 Jie Li1,2†
Jie Li1,2† Tao Wang1,2*†Wei Liu3†Danqing Yin4Zhengqing Lai1Guosong Zhang1Kai Zhang1,2
Tao Wang1,2*†Wei Liu3†Danqing Yin4Zhengqing Lai1Guosong Zhang1Kai Zhang1,2 Jie Ji1,2
Jie Ji1,2 Shaowu Yin1,2*
Shaowu Yin1,2*- 1College of Marine Science and Engineering, Jiangsu Province Engineering Research Center for Aquatic Animals Breeding and Green Efficient Aquacultural Technology, Nanjing Normal University, Nanjing, Jiangsu, China
- 2Co-Innovation Center for Marine Bio-Industry Technology of Jiangsu Province, Lianyungang, Jiangsu, China
- 3Institute of Fisheries Science of Nanjing, Nanjing, China
- 4School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, University of Hong Kong, Pokfulam, Hong Kong SAR, China
Pelteobagrus vachelli is a freshwater fish with high economic value, but the lack of genome resources has severely restricted its industrial development and population conservation. Here, we constructed the first chromosome-level genome assembly of P. vachelli with a total length of approximately 662.13 Mb and a contig N50 was 14.02 Mb, and scaffolds covering 99.79% of the assembly were anchored to 26 chromosomes. Combining the comparative genome results and transcriptome data under environmental stress (high temperature, hypoxia and Edwardsiella. ictaluri infection), the MAPK signaling pathway, PI3K-Akt signaling pathway and apelin signaling pathway play an important role in environmental adaptation of P. vachelli, and these pathways were interconnected by the ErbB family and involved in cell proliferation, differentiation and apoptosis. Population evolution analysis showed that artificial interventions have affected wild populations of P. vachelli. This study provides a useful genomic information for the genetic breeding of P. vachelli, as well as references for further studies on fish biology and evolution.
Highlights
1) The first high-quality genome of P. vachelli (contig N50 = 14.02 Mb).
2) Cell proliferation, differentiation and apoptosis play an important role in environmental adaptation of P. vachelli.
3) The environmental adaptation pathways and candidate genes of P. vachelli were identified.
4) Artificial interventions have affected wild populations of P. vachelli.
1 Introduction
Pelteobagrus vachelli (Siluriformes, Bagridae) is an omnivorous freshwater fish that is mainly distributed in the Yangtze River and its tributaries (Li et al., 2015; Zhang et al., 2016a). This fish has been one of the top consumer choices in the domestic freshwater fish market because of the less intermuscular spines, high nutrition, taste, and tenderness (Zhang et al., 2016a). The high market value and consumer demand have promoted the rapid industrialization of P. vachelli farming. This rapid scaling up of the product has helped to meet the market demand however has come up with several problems.
The artificial breeding program and high-density farming have been associated with physiological stresses like high temperature, hypoxia, and bacterial-borne diseases in the P. vachelli population, impacting the fish’s health, quality and quantity of the production (Zhang et al., 2016a; Zhang et al., 2016c; Qin et al., 2018). The natural living environment of flowing water is responsible for the intolerance of P. vachelli to hypoxia. In fact, continuous rainy weather or high breeding density will cause hypoxia death in pond aquaculture and remarkable economic losses in the farmer industry (Zhang et al., 2017a). Water pollution caused by intensive farming will lead to an increase in pathogenic bacteria, causing the outbreak of various diseases. In addition, the high temperature caused by global warming further exacerbates hypoxia and disease outbreaks in aquaculture. With the recent development of sequencing technology, transcriptomics (Zhang et al., 2017b; Qin et al., 2017) and proteomics (Zhang et al., 2017a; Li et al., 2019) have been used to explore the mechanisms of hypoxic stress and innate immunity in P. vachelli; however, the accuracy of omics annotation cannot be guaranteed due to a lack of high-quality reference genomes.
With the rapid development of the industrial scale of P. vachelli, the brood stock on the market can no longer meet the demand for artificial breeding. Prior to 2019, remarkable amounts of P. vachelli were caught in the Yangtze River for seed production, which severely damaged and disturbed its wild population. Studies have shown that the genetic diversity of P. vachelli in the Yangtze River has decreased in recent years (Zheng et al., 2020). Population evolution analysis based on whole genome sequencing (WGS) can help us understand the evolutionary history and current status of its population to formulate corresponding protection measures (Hu et al., 2021).
In this study, we reported the first chromosome-scale genome assembly of P. vachelli. To the best of our knowledge, except for that of Thamnaconus septentrionalis (contig N50 = 22.46 Mb) (Bian et al., 2020), the genome of P. vachelli (contig N50 = 14.02 Mb) has the highest quality among published fish genomes. The comparative genome and population evolution analyses performed herein provided insights into environmental adaptation and population history. Accordingly, the findings of this study can be used as a genetic basis for future biological research on P. vachelli, ultimately providing valuable resources for genetic breeding and population conservation.
2 Materials and methods
2.1 Sample collection and genome sequencing
The XX genotype female Darkbarbel catfish, P. vachelli, was collected from Meishan City, Sichuan Province. After dissecting the fish, muscle tissue was obtained and flash-frozen in liquid nitrogen and stored at −80°C. High-quality genomic DNA was extracted from the muscle using a modified CTAB method (Cota-Sanchez et al., 2006). RNase A (0.1 ml 100 mg/L RNase, 37°C for 30–60 min) was used to remove RNA contaminants. The quality of the DNA was checked using Agilent 2100 Bioanalyzer (California, United States) and high integrity DNA molecules were measured using 1% agarose gel electrophoresis. Sequencing libraries were generated using VAHTS Universal DNA Library Prep Kit for MGI (Vazyme, Nanjing, China) following the manufacturer’s instructions. Index codes were used to cross-index the sequences and samples, that was, the DNA samples were fragmented by sonication and then end-polished, A-tailed, and ligated with the full-length adapter for MGI sequencing followed by PCR amplifcation. The resulting PCR products were purifed (AMPure XP system) and the sequence libraries were analyzed for size distribution by Agilent 2100 Bioanalyzer and quantifed using real-time PCR. A total of one 300–500 bp short-insert libraries and one 40 kb long-insert libraries were prepared. Then, the MGISEQ 2000 platform (BGI, Shenzhen, China) (Chen et al., 2019) and PacBio Sequel II (Menlo Park, CA, United States) (Rhoads and Au, 2015) were employed for whole-genome sequencing. Short reads generated from the MGI platform were used to estimate the genome size, the level of heterozygosity, and repeat content of the genome. Long reads from the PacBio platform were used for genome assembly.
2.2 Estimation of genome features using the k-mer method
The short reads (300–500 bp) from the MGISEQ 2000 platform were quality filtered using HTQC (v1.92.310) (Yang et al., 2013) as described below. First, the adaptors were removed from the sequencing reads. Thereafter, read pairs were excluded if any one end had an average quality lower than 20. The ends of reads were trimmed if the average quality was lower than 20 in the sliding window size of 5 bp. Finally, read pairs with an end shorter than 75 bp were removed. Quality-filtered reads were used for genome size estimation. We generated the 17-mer occurrence distribution of sequencing reads from short libraries using the k-mer method (Guo et al., 2015). The proportion of repeat sequences and the heterozygosity rate of the genome were determined using the GEF (v1.02) (Liu et al., 2013).
2.3 Genome assembly using third-generation long reads
We generated 158.95 Gb subreads using one SMRT cell in the PacBio platform by removing adaptor sequences within sequences. The longest 150X subreads data were used for genome assembly of P. vachelli. The draft assembly of the genome was assembled using NextDenvo (seed_cutoff = 33348; read_cutoff = 1000) (Zhang J. et al., 2020). The assembly results were corrected by Arrow (v2.10) (Wang et al., 2010) using subreads and PILON (v1.2.2) using the second-generation data (Walker et al., 2014). Haplotigs (default parameter) (Kronenberg et al., 2021) was used to remove heterozygous redundancy after error correction.
2.4 Chromosome assembly using Hi-C technology
Muscle from the same sample was used to construct a Hi-C chromatin contact map to enable chromosome-level assembly. To construct the Hi-C library, samples were cross-linked with 1% formaldehyde for 10 min at room temperature and quenched with 0.125 M final concentration glycine for 5 min. The cross-linked cells were subsequently lysed. Endogenous nuclease were inactivated with 0.3% SDS, then chromatin DNA were digested by 100 U MboI (NEB), and marked with biotin-14-dCTP (Invitrogen) and then ligated by 50 U T4 DNA ligase (NEB). After reversing cross-links, the ligated DNA was extracted through QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to manufacturers’ instructions. Purified DNA was sheared to 300- to 500-bp fragments and were further blunt-end repaired, A-tailed and adaptor-added, followed by purification through biotin-streptavidin–mediated pull-down and PCR amplification. Finally, the Hi-C libraries were quantified and sequenced on the or MGI-seq platform. The final library was sequenced on the MGISEQ 2000 platform with 150 paired-ends. Thereafter, the clean read pairs from Hi-C library sequencing were mapped to the polished genome using BWA (v0.7.16) (Xie et al., 2022). Paired reads with mate mapped to a different contig were used to perform the Hi-C-associated scaffolding. Self-ligation, non-ligation, and other invalid reads, such as Start NearRsite, PCR amplification, random breaks, large small fragments, and extreme fragments, were filtered. Thereafter, Juicer (v2.0) (Durand et al., 2016b) and a 3D de novo assembly (3d-DNA, v170123) (Dudchenko et al., 2017) were used to determine the location and direction of each contig. Finally, JuiceBox (v1.1) (Durand et al., 2016a) was applied to correct the contig orientation and to remove suspicious fragments in the contig to unanchored groups by visual inspection.
2.5 Annotation of repetitive sequences
The following methods were combined to identify the repeat contents in the P. vachelli genome: homology-based and de novo prediction. Homology-based analysis: we identified the known TEs within the P. vachelli genome using RepeatMasker (v4.1.0) (Zhang et al., 2022) with the Repbase TE library (Jurka et al., 2005). RepeatProteinMask searches were also conducted using the TE protein database as a query library. De novo prediction: we constructed a de novo repeat library of the P. vachelli genome using RepeatModeler, which automatically executes two core de novo repeat-finding programs, namely RECON (v1.08) (Bao and Eddy, 2002) and RepeatScout (v1.0.5) (Price et al., 2005), to comprehensively conduct, refine, and classify consensus models of putative interspersed repeats for the P. vachelli genome. We also performed a de novo search for long terminal repeat (LTR) retrotransposons against the genome sequences using LTR_FINDER (v1.0.7) (Xu and Wang, 2007). Thereafter, we identified tandem repeats using the Tandem Repeat Finder (TRF) (https://tandem.bu.edu/trf/trf.html), including low-complexity repeats, satellites, and simple repeats. Finally, we merged the library files of the two methods and identify the repeat contents.
2.6 Functional annotation of protein-coding genes
We predicted protein-coding genes of the P. vachelli genome using three methods, including ab initio gene prediction, homology-based gene prediction and RNA-Seq-aided gene prediction. Prior to gene prediction, the assembled genome was hard and soft masked using RepeatMasker (v 4.1.0) (Zhang et al., 2022). We adopted Augustus (v3.2) (Ng et al., 2021) and Genescan (v3.1) (Vidal et al., 2021) to perform ab initio gene prediction. Models used for each gene predictor were trained from a set of high-quality proteins generated from homology annotation result. We used Exonerate (v2.2.0) (Chakraborty et al., 2021) to conduct homology-based gene prediction with the default parameters. To carry out RNA-Seq-aided gene prediction, we first mapped the clean RNA-Seq reads into reference sequences using TopHat (v2.1.1) (Trapnell et al., 2009), and the gene structures were built using Cufflinks (v2.2.1) (Hards et al., 2022). Finally, Maker (3.01.03) (Cantarel et al., 2008) was used to integrate the prediction results of the three methods to predict genes modles. The output included a set of consistent and non-overlapping sequence assemblies, which were used to describe the gene structures.
For gene function annotation, we used BLASTP to align the candidate sequences to the Swissport protein databases, TrEMBL and Kyoto Encyclopedia of Genes and Genomes (KEGG) database with an E-value threshold of 1E-5. The protein domains were annotated using PfamScan and InterProScan based on the InterPro protein databases (http://www.ebi.ac.uk/interpro/) (Sidhu et al., 2020). The motifs and domains within the gene models were identified using the PFAM databases (http://pfam.xfam.org/). Gene Ontology (GO) IDs for each gene were obtained from Blast2GO (Conesa et al., 2005).
2.7 Gene family identification and phylogenetic analysis
To cluster families from protein-coding genes, proteins from the longest transcripts of each gene from P. vachelli and other 16 closely related species (Supplementary Table S6) were extracted and aligned to each other using BLASTp with a maximal E-value of 1E-5. To exclude putative fragmented genes, we filtered out genes with identity less than 30%, coverage less than 50%, and genes encoding protein sequences shorter than 100 bp. The OrthoMCL (v2.0.9) (Li et al., 2003) method was used to cluster genes from these different species into gene families with the parameter of “-inflation 1.5.” The protein sequences of the single-copy ortholog genes were aligned with the MUSCLE (v3.8.31) (Edgar, 2004) program, and the corresponding coding DNA sequence (CDS) alignments were generated and concatenated with the guidance of protein alignment. RAxML (v8.2.11) (Stamatakis, 2014) was used to construct the phylogenetic tree using the maximum likelihood method. Thereafter, the MCMCtree program in the PAML package (v4.8) (Yang, 2007) was used to calculate the divergence time. Eight potential fossil records were downloaded from the TimeTree database (http://www.timetree.org) to calibrate the results.
2.8 Gene family expansion and contraction analysis
Based on the identified gene families and the constructed phylogenetic tree with predicted divergence time of those species, we used CAFE4 (Han et al., 2013) to analyze gene family expansion and contraction. A random birth and death model is proposed in CAFE to study gene gain or loss in gene families across a specified phylogenetic tree. Herein, a conditional p-value was calculated for each gene family, and families with conditional p-value less than 0.05 were considered to have an accelerated rate for gene gain or loss. Such expansion and contraction gene families in P. vachelli (p < 0.05) were mapped to KEGG pathways for functional enrichment analysis, which was conducted using enrichment methods. Functional enrichment analysis of expansion and contraction gene families was performed against the background of all KEGG-annotated genes in the P. vachelli genome. Hypergeometric test algorithms were implemented for the analysis, and the Q-value (FDR, false discovery rate) was calculated to adjust the p-value using the R method p. adjust (https://github.com/StoreyLab/qvalue).
2.9 Transcriptome sequencing of P. vachelli under environmental stress
The experimental fish (19 ± 1.77 g weight, 13 ± 1.36 cm length, 120 days after hatching) were temporarily raised in the circulating water tank (equipped with cooling and heating functions and a volume of 200 L and flow rate of 5 L/min) for 2 weeks, and the formal experiment was carried out after stopping feeding for 2 days. Bacterial infection methods are as follows: To determine the 50% lethal concentration (LC50) o f Edwardsiella ictaluri (Zhejiang Institute of Freshwater Fisheries, Huzhou, China) at 24 h, a total of 30 fish were intraperitoneally administered with 0.1 ml of 103, 104, 1 05, 106, or 107 CFU/ml (Colony Forming Unit, CFU) of E. ictaluri. Fish mortality was monitored every 2 h. The LC50 was determined to be 2.0 × 104 CFU/ml. The experimental group was intraperitoneally injected with 0.1 ml E. ictaluri at a density of 2.0 × 103 CFU/ml (1/10th LC50) and the control group was injected with an equal volume of phosphate buffer solution (PBS, ×1). At 24 h after E. ictaluri challenge, liver tissues were quickly dissected, and the samples included three control groups (BC1, BC2, BC3) and three treatment groups (BT1, BT2, BT3). The experimental methods of high temperature stress were as follows: control groups at 25°C, treatment groups at 33°C, after 72 h, the experimental fish were dissected and the liver tissues of the control groups (WC1, WC2, WC3) and the treatment groups (WT1, WT2, WT3) were obtained. The hypoxic transcriptome data was obtained from our previous experiment (Zhang et al., 2016b) with re-annotating based on the P. vachelli genome. The specific experimental process is as follows: based on the pre-experiment, we chose 0.7 mg/L as the oxygen concentration level for creating a hypoxic condition. Control fish (P0 a, P0 b, P0 c) were removed from three aquaria for immediate liver dissection. Next, the oxygen infiltration and recirculation systems in the three aquaria were closed to initiate the hypoxia experiments. The water was deoxygenated for 30–35 min by bubbling pure nitrogen gas in order to decrease oxygen concentration from 6.8 to 0.7 mg/L. After oxygen concentration was maintained for 4 h by continuous bubbling of nitrogen gas, the experimental fish (P4 a, P4 b, P4 c) were quickly removed for liver dissection. In all of these experiments, the samplings of control fish and experimental fish had three biological replicates, each made up of three different individual liver tissues.
Total RNA was extracted using Trizol reagent (Invitrogen, CA, United States) following the manufacturer’s instructions. RNA quantity and purity were analyzed with Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent, CA, United States) with RIN number >7.0. For cDNA library constructions, Poly(A) RNA was purified from total RNA (5 μg) using poly-T oligo-attached magnetic beads using two rounds of purification. Following purification, the mRNA was fragmented into small pieces using divalent cations under elevated temperature. Then the cleaved RNA fragments were reverse-transcribed to create the final cDNA library in accordance with the protocol for the TruSeq Stranded mRNA Sample Prep Kit (Illumina, San Diego, United States), the average insert size for the paired-end libraries was 300 bp (±50 bp). We then performed the paired-end sequencing on an Illumina Hiseq 4000 (LC Sciences, United States) following the vendor’s recommended protocol. The filtered Clean Data was assembled and annotated based on the P. vachelli genome.
2.10 Population evolution
2.10.1 Sample collection and genome resequencing
Genome resequencing samples were collected from three populations in the upper (SC), middle (HB), and lower reaches (JS) of the Yangtze River (Figure 3A), with five individuals per population. DNA was extracted from the muscle tissue and subjected to 0.8% agarose gel electrophoresis. The qualified DNA was randomly broken into fragments of 350 bp in length, and the 1 µg DNA per sample was used as the input material and sequencing libraries were generated using the VAHTS Universal DNA Library Prep Kit for MGI (Vazyme, Nanjing, China) following the manufacturer’s. The DNA fragments were repaired by ends. Thereafter, poly A tails followed by sequencing adapters were added. The DNA fragments were purified and amplified by PCR to complete the entire library preparation. After the library was constructed, Qubit 2.0 (Invitrogen Ltd., Paisley, United Kingdom) was used for preliminary quantification, and the library was diluted to 1.5 ng/μl. Then Agilent 2100 BioAnalyzer (California, United States) was used to detect the insert size of the library. qRT-PCR was used to accurately quantify the effective concentration of the library to ensure the quality of the library. After the library quality was verified, the MGISEQ 2000 platform was used for PE150 sequencing. The following four main steps comprise the raw data filtering method: 1) filter out reads containing the adapter sequence; 2) remove leading and trailing bases with base quality less than 20; 3) delete reads less than 50 bp in length; and 4) retain only paired reads.
2.10.2 Variation detection and annotation
Clean reads were compared to the reference genome using BWA (v0.7.16) (Xie et al., 2022), and duplicate reads were processed using Picard software (v1.87) (Kambouris et al., 2014). The mapping rate, coverage, and sequencing depth of the filtered data were analyzed. We further filtered the calls using GATK VariantFiltration (Lin et al., 2022) with the following parameters: QD < 2.0, FS > 60.0, MQ < 40.0, MQRankSum < −12.5, ReadPosRankSum < −8.0. Mutational positions, genomic regions, and potential amino acid changes were assessed using ANNOVAR (v2019). The SNP calls for each sample were then combined using GATK CombineGVCFs under default settings. Each SNP position were further assessed and filtered if: 1) minor allele frequency <0.01, 2) samples with missing genotypes >0.2, and 3) sequencing depth <4.
2.10.3 Population genetic structure analysis
All the SNPs that passed filtering in the population were used to construct a phylogenetic tree using treebest (http://treesoft.sourceforge.net/treebest.shtml) based one Neighbor-Joining Algorithm. The final phylogentic tree was plotted using Itol (https://itol.embl.de). Principal Component analysis was performed using GCTA (v1.91.4) (Yang et al., 2011). The input files for GCTA was converted from VCF files using Plink (v1.9) (Purcell et al., 2007). Admixture (v1.3.0) (Ioannidis et al., 2020) was employed to analyze the population structure based on the Bayesian mathematical model and SNP markers to determine the total population structure. The best K value was determined by calculating the cross-validation error, and finally the optimal number of groups in the population.
2.10.4 Analysis of effective population size and population history dynamics
Based on the SNP markers, the linkage imbalance (LD) between markers can be used to accurately estimate the effective population size. The LD level was evaluated using a pair-wise algorithm, and PopLDdecay (v3.3.1) (Wu et al., 2022) was used to plot the LD decay graph. The demographic history of P. vachelli was analysed with the MSMC (v2.0.0) (Schiffels and Wang, 2020). The synonymous mutation rate per base per year was inferred based on formula T = ks/(2λ).
3 Results and discussion
3.1 Genome sequencing, assembly, and annotation
3.1.1 Genome sequencing and assembly
A total of 80.2 Gb clean reads were produced using the MGISEQ 2000 platform (Table 1). Genome size, heterozygosity, and repeat content of the genome are crucial for designing the strategy for PacBio long-read genome sequencing and assembly. Based on short read data and 17-mer analysis (Supplementary Figure S1), we estimated a genome size of 675.72 Mb, a heterozygosity rate of 0.45%, and repeat ratio of 43.31%. Thereafter, ∼158 Gb subreads data were produced for reference genome construction using the PacBio platform (Table 1). After assembly based on subreads data, a ∼663.50 Mb high-quality genome was obtained, with a contig N50 of 16.67 Mb. We also obtained ∼69 Gb Hi-C clean reads, and genome assembly spanned 662.13 Mb with a contig N50 = 14.02 Mb (Table 2) and GC content of 39.5% (Supplementary Figure S2), thereby accounting for 99.79% of the original genome length. Based on Hi-C scaffolding, we anchored the genome to 26 chromosomes (Figure 1B, Supplementary Table S1), which was congruent with the P. vachelli karyotype (2n = 52) (Zhang et al., 2020a). Finally, the assembled genome was evaluated by BUSCO pipeline, the assembly contained 92.7% complete and 1.4% fragmented conserved single-copy ortholog genes (Supplementary Table S2). Such finding indicates that the genome assembly has high coverage and completeness.
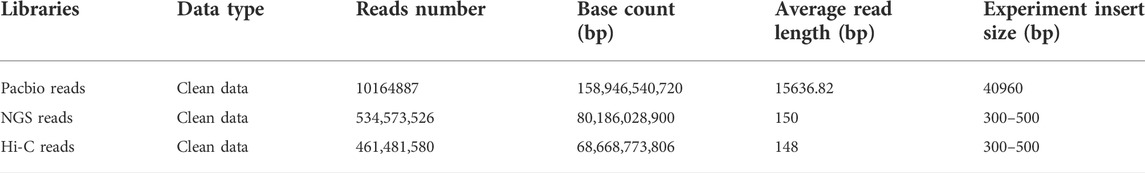
TABLE 1. Statistics of the genome sequencing data.

TABLE 2. Statistics of the P. vachelli genome before and after chromosome level assembly.
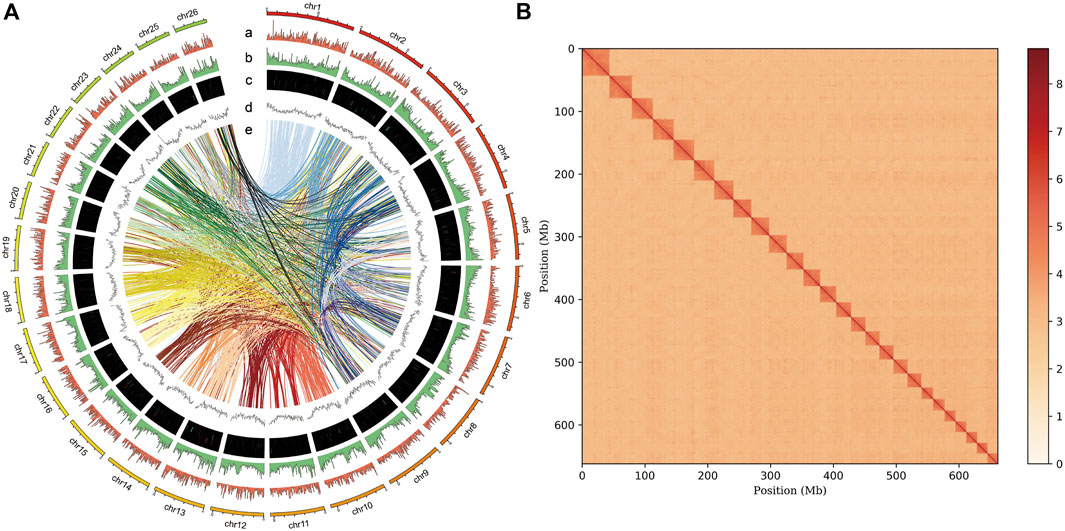
FIGURE 1. Genome assembly of P. vachelli. (A) Characteristics of 26 chromosomes of P. vachelli. Tracks from the outside to the inside represent distribution of gene density (a), repetitive sequence density (b), distribution of non-codling RNAs (ncRNAs) (c), GC content (d) and gene collinearity (e). Band width is proportional to the syntenic block size. (B) Interaction between chromosomes of the P. vachelli scaffolds. The blocks represent the contacts between one location and another. The color reflects the intensity of each contact, with deeper colors representing higher intensity.
3.1.2 Genome annotation
A total of 231.60 Mb of repetitive elements were identified, accounting for 34.90% of the P. vachelli genome (Figure 1A). Notably, this value was lower than that of Lethenteron reissneri (57.25%), Epinephelus lanceolatus (41.01%), and Danio rerio (52.50%), and markedly higher than that of Gasterosteus aculeatus (25.20%), Larimichthys crocea (18.1%), and Oryzias latipes (17.5%). The most abundant TEs were DNA transposons (19.13%), followed by long interspersed elements (LINEs, 8.63%) and long terminal repeats (LTRs, 7.53%) (Supplementary Table S3).
A protein-coding gene set comprising 21,974 genes was predicted by integrating de novo and homology searching methods (Supplementary Table S4). Approximately 97.87% of the protein-coding genes exhibited a significant sequence-level similarity to entries of other species using at least one public database, the quality of which was comparable to that of published high-quality gene sets (Supplementary Table S5). By comparing the distribution of genes, coding sequences (CDS), exon and intron lengths, exon and intron numbers, and the gene and CDS gene content of P. vachelli to those of closely related species, we found that their distributions in the P. vachelli genome were comparable to those of other teleosts (Supplementary Figure S3). Furthermore, 91.7% complete BUSCO genes were successfully identified (Supplementary Table S2). The above results show that our genome annotation is of high integrity and quality for further analysis.
3.2 Comparative genomics analysis
To investigate the phylogenetic relationship of P. vachelli with other species, we compared the genomes of P. vachelli and other published vertebrate species (Figure 2C). Gene family UpSet plot showed the interactive genes between P. vachelli and its relatives fish species, the number of gene families shared by all species was the largest, and P. vachelli had 79 unique gene families (Figure 2A). Further, we performed an intergenomic co-linearity analysis of P. vachelli and its closest relatives, and the results showed the homology of the genomes of P. vachelli, P. fulvidraco, and I. punctatus (Supplementary Figure S4). Additionally, a phylogenetic tree based on single-copy orthologs was constructed (Figure 2C), in which the divergence time of P. fulvidraco and I. punctatus (96.4 Mya) were similar to those presented in other reports (Gong et al., 2018). The phylogenetic tree showed that the divergence time between Pelteobagrus and Ictalurus was approximately 166.9 (155.0–176.6) million years ago (Mya). In addition, the P. fulvidraco was the most closely related to P. vachelli with a divergence time around 40.0 (28.5–54.0) Mya.
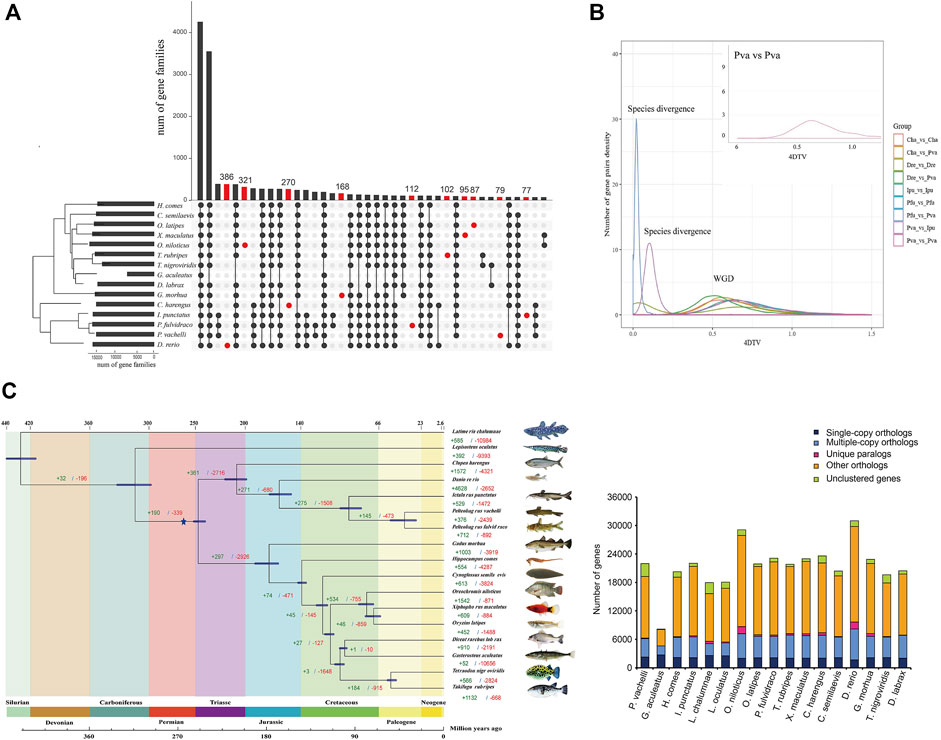
FIGURE 2. Comparative genomics analysis of P. vachelli. (A) An UpSet plot of gene families in P. vachellia and other fishes. The UpSet Plot was drawn using the R package ‘UpSet’. The abscissa of the horizontal bar chart represents the total number of gene families in each species, the ordinate represents the size of gene families, the red dots represent unique gene families, and the line segment represents the intersection relationship. (B) 4DTv prediction of the timing of WGD. Different colors indicate the comparisons between different species. Cha = Clupea harengus, Dre = Danio rerio, Ipu = Ictalurus punctatus, Pfu = Pelteobagrus fulvidraco, Pva = Pelteobagrus vachelli. (C) Inferred phylogenetic tree with 1,106 single-copy orthologs of 17 species identified by RAxML. Blue star represented whole-genome duplication (WGD) events. The green numbers represent the expansion gene familys and red numbers represent the contraction gene familys.
Whole-genome duplication (WGD) events are an important driving force for species evolution (Walden et al., 2020). Ray-finned fish evolution spanned more than 400 million years (Near et al., 2012). In addition to the two rounds of WGD that occurred at the root of the vertebrate lineage (VGD1 and VGD2, 320–350 Mya) (Dehal and Boore, 2005), teleost fish experienced a third round of WGD (TSGD) (Bayes et al., 2017). Additional WGDs have also been described in the teleost lineage including the salmonid-specific WGD (SaGD) that occurred about 100 Mya (Larhammar and Risinger, 1994). Previous studies have shown that the peak around 0.6 in 4DTv represents the TSGD, such as zebrafish and carp (4DTv = 0.58) (Xu et al., 2014). In this study, the 4DTv results showed a peak around 0.6 (Figure 2B), which was consistent with the 4DTv of TSGD. Furthermore, the WGD (Figure 2C) in this study (around 270 Mya) was consistent with the TSGD time (226–316 Mya) (Pasquier et al., 2016) reported in previous studies, indicating the WGD belongs to TGSD.
3.3 Analysis of environmental adaptation evolution in P. vachelli
3.3.1 Gene family comparison
The expansion and contraction of gene families may be one of the most important factors for phenotypic diversity and evolutionary adaptation to the environment (Harris and Hofmann, 2015). Based on the phylogenetic tree, a gene family comparison analysis was performed, and 386 expanded and 245 contracted gene families were identified in P. vachelli (Supplementary Table S7). In the expanded gene families, KEGG enrichment showed that environmental information processing related pathways were significantly enriched (p < 0.05), such as ErbB signaling pathway, MAPK signaling pathway, PI3K-Akt signaling pathway and apelin signaling pathway (Supplementary Table S7). In addition, ErbB (receptor tyrosine-protein kinase erbB) family (EGFR, ErbB2, ErbB3, ErbB4) were repeatedly enriched in these pathways. Further, we performed an evolutionary analysis of the ErbB family in P. vachelli and other fishes according to the expansion gene family (Figure 3A). The results showed that only the P. vachelli and D. rerio had completed ErbB family in the evolution process, while other species had different degrees of deletion. There were also differences in gene copy number across species, which might be due to differences in the pressures they face to survive.
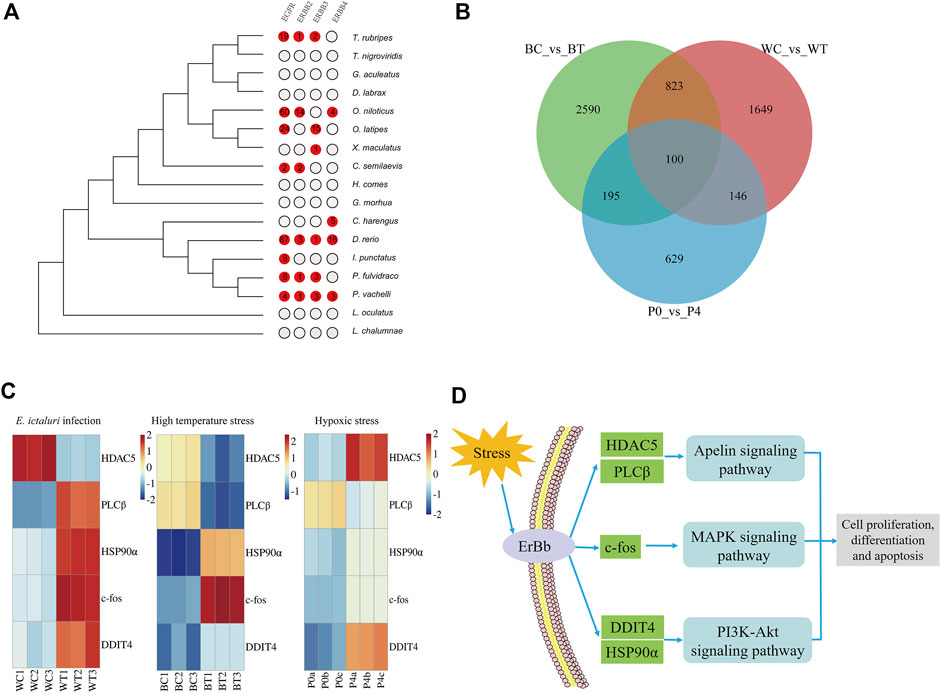
FIGURE 3. Evolutionary analysis of environmental adaptation in P. vachelli. (A) Evolution of the ErbB family in P. vachelli and other fishes. (B) Veen diagram of significantly differentially expressed genes in all transcriptomes. BC vs. BT represent control vs. treatment groups in E. ictaluri infection transcriptome, WC vs. WT represent control vs. treatment groups in high temperature stress transcriptome, P0 vs. P4 represent control vs. treatment groups in hypoxic stress transcriptome. (C) Transcriptome expression of candidate genes for environmental adaptation evolution in P. vachelli. Color gradients represent gene expression level of control group and treatment group. (D) Candidate genes and pathways for environmental adaptation evolution in P. vachelli.
3.3.2 Transcriptome analysis of environmental stress
We performed transcriptome sequencing of P. vachelli under bacterial infection, hypoxia and high temperature stress respectively (Supplementary Figure S5). In bacterial infection transcriptome, a total of 18,629 unique genes were identified from six complementary DNA libraries (BC1, BC2, BC3, BT1, BT2, BT3), including 3,708 differentially expressed genes (DEGs) (foldchange > 2, p < 0.05). KEGG analysis showed that 19 pathways were significantly enriched (FDR <0.05), which were mainly involved in immunity and disease, proteolysis and synthesis, and metabolism (Supplementary Table S8). In hypoxia stress transcriptome, 18,634 unique genes were identified from six DNA libraries (P0a, P0b, P0c, P4a, P4b, P4c), including 1,070 DEGs (foldchange >2, p < 0.05). KEGG analysis showed that 14 pathways were significantly enriched (FDR <0.05), which were mainly involved in carbohydrate metabolism, endocrine system, endocrine and metabolic disease (Supplementary Table S8). Notably, the HIF-1 signaling pathway, a key pathway in the hypoxia response, was also significantly enriched (Li et al., 2022). In high temperature stress transcriptome, 18,832 unique genes were identified from six DNA libraries (WC1, WC2, WC3, WT1, WT2, WT3), including 2,718 DEGs (foldchange >2, p < 0.05). KEGG analysis showed that 14 pathways were significantly enriched (FDR < 0.05), which were mainly involved in carbohydrate metabolism, immunity and disease, proteolysis and synthesis (Supplementary Table S8). In conclusion, based on the KEGG analysis of DEGs, we found that the response pathways of P. vachelli under different environmental stresses was specific. The response pathways under bacterial infection and high temperature stress were similar (mainly involved in immunity and diseases, proteolysis and synthesis), while endocrine related pathways under hypoxia stress may played a role in hypoxia response. Endocrine is an important and tightly regulated system for maintaining body homeostasis. Current studies have shown that hypoxia can induce the dysfunction of endocrine organs, resulting in dysregulation of body homeostasis.
To further explore candidate genes for environmental adaptation, we conducted a venn diagram analysis based on the DEGs of the three transcriptomes, the results showed that they shared 100 genes. (Figure 3B, Supplementary Table S8). Further analysis revealed that among the 100 genes, only five genes [histone deacetylase 5 (HDAC5), phosphatidylinositol phospholipase C beta (PLCβ), proto-oncogene c-Fos (c-fos), DNA damage-inducible transcript 4 protein-like (DDIT4), heat shock protein HSP 90-alpha (HSP90α)] were functional genes in the key KEGG pathway of above expanded gene families (Figure 3C). HDAC5 and PLCβ were involved in apelin signaling pathway, c-fos was involved in MAPK signaling pathway, DDIT4 and HSP90α were involved in PI3K-Akt signaling pathway. According to the KEGG database, it was worth noting that these pathways were interconnected by the ErbB family and involved in the regulation of cell proliferation, differentiation and apoptosis (Figure 3D) (Yarden and Sliwkowski, 2001; Liu et al., 2015; Zhang P et al., 2020), indicating these physiological processes play a role in environmental adaptation of P. vachelli. In addition, these five genes had different expression patterns under bacterial infection, hypoxia and high temperature stress, suggesting that unique adaptive mechanisms to different kinds of environmental stress in P. vachelli (Figure 3C).
3.4 Evolution analysis of the geographical population of P. vachelli
Population evolution research is used to analyze population genetic diversity, genetic structure, gene exchange, species formation mechanism, population evolution dynamics, and other biological issues by obtaining SNP variation information for each subgroup of natural population (Gui et al., 2020; Nematollahi et al., 2021). Based on the natural geographical distribution characteristics of P. vachelli, we conducted population evolution analysis of the populations in the upper reaches (SC), middle reaches (HB), and lower reaches (JS) of the Yangtze River (Figure 4A). Based on the analysis of effective population size and population history dynamics, during the evolution process, a difference was found in the effective population size of different geographic populations of P. vachelli (Figure 4B, Supplementary Figure S6). The population size of JS and SC declined sharply in 100–200 and 40–50 thousand years ago, respectively, while that of HB did not fluctuate significantly in the evolutionary process. The selection pressure caused by the change of natural environment is an important reason affecting the population size. According to relevant information, a sea level rise event occurred in the lower reaches of the Yangtze River approximately 130 thousand years ago (Otto-Bliesner et al., 2006). We speculate that the rising sea level disrupted the habitat of P. vachelli in the lower reaches of the Yangtze River, leading to a significant reduction in its population size.
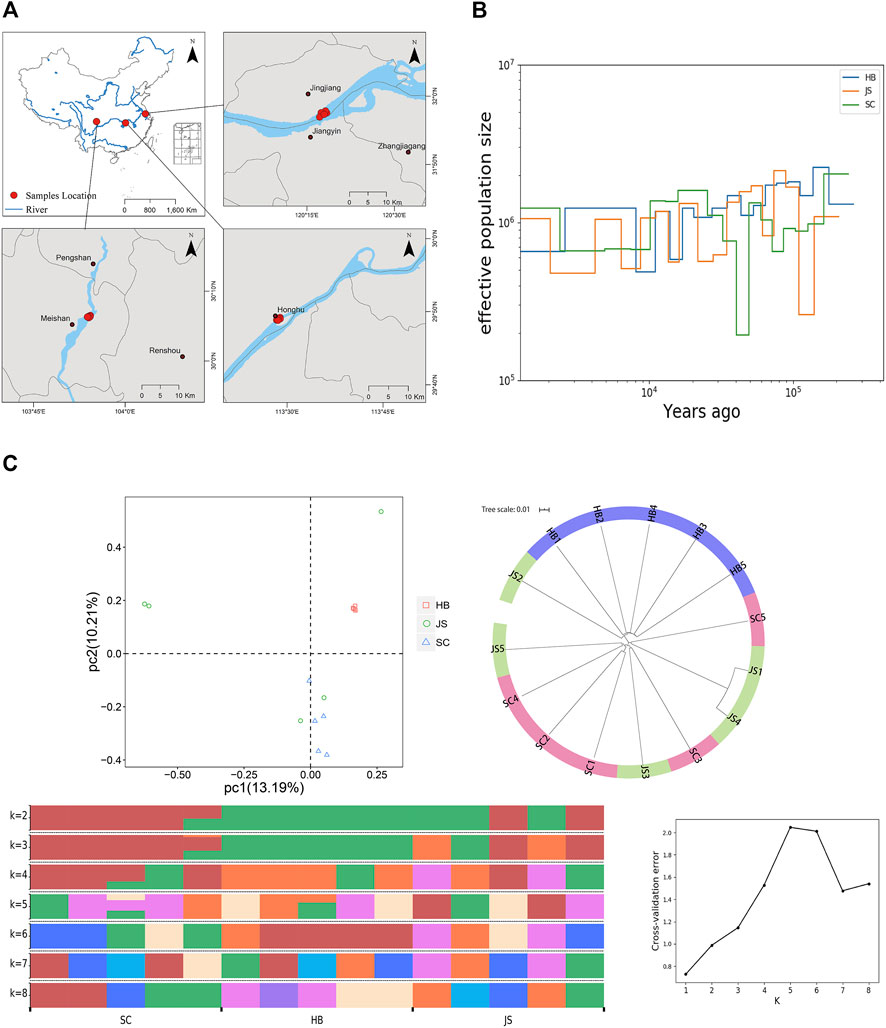
FIGURE 4. Population evolution of P. vachelli. (A) Geographical distribution of P. vachelli. (B) Demographic histories of P. vachelli. The blue line represents Hubei P. vachelli population (HB), the orange line represents Jiangsu P. vachelli population (JS), and the green line represents Sichuan P. vachelli population (SC). (C) Population structure of P. vachelli, including principal component analysis (PCA), phylogenetic tree and genetic structure analysis (different colors indicate different genetic information).
Principal component analysis (PCA) showed that all HB individuals were clustered together, where SC groups and part of JS individuals clustered together (Figure 4C). In addition, the phylogenetic tree analysis also showed that the SC population and JS population had a close genetic relationship (Figure 4C). Furthermore, we conducted population genetic structure analysis (best K value = 5), and the results showed that JS population was mainly composed of SC and HB population, which was especially obvious when K = 3 or K = 2 (Figure 4C). In conclusion, the genetic relationship among the P. vachelli populations contradicted their natural geographical distribution, populations in the upper (SC) and lower reaches (JS) of the Yangtze River showed closer genetic relationship. In recent years, the breeding parents and released fry of P. vachelli in the lower reaches of the Yangtze River are mainly sourced from Sichuan, China. Therefore, we speculated that the artificial intervention had affected the P. vachelli population genetic structure in the Yangtze River.
4 Conclusion
In this study, we established the first high-quality reference genome for P. vachelli, which can serve as a basic database for the protection of its germplasm resources. Based on the 4DTv analysis, we found a WGD at about 270 Mya, which was consistent with the reported TSGD time. The MAPK signaling pathway, PI3K-Akt signaling pathway and apelin signaling pathway play an important role in the evolution of environmental adaptation in P. vachelli, which were interconnected by the ErbB family and involved in the regulation of cell proliferation, differentiation and apoptosis. Population evolution analysis showed that artificial interventions have affected P. vachelli wild populations.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal study was reviewed and approved by the All experiments were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals in China. The protocols of this study were approved by the Institutional Animal Care and Use Committee of Nanjing Normal University (Grant No. SYXK 2015-0028, Jiangsu).
Author contributions
T.W. and S.Y. conceived and designed the study. All experiments and data analysis were performed by J.L, T.W, W.L, D.Y, Z.L, G.Z, K.Z, J.J. The manuscript wrote by J.L. and T.W. S.Y. and W.L. advised on development related content. All authors read and approved the final manuscript.
Funding
This study was supported by the “JBGS” Project of Seed Industry Revitalization in Jiangsu Province (JBGS(2021)034). Creation Project of Major New Species of Agriculture in Jiangsu Province (PZCZ201742). National Natural Science Foundation of China (32102760). Natural Science Foundation of the Higher Education Institutions of Jiangsu Province, China (No.18KJB240001, 20KJD240001). The Earmarked Fund for Jiangsu Agricultural Industry Technology System (JATS(2021)417). Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX21_1362).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1050192/full#supplementary-material
References
Bao, Z. R., and Eddy, S. R. (2002). Automated de novo identification of repeat sequence families insequenced genomes. Genome Res. 12 (8), 1269–1276. doi:10.1101/gr.88502
Bayes, A., Collins, M. O., Reig-Viader, R., Gou, G., Goulding, D., Izquierdo, A., et al. (2017). Evolution of complexity in the zebrafish synapse proteome. Nat. Commun. 8, 14613. doi:10.1038/ncomms14613
Bian, L., Li, F. H., Ge, J. L., Wang, P. F., Chang, Q., Zhang, S. N., et al. (2020). Chromosome-level genome assembly of the greenfin horse-faced filefish (Thamnaconus septentrionalis) using Oxford Nanopore PromethION sequencing and Hi-C technology. Mol. Ecol. Resour. 20 (4), 1069–1079. doi:10.1111/1755-0998.13183
Cantarel, B. L., Korf, I., Robb, S. M. C., Parra, G., Ross, E., Moore, B., et al. (2008). Maker: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18 (1), 188–196. doi:10.1101/gr.6743907
Chakraborty, A., Mahajan, S., Jaiswal, S. K., and Sharma, V. K. (2021). Genome sequencing of turmeric provides evolutionary insights into its medicinal properties. Commun. Biol. 4 (1), 1193. doi:10.1038/s42003-021-02720-y
Chen, J. Y., Li, X. S., Zhong, H. B., Meng, Y. H., and Du, H. L. (2019). Systematic comparison of germline variant calling pipelines cross multiple next-generation sequencers. Sci. Rep. 9, 9345. doi:10.1038/s41598-019-45835-3
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21 (18), 3674–3676. doi:10.1093/bioinformatics/bti610
Cota-Sanchez, J. H., Remarchuk, K., and Ubayasena, K. (2006). Ready-to-use DNA extracted with a CTAB method adapted for herbarium specimens and mucilaginous plant tissue. Plant Mol. Biol. Rep. 24 (2), 161–167. doi:10.1007/bf02914055
Dehal, P., and Boore, J. L. (2005). Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 3 (10), e314–e1708. doi:10.1371/journal.pbio.0030314
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356 (6333), 92–95. doi:10.1126/science.aal3327
Durand, N. C., Robinson, J. T., Shamim, M. S., Machol, I., Mesirov, J. P., Lander, E. S., et al. (2016a). Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3 (1), 99–101. doi:10.1016/j.cels.2015.07.012
Durand, N. C., Shamim, M. S., Machol, I., Rao, S. S. P., Huntley, M. H., Lander, E. S., et al. (2016b). Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3 (1), 95–98. doi:10.1016/j.cels.2016.07.002
Edgar, R. C. (2004). Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32 (5), 1792–1797. doi:10.1093/nar/gkh340
Gong, G. R., Dan, C., Xiao, S. J., Guo, W. J., Huang, P. P., Xiong, Y., et al. (2018). Chromosomal-level assembly of yellow catfish genome using third-generation DNA sequencing and Hi-C analysis. Gigascience 7 (11), 1–9. doi:10.1093/gigascience/giy120
Gui, F. R., Lan, T. M., Zhao, Y., Guo, W., Dong, Y., Fang, D. M., et al. (2020). Genomic and transcriptomic analysis unveils population evolution and development of pesticide resistance in fall armyworm Spodoptera frugiperda. Protein Cell 13 (7), 513–531. doi:10.1007/s13238-020-00795-7
Guo, L. T., Wang, S. L., Wu, Q. J., Zhou, X. G., Xie, W., and Zhang, Y. J. (2015). Flow cytometry and K-mer analysis estimates of the genome sizes of Bemisia tabaci B and Q (Hemiptera: Aleyrodidae). Front. Physiol. 6, 144. doi:10.3389/fphys.2015.00144
Han, M. V., Thomas, G. W. C., Lugo-Martinez, J., and Hahn, M. W. (2013). Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30 (8), 1987–1997. doi:10.1093/molbev/mst100
Hards, K., Cheung, C. Y., Waller, N., Adolph, C., Keighley, L., Tee, Z. S., et al. (2022). An amiloride derivative is active against the F1Fo-ATP synthase and cytochrome bd oxidase of Mycobacterium tuberculosis. Commun. Biol. 5 (1), 166. doi:10.1038/s42003-022-03110-8
Harris, R. M., and Hofmann, H. A. (2015). Seeing is believing: Dynamic evolution of gene families. Proc. Natl. Acad. Sci. U. S. A. 112 (5), 1252–1253. doi:10.1073/pnas.1423685112
Hu, Y., Colantonio, V., Muller, B. S. F., Leach, K. A., Nanni, A., Finegan, C., et al. (2021). Genome assembly and population genomic analysis provide insights into the evolution of modern sweet corn. Nat. Commun. 12 (1), 1227. doi:10.1038/s41467-021-21380-4
Ioannidis, A. G., Blanco-Portillo, J., Sandoval, K., Hagelberg, E., Miquel-Poblete, J. F., Moreno-Mayar, J. V., et al. (2020). Native American gene flow into Polynesia predating Easter Island settlement. Nature 583 (7817), 572–577. doi:10.1038/s41586-020-2487-2
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., and Walichiewicz, J. (2005). Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110 (1-4), 462–467. doi:10.1159/000084979
Kambouris, M., Maroun, R. C., Ben-Omran, T., Al-Sarraj, Y., Errafii, K., Ali, R., et al. (2014). Mutations in zinc finger 407 ZNF407 cause a unique autosomal recessive cognitive impairment syndrome. Orphanet J. Rare Dis. 9, 80. doi:10.1186/1750-1172-9-80
Kronenberg, Z. N., Rhie, A., Koren, S., Concepcion, G. T., Peluso, P., Munson, K. M., et al. (2021). Extended haplotype-phasing of long-read de novo genome assemblies using Hi-C. Nat. Commun. 12 (1), 1935. doi:10.1038/s41467-020-20536-y
Larhammar, D., and Risinger, C. (1994). Molecular genetic aspects of tetraploidy in the common carp Cyprinus carpio. Mol. Phylogenet. Evol. 3 (1), 59–68. doi:10.1006/mpev.1994.1007
Li, J., Zhang, X. Y., Xu, J. J., Pei, X. Y., Wu, Z. W., Wang, T., et al. (2019). iTRAQ analysis of liver immune-related proteins from darkbarbel catfish (Pelteobagrus vachelli) infected with Edwardsiella ictaluri. Fish. Shellfish Immunol. 87, 695–704. doi:10.1016/j.fsi.2019.01.036
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13 (9), 2178–2189. doi:10.1101/gr.1224503
Li, M., Chen, L. Q., Li, E. C., Yu, N., Ding, Z. L., Chen, Y. L., et al. (2015). Growth, immune response and resistance to Aeromonas hydrophila of darkbarbel catfish, Pelteobagrus vachelli (Richardson), fed diets with different linolenic acid levels. Aquac. Res. 46 (4), 789–800. doi:10.1111/are.12236
Li, X. H., Ling, C., Wang, Q. X., Feng, C., Luo, X. Z., Sha, H., et al. (2022). Hypoxia stress induces tissue damage, immune defense, and oxygen transport change in gill of silver carp (Hypophthalmichthys molitrix): Evaluation on hypoxia by using transcriptomics. Front. Mar. Sci. 9, 900200. doi:10.3389/fmars.2022.900200(
Lin, Y. L., Chang, P. C., Hsu, C., Hung, M. Z., Chien, Y. H., Hwu, W. L., et al. (2022). Comparison of GATK and DeepVariant by trio sequencing. Sci. Rep. 12 (1), 1809. doi:10.1038/s41598-022-05833-4
Liu, B., Shi, Y., Yuan, J., Hu, X., Zhang, H., Li, N., et al. (2013). Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quant. Biol. 35, 62–67. doi:10.48550/arXiv.1308.2012
Liu, Q. F., Yu, H. W., Sun, L. L., You, L., Tao, G. Z., and Qu, B. Z. (2015). Apelin-13 upregulates Egr-1 expression in rat vascular smooth muscle cells through the PI3K/Akt and PKC signaling pathways. Biochem. Biophys. Res. Commun. 468 (4), 617–621. doi:10.1016/j.bbrc.2015.10.171
Near, T. J., Eytan, R. I., Dornburg, A., Kuhn, K. L., Moore, J. A., Davis, M. P., et al. (2012). Resolution of ray-finned fish phylogeny and timing of diversification. Proc. Natl. Acad. Sci. U. S. A. 109 (34), 13698–13703. doi:10.1073/pnas.1206625109
Nematollahi, S., Panahborhani, N., and Koolivand, D. (2021). Molecular characterization and population evolution analysis of Watermelon mosaic virus isolates on cucurbits of Northwest Iran. 3 Biotech. 11 (2), 43. doi:10.1007/s13205-020-02609-3
Ng, K. K. S., Kobayashi, M. J., Fawcett, J. A., Hatakeyama, M., Paape, T., Ng, C. H., et al. (2021). The genome of Shorea leprosula (Dipterocarpaceae) highlights the ecological relevance of drought in aseasonal tropical rainforests. Commun. Biol. 4 (1), 1166. doi:10.1038/s42003-021-02682-1
Otto-Bliesner, B. L., Marsha, S. J., Overpeck, J. T., Miller, G. H., Hu, A. X., and Mem, C. L. I. P. (2006). Simulating arctic climate warmth and icefield retreat in the last interglaciation. Science 311 (5768), 1751–1753. doi:10.1126/science.1120808
Pasquier, J., Cabau, C., Nguyen, T., Jouanno, E., Severac, D., Braasch, I., et al. (2016). Gene evolution and gene expression after whole genome duplication in fish: The PhyloFish database. Bmc Genomics 17, 368. doi:10.1186/s12864-016-2709-z
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, I351–I358. doi:10.1093/bioinformatics/bti1018
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M., Bender, D., et al. (2007). Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi:10.1086/519795
Qin, C. J., Gong, Q., Wen, Z. Y., Yuan, D. Y., Shao, T., and Li, H. T. (2018). Molecular characterization and expression of toll-like receptor 5 genes from Pelteobagrus vachellii. Fish. Shellfish Immunol. 75, 198–207. doi:10.1016/j.fsi.2018.02.002
Qin, C. J., Gong, Q., Wen, Z. Y., Yuan, D. Y., Shao, T., Wang, J., et al. (2017). Transcriptome analysis of the spleen of the darkbarbel catfish Pelteobagrus vachellii in response to Aeromonas hydrophila infection. Fish. Shellfish Immunol. 70, 498–506. doi:10.1016/j.fsi.2017.09.042
Rhoads, A., and Au, K. F. (2015). PacBio sequencing and its applications. Genomics Proteomics Bioinforma. 13 (5), 278–289. doi:10.1016/j.gpb.2015.08.002
Schiffels, S., and Wang, K. (2020). MSMC and MSMC2: The multiple sequentially markovian coalescent. Methods Mol. Biol. 2090, 147–166. doi:10.1007/978-1-0716-0199-0_7
Sidhu, N. S., Pruthi, G., Singh, S., Bishnoi, R., and Singla, D. (2020). Genome-wide identification and analysis of GRAS transcription factors in the bottle gourd genome. Sci. Rep. 10 (1), 14338. doi:10.1038/s41598-020-71240-2
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi:10.1093/bioinformatics/btu033
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: Discovering splice junctions with RNA-seq. Bioinformatics 25, 1105–1111. doi:10.1093/bioinformatics/btp120
Vidal, M., Konigseder, F., Giehr, J., Schrempf, A., Lucas, C., and Heinze, J. (2021). Worker ants promote outbreeding by transporting young queens to alien nests. Commun. Biol. 4 (1), 515. doi:10.1038/s42003-021-02016-1
Walden, N., German, D. A., Wolf, E. M., Kiefer, M., Rigault, P., Huang, X. C., et al. (2020). Nested whole-genome duplications coincide with diversification and high morphological disparity in Brassicaceae. Nat. Commun. 11 (1), 3795. doi:10.1038/s41467-020-17605-7
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. Plos One 9 (11), e112963. doi:10.1371/journal.pone.0112963
Wang, K., Li, M. Y., and Hakonarson, H. (2010). Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Wu, D. Y., Shen, E. H., Jiang, B. W., Feng, Y., Tang, W., Lao, S. T., et al. (2022). Genomic insights into the evolution of Echinochloa species as weed and orphan crop. Nat. Commun. 13 (1), 689. doi:10.1038/s41467-022-28359-9
Xie, P., Tang, S., Chen, C., Zhang, H., Yu, F., Li, C., et al. (2022). Natural variation in Glume Coverage 1 causes naked grains in sorghum. Nat. Commun. 13 (1), 1068. doi:10.1038/s41467-022-28680-3
Xu, P., Zhang, X. F., Wang, X. M., Li, J. T., Liu, G. M., Kuang, Y. Y., et al. (2014). Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat. Genet. 46 (11), 1212–1219. doi:10.1038/ng.3098
Xu, Z., and Wang, H. (2007). LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi:10.1093/nar/gkm286
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). Gcta: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. doi:10.1016/j.ajhg.2010.11.011
Yang, X., Liu, D., Liu, F., Wu, J., Zou, J., Xiao, X., et al. (2013). Htqc: A fast quality control toolkit for Illumina sequencing data. Bmc Bioinforma. 14, 33. doi:10.1186/1471-2105-14-33
Yang, Z. H. (2007). Paml 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24 (8), 1586–1591. doi:10.1093/molbev/msm088
Yarden, Y., and Sliwkowski, M. X. (2001). Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2 (2), 127–137. doi:10.1038/35052073
Zhang, G. S., Li, J., Zhang, J. J., Liang, X., Wang, T., and Yin, S. W. (2020a). A high-density SNP-based genetic map and several economic traits-related loci in Pelteobagrus vachelli. Bmc Genomics 21 (1), 700. doi:10.1186/s12864-020-07115-7
Zhang, G. S., Mao, J. Q., Liang, F. F., Chen, J. W., Zhao, C., Yin, S. W., et al. (2016a). Modulated expression and enzymatic activities of Darkbarbel catfish, Pelteobagrus vachelli for oxidative stress induced by acute hypoxia and reoxygenation. Chemosphere 151, 271–279. doi:10.1016/j.chemosphere.2016.02.072
Zhang, G. S., Yin, S. W., Mao, J. Q., Liang, F. F., Zhao, C., Li, P., et al. (2016b). Integrated analysis of mRNA-seq and miRNA-seq in the liver of Pelteobagrus vachelli in response to hypoxia. Sci. Rep. 6, 22907. doi:10.1038/srep22907
Zhang, G. S., Yin, S. W., Wang, Y. Y., Li, L., Wang, X. L., Ding, Y. D., et al. (2016c). The effects of water temperature and stocking density on survival, feeding and growth of the juveniles of the hybrid yellow catfish from Pelteobagrus fulvidraco (♀) × Pelteobagrus vachelli (♂). Aquac. Res. 47 (9), 2844–2850. doi:10.1111/are.12734
Zhang, G. S., Zhang, J. J., Wen, X., Zhao, C., Zhang, H. Y., Li, X. R., et al. (2017a). Comparative iTRAQ-based quantitative proteomic analysis of Pelteobagrus vachelli liver under acute hypoxia: Implications in metabolic responses. Proteomics 17 (17-18), 1700140. doi:10.1002/pmic.201700140
Zhang, G. S., Zhao, C., Wang, Q. T., Gu, Y. C., Li, Z. C., Tao, P. F., et al. (2017b). Identification of HIF-1 signaling pathway in Pelteobagrus vachelli using RNA-seq: Effects of acute hypoxia and reoxygenation on oxygen sensors, respiratory metabolism, and hematology indices. J. Comp. Physiol. B 187 (7), 931–943. doi:10.1007/s00360-017-1083-8
Zhang, J., Fu, X. X., Li, R. Q., Zhao, X., Liu, Y., Li, M. H., et al. (2020). The hornwort genome and early land plant evolution. Nat. Plants 6 (2), 107–118. doi:10.1038/s41477-019-0588-4
Zhang, L., He, J., Tan, P. P., Gong, Z., Qian, S. Y., Miao, Y. Y., et al. (2022). The genome of an apodid holothuroid (Chiridota heheva) provides insights into its adaptation to a deep-sea reducing environment. Commun. Biol. 5 (1), 224. doi:10.1038/s42003-022-03176-4
Zhang, P., Wang, A. P., Yang, H. P., Ai, L., Zhang, H. J., Wang, Y. M., et al. (2020). Apelin-13 attenuates high glucose-induced calcification of MOVAS cells by regulating MAPKs and PI3K/AKT pathways and ROS-mediated signals. Biomed. Pharmacother. 128, 110271. doi:10.1016/j.biopha.2020.110271
Keywords: Pelteobagrus vachelli, genomics, chromosomal assembly, population history, environmental adaptation
Citation: Li J, Wang T, Liu W, Yin D, Lai Z, Zhang G, Zhang K, Ji J and Yin S (2022) A high-quality chromosome-level genome assembly of Pelteobagrus vachelli provides insights into its environmental adaptation and population history. Front. Genet. 13:1050192. doi: 10.3389/fgene.2022.1050192
Received: 21 September 2022; Accepted: 01 November 2022;
Published: 14 November 2022.
Edited by:
Wenqin Wang, Shanghai Normal University, ChinaReviewed by:
Wanfei Liu, Agricultural Genomics Institute at Shenzhen (CAAS), ChinaBaohua Chen, Zhejiang University, China
Upendra Raj Bhattarai, Harvard University, United States
Copyright © 2022 Li, Wang, Liu, Yin, Lai, Zhang, Zhang, Ji and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Wang,c2Vhd2F0ZXI4OEAxMjYuY29tShaowu Yin,eWluc2hhb3d1QDE2My5jb20=
†These authors have contributed equally to this work