normal in the proband. Two novel variants in CEP152, c.1060C>T (p.Arg354*) and c.1414-14A>G, were identified in the proband through trio-WES. The qPCR results showed that the total CEP152 mRNA expression levels were significantly reduced in c.1060C>T (p.Arg354*) and c.1414-14A>G compared with healthy control individuals. Moreover, aberrant skipping of exon 12 due to the non-canonical splice-site variant was revealed by RT-PCR and Sanger sequencing.
Conclusion: Our findings expanded pathogenic variant spectra in SCKL and offered new insights into the pathogenicity of a non-classical splice-site variant in CEP152, which provided additional information for helping the family improve pregnancy plans in the future.
1 Introduction
Seckel syndrome (SCKL) is a rare autosomal recessive inherited disorder with high clinical heterogeneity and is mainly characterized by a typical “bird-head” facial appearance, severe microcephaly, intellectual disability, and a proportionate short stature (Seckel, 1960; Takikawa et al., 2008; Khojah et al., 2021). Other clinical symptoms, including low birth weight (Majewski and Goecke, 1982), cardiovascular anomalies (Khojah et al., 2021; Donmez et al., 2022), postnatal growth restriction (O’driscoll et al., 2004), and myelodysplasia (Verloes et al., 2013), were also occasionally reported.
So far, with the insight into the genetic underpinnings, this severe inherited disorder has been proved to be associated with pathogenic variants involved in DNA damage response pathways (O’driscoll et al., 2003), initiation of mitosis at cell cycle checkpoints (Casper et al., 2004), and centrosome duplication (Fu et al., 2016; Zheng et al., 2016). To date, nine classified subtypes of SCKL have been reported, according to different genetic causes, and at least 11 genes are responsible for the Seckel syndrome subtypes listed on Online Mendelian Inheritance in Man (OMIM).
Seckel syndrome 5 (MIM#613823, SCKL5) is caused by homozygous or compound heterozygous pathogenic variants in CEP152, located at 15q21.1 and contains 27 exons in total. CEP152 codes a core protein of the centrosome with a molecular mass of 152 kDa, which is crucial in cell division. Meanwhile, it was proved to be involved in the maintenance of genomic integrity and is capable of responding to DNA damage (Andersen et al., 2003; Dinçer et al., 2017). Since the first identification of SCKL5 by Kalay et al. (2011) until now, only 13 cases have been reported worldwide. Hence, more patients and further studies are required to understand this rare disease better.
In this study, we described a 7-year-old boy with suspected Seckel syndrome. Further genetic testing, including karyotype analysis, copy number variation sequencing (CNV-seq), and whole-exome sequencing (WES), were performed to verify the diagnosis of this proband. All the tests and subsequent functional experiments revealed that novel compound heterozygous variants in CEP152 would be the genetic cause for the phenotype observed.
2 Materials and methods
2.1 Subjects
The proband was a boy aged 7 years in 2018 from a non-consanguineous Chinese Han family (II-1, Figure 1A), who was mainly presented with microcephaly, short stature, intellectual disability, and dysphasia. His mother got pregnant again in 2013. The ultrasound showed that the fetus had intrauterine growth retardation, and they chose to terminate the pregnancy afterward without genetic testing.
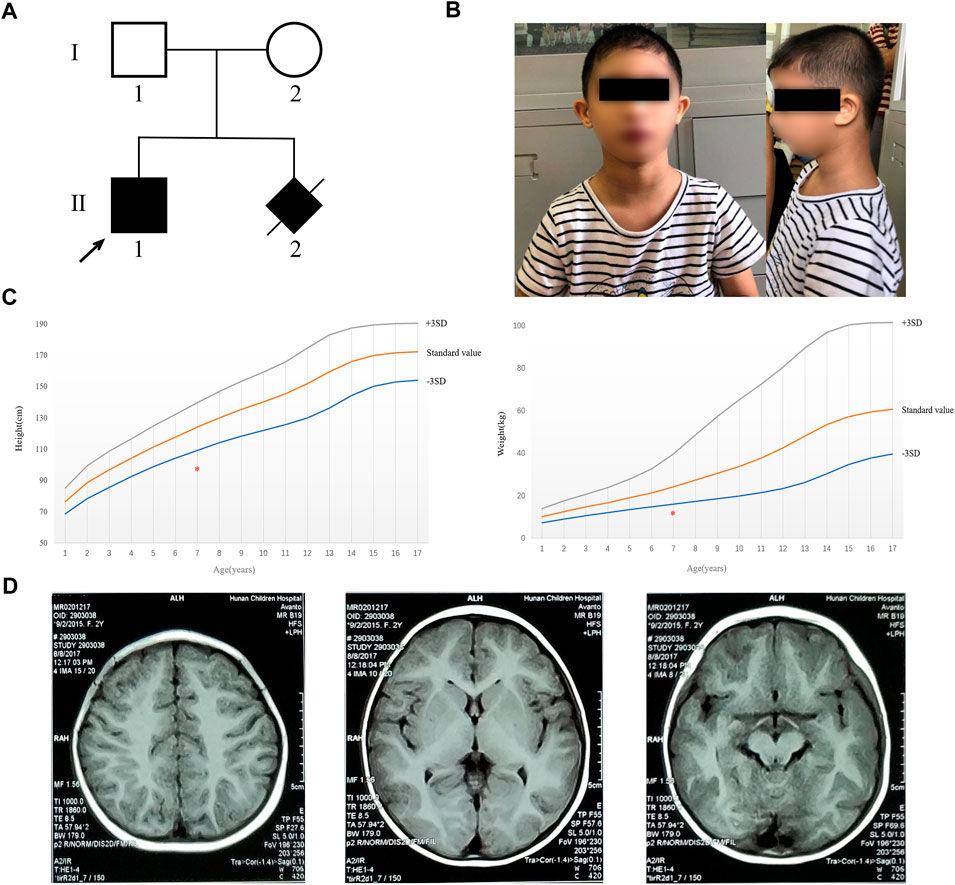
FIGURE 1. Pedigree of the family and clinical characterization of the proband. (A) Pedigree of the family. Filled black symbols indicate the affected subjects. The arrow indicates the proband in our study. (B) Clinical characteristics of the proband (including microcephaly, short stature, sloping forehead, high nasal bridge, beaked nose, and retrognathia). (C) Height and weight of the proband are marked with the red asterisk and were less than 3 SD of the standard value of people of the same age. (D) MRI result demonstrated that no abnormality was found.
This study was approved by the Ethics Board of School of Life Sciences (No. 2021-1-12), Central South University, in accordance with the approved ethical guidelines. Informed consent was obtained from the patient of this family. The patient was not involved in the development of the research, and there were no core outcome sets used in this study.
2.2 Karyotype analysis and copy number variation sequencing
Karyotype analysis was conducted by G-banding where the metaphase peripheral blood of the proband was prepared using standard procedures (McGowan-Jordan et al., 2016), and a level of 550 bands was carried out using these metaphase spreads. At least 50 metaphase spreads were examined.
Genome-wide CNV-seq of the proband was performed using a chromosome copy number variation detection kit (Berry Genomics, Hangzhou, Zhejiang, China). After library construction and purification, library sequencing was performed on a NextSeq 500 sequencer (Illumina, San Diego, CA, United States). Data analysis was performed by chromosome copy number variation data analysis software V3 (Berry Genomics, Beijing, China).
2.3 Whole-exome sequencing
DNA was extracted following peripheral blood and stored for genetic testing. WES in the family trio was performed using stored DNA. The exomes were hybridized and captured by the xGen® Exome Panel v1.0 (Integrated DNA Technologies, Coralville, Iowa, United States), and 100-bp paired-end reads were generated on a HiSeq 2000 system (Illumina, San Diego, CA, United States). Data analysis was performed according to the protocols described previously (Luo et al., 2019), and potential disease-causing variants were confirmed by following Sanger sequencing.
2.4 RNA extraction
The TRIzol reagent (Invitrogen, Carlsbad, CA, United States) was used following the manufacturer’s instructions to extract the total RNA from peripheral blood lymphocyte cells of the family members and healthy control individuals, including two males (21 and 34 years old), two females (26 and 28 years old), and two boys (8 and 11 years old). The isolated RNA was reverse-transcribed into cDNA using a Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Carlsbad, CA, United States) for the following real-time quantitative PCR and reverse transcription PCR experiments.
2.5 Real-time quantitative PCR and reverse transcription PCR
Real-time quantitative PCR (qPCR) assay was carried out on the LightCycler platform (Roche, Boston, MA, United States) with the Maxima SYBR® Green Master Mix (Thermo Fisher Scientific, Carlsbad, CA, United States). The forward primer of CEP152 was 5′-AACTTCGTGGGCAGTACATT-3′, and the reverse primer was 5′-TTCGGGCGGTTTCTTGAC-3’. Relative gene expression levels were determined using the 2-ΔΔCt method after normalizing the mRNA level of UBE2D2 amplified by the primer pair of 5′-GATCACAGTGGTCTCCAGCA-3′ and 5′-CGAGCAATCTCAGGCACTAA-3’. Three replicates were analyzed.
Reverse transcription PCR (RT-PCR) was performed by the forward primer (5′-ATTTGGATGCCACAGTCAC-3′) in exon 9 and the reverse primer (5′-TGGTGCTGCTGATAAGTCC-3′) in exon 16. Agarose gel electrophoresis was used to ascertain the effect of the novel CEP152 non-classical splice-site variant on the mRNA splicing pattern. After gel electrophoresis, PCR products were purified by excision and purification of the bands, and then sent for Sanger sequencing.
3 Results
3.1 Clinical characteristics
Physical examination: The 7-year-old boy (the proband) presented with microcephaly (head circumference 31 cm, <–3SD, and with bird-headed profile), short stature, slanting forehead, high bridge of the nose, beak nose, and micrognathia (Figure 1B). His height was 93 cm (<-3SD) and weight was 12 kg (<-3SD) (Figure 1C). He had multiple oral caries, pigeon toes, and the fifth finger clinodactyly. The detailed phenotypes are shown in Table 1.
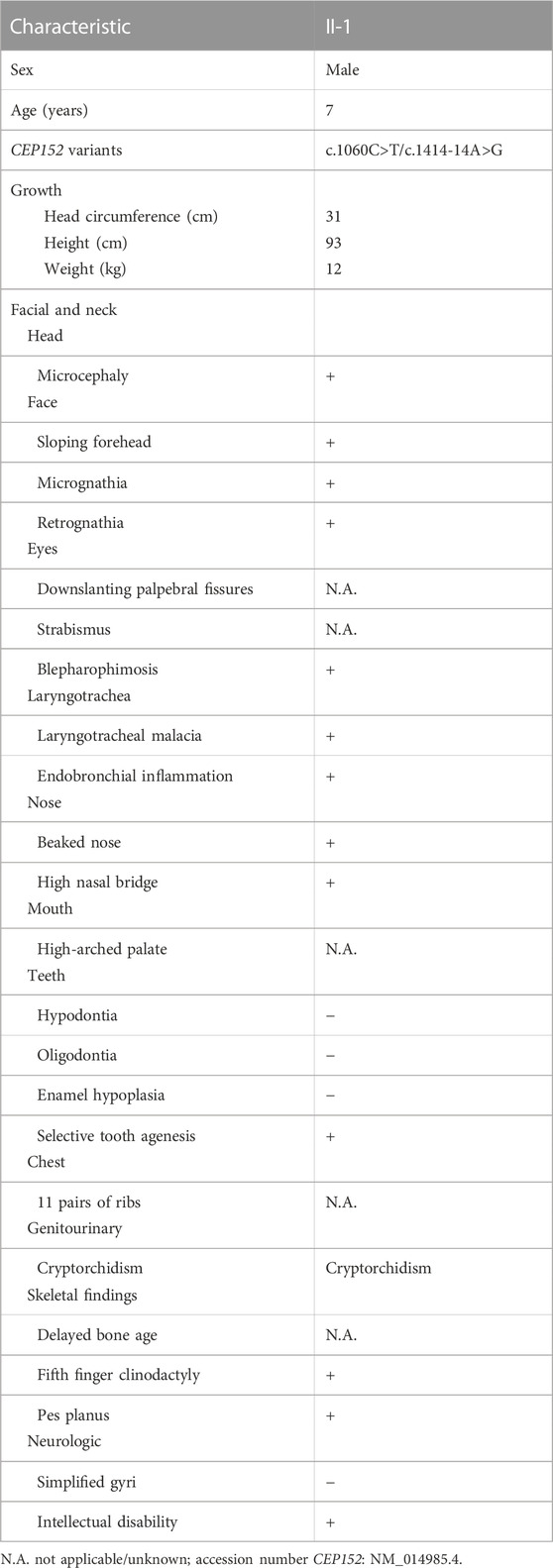
TABLE 1. Clinical findings in the SCKL5 proband described in this study.
Intelligence test: The patients were tested with the Chinese Wechsler Intelligence Scale for Children (C-WISC) and scored 57, 68, and 59 on the verbal intelligence quotient (VIQ), performance intelligence quotient (PIQ), and full-scale intelligence quotient (FIQ), respectively, which suggested intellectual disability.
Auxiliary examination: Brain magnetic resonance imaging (MRI) indicated no obvious abnormality (Figure 1D). The levels of amino acid and acylcarnitine spectrum analysis were in the normal ranges (Supplementary Figure S1). Fiberoptic bronchoscope examination showed that the proband had laryngotracheal malacia and endobronchial inflammation (Supplementary Figure S2). Electroencephalogram (EEG) and brain electrical activity mapping (BEAM) were recorded during sleep. The background activity of the EEG in the proband mainly comprised intermittent rhythmic d and θ activities within each brain region. These waveforms were irregular with sporadic low-amplitude β-rhythm (<10 μV). The results showed no obvious abnormality in EEG during sleep, and no seizure wave was found in the whole range.
3.2 Identification of two novel variants in CEP152
G-banding karyotype analysis of the proband revealed a normal male karyotype of 46, XY. The genome-wide CNV-seq analysis showed that no chromosomal aneuploidy or known genome copy number variation above 100 kb was detected. We identified a novel compound heterozygous variant in CEP152 by using trio-WES, which was composed of a paternally inherited c.1414-14A>G splice-site variant in intron 12 and a maternally inherited c.1060C>T (p.Arg354*) non-sense variant in exon 9 (Supplementary Figure S3A). The two variants have not been reported previously in public databases. The online software application speculated that the non-sense variant led to the loss of nearly four-fifths of the CEP152 protein and was considered to be damaging (http://varcards.biols.ac.cn/). According to the standards and guidelines for the interpretation of sequence variants proposed by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) (Richards et al., 2015), the variant was evaluated as “likely pathogenic”. Since the splice-site variant (c.1414-14A>G) detected in our study was not in the 1–2 nucleotide site near the junction of an exon and intron, the variant was evaluated as “uncertain significance (VUS)” (Supplementary Figures S3B, C). The positions of these two variants are shown in Figure 2.
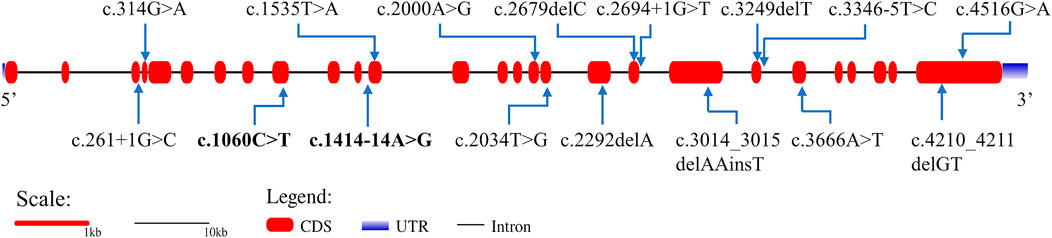
FIGURE 2. Gene structure of CEP152 and the distribution of variants that have been reported to cause SCKL. Bold variants are those identified in this study. The red boxes represent the coding sequence (CDS). The blue gradient boxes represent the 5′ and 3′ untranslated regions (UTR).
3.3 Total CEP152 mRNA expression analysis and conformation for skipping exon 12
To further evaluate the potential effect of the non-classical splice-site variant c.1414-14A>G of CEP152, qPCR and RT-PCR were performed. The qPCR revealed that the expression level of CEP152 decreased drastically (proband: 83.4%, father: 64.0%, and mother: 62.7%) in the family compared to a healthy control individual (Figure 3A). Moreover, splicing patterns of the proband and his parents were analyzed by spanning exon amplification, agarose gel electrophoresis, and Sanger sequencing. In the lanes of the proband and his father, an additional 743 bp amplification product was observed, which is absent in his mother and the healthy control individual (Figure 3B). The left half of Figure 3C displayed a single exon 12 skipping, including 164 bp nucleotides, that was found in the 743-bp PCR product by Sanger sequencing, which led to the c.1414_1577del (p.Glu472_Ser526delfs*9) frameshift variant of CEP152. The right half showed the schematic representation of the normal and aberrant transcripts, respectively. Thus, our study further demonstrated the abnormality of the splicing pattern.
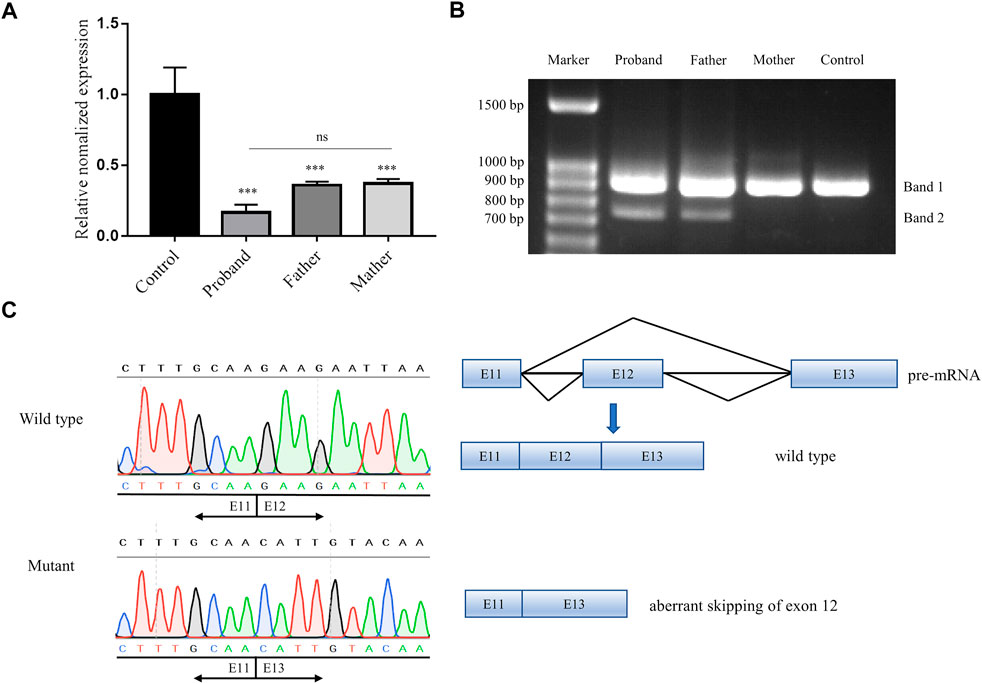
FIGURE 3. Pathogenicity verification of a novel splice-site variant in our study. (A) qPCR result showed the relative expression of CEP152 of the proband and his parents to be 16.6%, 36.0%, and 37.3%, respectively. ***p < 0.001 (t-test). (B) Agarose gel electrophoresis showed that spanning exon amplification products in the proband and his father, who also carried the splice-site variant, were compared with his mother and the healthy control individual. All subjects displayed a common 907-bp band (band 1, wild type), while the proband and his father had an additional 743-bp band (band 2, mutant). (C) Left half is Sanger sequencing showing the 743-bp band deleted exon 12 (164 bp nucleotides of CEP152 cDNA) compared to the 907-bp band (matching to the reference sequence). The right half is the schematic representation of the normal and aberrant transcripts.
4 Discussion
SCKL has been mainly classified into nine subtypes caused by different genes (Table 2). Biallelic pathogenic variants in CEP152 have been mainly described in patients with autosomal recessive primary microcephaly-9 (MIM#614852, MCPH9) and Seckel syndrome 5. Based on the Human Gene Mutation Database (HGMD), 32 variants in CEP152 have been identified in patients and have reported to be associated with different kinds of diseases including microcephaly, Seckel syndrome, atrioventricular septum defect, and autism spectrum disorder. Nevertheless, the pathogenicity of some variants is still unknown. So far, only 14 variants involved in six literature studies were reported to cause SCKL 5 (Figure 2; Supplementary Table S1).
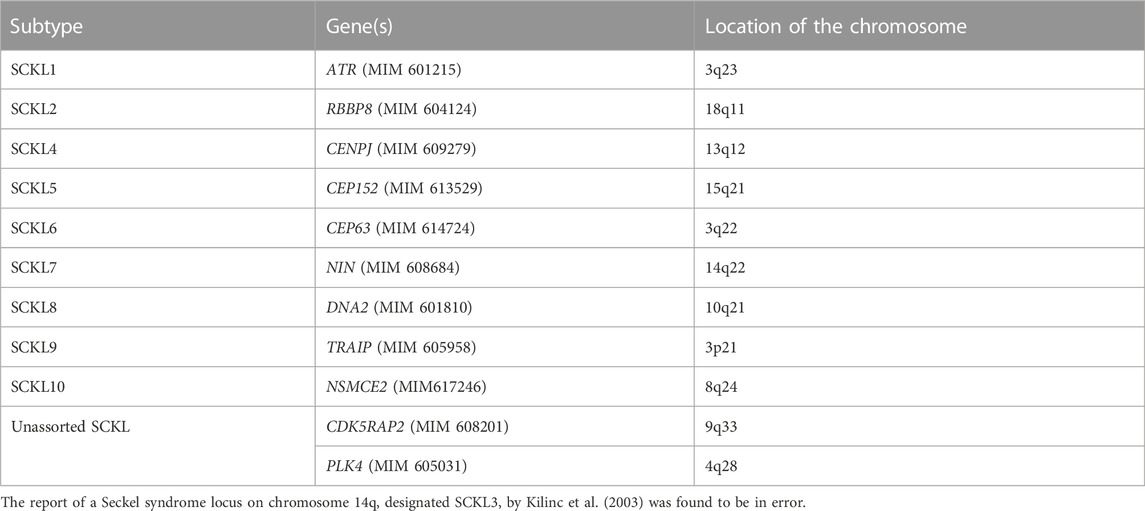
TABLE 2. Correlation between genes and subtypes of SCKL.
Although all the cases, including the case in our study, reported proportionate short stature, disproportionate head circumference in growth, sloping forehead, micrognathia, beaked nose in facial dysmorphism, and intellectual disability, some phenotypic heterogeneity was also displayed among different populations and pathogenic variants. For example, four Turkish patients with homozygous c.261 + 1G>C pathogenic variants in CEP152 showed high-arched palate, selective tooth agenesis, enamel hypoplasia, fifth finger clinodactyly, genital anomalies, and simplified gyri. A French individual with the same genotype, however, did not manifest such phenotypes (Kalay et al., 2011). Furthermore, different variants may associate with various other exceptional phenotypes. Compound heterozygous c.1535T>A and c.3346-5T>C results in the additional phenotype of the cleft palate (Hong et al., 2019), while c.2000 A>G/c.4210-4211delGT leads to scoliosis (Kalay et al., 2011). In our case, laryngotracheal malacia and endobronchial inflammation have not been reported previously. Since CEP152 is necessary for centrosome duplication (Kodani et al., 2015), centriole formation (Cizmecioglu et al., 2010), and G2/M transition of the mitotic cell cycle, and has also been reported to interact with CDK5RAP2, WDR62, CEP63, CEP131, PLK4, and CENPJ (Sir et al., 2011), phenotypic heterogeneity in patients with SCKL may be due to diverse proteins affected by specific variants of CEP152. Therefore, the correlation between these phenotypes and the location of the variants in CEP152 along with the associated pathways needs to be further verified by more case analysis and functional studies.
Meanwhile, since the offspring of an affected family has a 25% chance of recurrence risk, it is important for the family of SCKL to undertake genetic testing, counseling, and prenatal diagnosis for the subsequent pregnancy (Gupta et al., 2014; Akkurt et al., 2019). In 2019, Capalbo et al. found four deleterious variants in CEP152 detected by exome sequencing (ES) in 14,125 gamete donors and couples with no genetic diseases in their family history who underwent in vitro fertilization (IVF) treatment, suggesting the carrying rate of the pathogenic variant in CEP152 can be as high as 0.28% (Capalbo et al., 2019). With advances in next-generation sequencing and decreasing costs of this technology, more and more gene panels are applied to preconception/prenatal carrier screening. Considering the severity and high carrier rate of the SCKL, it is necessary to include SCKL-related genes in the gene list for carrier screening. Once screening shows that both of the couple carry the pathogenic variants in the same gene, preimplantation genetic testing for monogenic disorder (PGT-M) or prenatal diagnosis can be used to provide a basis for the selection and clinical management of the couple’s pregnancy. In this study, we recruited a family with suspected SCKL and collected their history of adverse pregnancy. The proband had typical SCKL phenotypes: a typical ‘bird-head’ facial appearance, severe microcephaly, short stature, intellectual disability, dysphasia, caries, and fifth finger clinodactyly. Unfortunately, due to the difficulty in obtaining detailed ultrasound information on the affected fetus, the analysis of the relationship between the genotype and ultrasound phenotype could not be conducted. Here, we present genetic evidence by trio-WES where biallelic pathogenic variants in CEP152 causing Seckel syndrome 5 were detected. In addition, the pathogenicity of the c.1414-14A>G variant was upgraded from VUS to pathogenic by adding functional evidence (applicable to PVS1 null variant interpretation of the ACMG guideline) of single exon 12 skipping, a frameshift variant of the c.1414_1577del (p.Glu472_Ser526delfs*9) of CEP152, which caused the exposure of premature termination codons (PTCs), and was predicted to be degraded by the non-sense-mediated mRNA decay (NMD) pathway because of locating PTC at more than 50–55 nucleotides upstream of the exon–exon junction triggering efficient NMD (Abou Tayoun et al., 2018; Dyle et al., 2020). It was worth noting that the splice-site variant was not located in the classical ±1 or ±2 position, and further splicing analysis confirmed its pathogenicity. All these results provided new insights into splice-site variants and a basis for genetic counseling for the couple’s subsequent pregnancy.
In conclusion, our study identified two novel variants, c.1060C>T and c.1414-14A>G, in a compound heterozygotic form of CEP152 and proved them to be the likely pathogenic factor for the phenotype of the affected family. The non-classical splice-site variant was confirmed to reduce the mRNA expression level and alter the splicing pattern by qPCR and RT-PCR, respectively. These findings expand the pathogenic variant spectrum of CEP152 and provide more information for understanding SCKL and give evidence for the genetic and reproductive counseling of the family.
Data availability statement
The data presented in the study are deposited in the open database Genome Sequence Archive (Chen et al., 2021) in National Genomics Data Center (CNCB-NGDC Members and Partners, 2022), and the accession number is HRA003542.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Board of School of Life Sciences (No. 2021-1-12), Central South University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s) and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
LZ: conceptualization, methodology, and writing—original draft. YT: methodology, data curation, validation, and writing—original draft. HH: visualization and validation. HZ and JW: investigation and resources. DL: funding acquisition and supervision. ZL: writing—review and editing and project administration. LW: funding acquisition and supervision. All authors listed approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (81974240, 81970829, and 82171711), the Science Technology Innovation Program of Hunan Province (2019SK1010 and 2019SK1014), and the Key Grant of Research and Development in Hunan Province (2020DK 2002).
Acknowledgments
The authors deeply thank the patients and family members who participated in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1052915/full#supplementary-material
References
Abou Tayoun, A. N., Pesaran, T., Distefano, M. T., Oza, A., Rehm, H. L., Biesecker, L. G., et al. (2018). Recommendations for interpreting the loss of function pvs1 acmg/amp variant criterion. Hum. Mutat. 39 (11), 1517–1524. doi:10.1002/humu.23626
Akkurt, M. O., Pakay, K., Akkurt, I., Temur, M., and Korkmazer, E. (2019). Prenatal diagnosis of seckel syndrome at 21 weeks' gestation and review of the literature. J. Matern. Fetal. Neonatal Med. 32 (11), 1905–1908. doi:10.1080/14767058.2017.1419467
Andersen, J. S., Wilkinson, C. J., Mayor, T., Mortensen, P., Nigg, E. A., and Mann, M. (2003). Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426 (6966), 570–574. doi:10.1038/nature02166
Capalbo, A., Valero, R. A., Jimenez-Almazan, J., Pardo, P. M., Fabiani, M., Jiménez, D., et al. (2019). Optimizing clinical exome design and parallel gene-testing for recessive genetic conditions in preconception carrier screening: Translational research genomic data from 14, 125 exomes. PLoS Genet. 15 (10), e1008409. doi:10.1371/journal.pgen.1008409
Casper, A. M., Durkin, S. G., Arlt, M. F., and Glover, T. W. (2004). Chromosomal instability at common fragile sites in seckel syndrome. Am. J. Hum. Genet. 75 (4), 654–660. doi:10.1086/422701
Chen, T., Chen, X., Zhang, S., Zhu, J., Tang, B., Wang, A., et al. (2021). The genome sequence archive family: Toward explosive data growth and diverse data types. Genom. Proteom. Bioinf. 19 (4), 578–583. doi:10.1016/j.gpb.2021.08.001
Cizmecioglu, O., Arnold, M., Bahtz, R., Settele, F., Ehret, L., Haselmann-Weiss, U., et al. (2010). Cep152 acts as a scaffold for recruitment of plk4 and cpap to the centrosome. J. Cell. Biol. 191 (4), 731–739. doi:10.1083/jcb.201007107
CNCB-NGDC Members and Partners (2022). Database resources of the national genomics data center, china national center for bioinformation in 2022. Nucleic Acids Res. 50 (D1), D27–D38. doi:10.1093/nar/gkab951
Dinçer, T., Yorgancıoğlu-Budak, G., Ölmez, A., Er, İ., Dodurga, Y., Özdemir Ö, M., et al. (2017). Analysis of centrosome and DNA damage response in plk4 associated seckel syndrome. Eur. J. Hum. Genet. 25 (10), 1118–1125. doi:10.1038/ejhg.2017.120
Donmez, Y. N., Giray, D., Epcacan, S., Goktas, E., and Aypar, E. (2022). Cardiovascular anomalies in seckel syndrome: Report of two patients and review of the literature. Cardiol. Young 32 (3), 487–490. doi:10.1017/s1047951121003097
Dyle, M. C., Kolakada, D., Cortazar, M. A., and Jagannathan, S. (2020). How to get away with nonsense: Mechanisms and consequences of escape from nonsense-mediated rna decay. Wiley Interdiscip. Rev. RNA 11 (1), 1560. doi:10.1002/wrna.1560
Fu, J., Lipinszki, Z., Rangone, H., Min, M., Mykura, C., Chao-Chu, J., et al. (2016). Conserved molecular interactions in centriole-to-centrosome conversion. Nat. Cell. Biol. 18 (1), 87–99. doi:10.1038/ncb3274
Gupta, A., Fazal, T. S., and Arora, R. (2014). Antenatal diagnosis of seckel syndrome. J. Obstet. Gynaecol. India 64 (1), 6–8. doi:10.1007/s13224-012-0339-1
Hong, L., Liu, J., and Wu, B. (2019). Analysis of clinical feature and genetic mutation in a Chinese family affected with seckel syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 36 (6), 595–597. doi:10.3760/cma.j.issn.1003-9406.2019.06.016
Kalay, E., Yigit, G., Aslan, Y., Brown, K. E., Pohl, E., Bicknell, L. S., et al. (2011). Cep152 is a genome maintenance protein disrupted in seckel syndrome. Nat. Genet. 43 (1), 23–26. doi:10.1038/ng.725
Khojah, O., Alamoudi, S., Aldawsari, N., Babgi, M., and Lary, A. (2021). Central nervous system vasculopathy and seckel syndrome: Case illustration and systematic review. Childs Nerv. Syst. 37 (12), 3847–3860. doi:10.1007/s00381-021-05284-8
Kodani, A., Yu, T. W., Johnson, J. R., Jayaraman, D., Johnson, T. L., Al-Gazali, L., et al. (2015). Centriolar satellites assemble centrosomal microcephaly proteins to recruit cdk2 and promote centriole duplication. Elife 4, e07519. doi:10.7554/eLife.07519
Luo, D., Linpeng, S., Zeng, L., Tan, H., Li, Z., and Wu, L. (2019). Molecular genetic study of 59 Chinese oculocutaneous albinism families. Eur. J. Med. Genet. 62 (10), 103709. doi:10.1016/j.ejmg.2019.103709
Majewski, F., and Goecke, T. (1982). Studies of microcephalic primordial dwarfism i: Approach to a delineation of the seckel syndrome. Am. J. Med. Genet. 12 (1), 7–21. doi:10.1002/ajmg.1320120103
McGowan-Jordan, J., Simons, A., and Schmid, M. (2016). ISCN: An International system for human cytogenomic nomenclature. Basel; New York: Karger.
O'driscoll, M., Gennery, A. R., Seidel, J., Concannon, P., and Jeggo, P. A. (2004). An overview of three new disorders associated with genetic instability: Lig4 syndrome, rs-scid and atr-seckel syndrome. DNA Repair (Amst) 3 (8-9), 1227–1235. doi:10.1016/j.dnarep.2004.03.025
O'driscoll, M., Ruiz-Perez, V. L., Woods, C. G., Jeggo, P. A., and Goodship, J. A. (2003). A splicing mutation affecting expression of ataxia-telangiectasia and rad3-related protein (atr) results in seckel syndrome. Nat. Genet. 33 (4), 497–501. doi:10.1038/ng1129
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Seckel, H. P. G. (1960). Bird-headed dwarfs: Studies in developmental anthropology including human proportions. first ed. New York: S. Karger.
Sir, J. H., Barr, A. R., Nicholas, A. K., Carvalho, O. P., Khurshid, M., Sossick, A., et al. (2011). A primary microcephaly protein complex forms a ring around parental centrioles. Nat. Genet. 43 (11), 1147–1153. doi:10.1038/ng.971
Takikawa, K. M., Kikuchi, A., Yokoyama, A., Ono, K., Iwasawa, Y., Sunagawa, S., et al. (2008). Perinatal findings of seckel syndrome: A case report of a fetus showing primordial dwarfism and severe microcephaly. Fetal diagn. Ther. 24 (4), 405–408. doi:10.1159/000167269
Verloes, A., Drunat, S., Gressens, P., and Passemard, S. (2013). “Primary autosomal recessive microcephalies and seckel syndrome spectrum disorders – retired chapter, for historical reference only,” in Gene Reviews® [internet]. Editors M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle, WA: University of Washington), 1993–2022.
Zheng, Y., Mennella, V., Marks, S., Wildonger, J., Elnagdi, E., Agard, D., et al. (2016). The seckel syndrome and centrosomal protein ninein localizes asymmetrically to stem cell centrosomes but is not required for normal development, behavior, or DNA damage response in drosophila. Mol. Biol. Cell. 27 (11), 1740–1752. doi:10.1091/mbc.E15-09-0655
Keywords: Seckel syndrome, microcephaly, CEP152, whole-exome sequencing, mRNA splicing
Citation: Zhang L, Teng Y, Hu H, Zhu H, Wen J, Liang D, Li Z and Wu L (2023) Two novel variants in CEP152 caused Seckel syndrome 5 in a Chinese family. Front. Genet. 13:1052915. doi: 10.3389/fgene.2022.1052915
Received: 24 September 2022; Accepted: 25 November 2022;
Published: 04 January 2023.
Edited by:
Kevin Slep, University of North Carolina at Chapel Hill, United StatesReviewed by:
Afagh Alavi, University of Social Welfare and Rehabilitation Sciences, IranCelia Badenas, Hospital Clinic of Barcelona, Spain
Copyright © 2023 Zhang, Teng, Hu, Zhu, Wen, Liang, Li and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingqian Wu, d3VsaW5ncWlhbkBza2xtZy5lZHUuY24=; Zhuo Li, bGl6aHVvQHNrbG1nLmVkdS5jbg==
†These authors have contributed equally to this work