
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 04 January 2023
Sec. Genomics of Plants and the Phytoecosystem
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1050040
 Dan Liu1,2†
Dan Liu1,2† Kai Qu2†
Kai Qu2† Yangchen Yuan3,8†Zhiheng Zhao4Ying Chen5
Yangchen Yuan3,8†Zhiheng Zhao4Ying Chen5 Biao Han1
Biao Han1 Wei Li2
Wei Li2 Yousry A. El-Kassaby6Yangyang Yin7
Yousry A. El-Kassaby6Yangyang Yin7 Xiaoman Xie1*Boqiang Tong1*Hongshan Liu8*
Xiaoman Xie1*Boqiang Tong1*Hongshan Liu8*Clematis is one of the large worldwide genera of the Ranunculaceae Juss. Family, with high ornamental and medicinal value. China is the modern distribution centre of Clematis with abundant natural populations. Due to the complexity and high morphological diversity of Clematis, the genus is difficult to classify systematically, and in particular, the phylogenetic position of the endangered Clematis acerifolia is highly controversial. The use of the mitochondrial complete genome is a powerful molecular method that is frequently used for inferring plants phylogenies. However, studies on Clematis mitogenome are rare, thus limiting our full understanding of its phylogeny and genome evolution. Here, we sequenced and annotated the C. acerifolia mt genome using Illumina short- and Nanopore long-reads, characterized the species first complete mitogenome, and performed a comparative phylogenetic analysis with its close relatives. The total length of the C. acerifolia mitogenome is 698,247 bp and the main structure is multi-branched (linear molecule 1 and circular molecule 2). We annotated 55 genes, including 35 protein-coding, 17 tRNA, and 3 rRNA genes. The C. acerifolia mitogenome has extremely unconserved structurally, with extensive sequence transfer between the chloroplast and mitochondrial organelles, sequence repeats, and RNA editing. The phylogenetic position of C. acerifolia was determined by constructing the species mitogenome with 24 angiosperms. Further, our C. acerifolia mitogenome characteristics investigation included GC contents, codon usage, repeats and synteny analysis. Overall, our results are expected to provide fundamental information for C. acerifolia mitogenome evolution and confirm the validity of mitochondrial analysis in determining the phylogenetic positioning of Clematis plants.
Clematis acerifolia Maxim of the Clematis genus in the Ranunculaceae family is a perennial shrub endemic to Beijing area and Taihang Mountains, China (Editorial Committee of flora of China, 1980). The species is characterized by its unique flowering (early spring flowering with large beautiful flowers) and habitat (growing on cliff faces, hence the name cliff flower). The species has narrow distribution with sparse populations and is threatened by Parthenocissus quinquefolia infestation and a range of human influences (Pang et al., 2022), that made it endangered and listed as a Grade II protected wild plant in 2021 (National Forestry and Grassland Administration (NFGA), 2021). The species unique flowing characteristics and its rare occurrence increased its ornamental and scientific values (Yuan et al., 2021). C. acerifolia was discovered by Karl Maximovich in the late 19th century (Maximowicz, 1879); however, the species suffers from limited research efforts. As the national germplasm survey continued to advance and the ornamental value of C. acerifolia continued to be explored, the species has gradually gained scholars attention with a series of morphological classification (Wang and Li, 2005), community characteristics (Yuan et al., 2021), population genetics (Lopez-Pujol et al., 2003), and ecological niches (Pang et al., 2022) studies.
Clematis is one of the world’s great genera, with over 350 known species, widely distributed on all continents except Antarctica and in the five major temperature zones, with greatest diversity in warm temperate and mountainous regions (Tamura, 1995). Clematis has been cultivated since the 16th century and more than 3, 500 horticultural cultivars have been named (Tamura, 1987; Grey-Wilson, 2000; Johnson, 2001; Lehtonen et al., 2016), occupying an important place in Japanese and Western gardens due to its high ornamental value (Christenhusz, 2000), while in China the species is widely known for its medicinal value (Zhang et al., 2018). However, the Clematis genera classification has been notoriously difficult, as its species are morphologically variable due to the genus complexity as well as the different characteristics emphasized by each classification system (Xie et al., 2011). In 1879, Maximovich published the original description of C. acerifolia taxonomy (Maximowicz, 1879). Based on the species flowering and leaf clusters traits, Volume 28 of the Flora of China classified C. acerifolia into Sect. Cheiropsis as a single species within Subsect. Acerifoliae; however, other morphological traits were different from those of Sect. Cheiropsis (Editorial Committee of flora of China, 1980; Yan et al., 2016). Geographically, plants of Sect. Cheiropsis are distributed in China, occurring in the south-western to central-western parts, whereas the range of C. acerifolia is far from Sect. Cheiropsis species, suggesting that C. acerifolia may not be closely related to Sect. Cheiropsis (Mu and Xie, 2011). Thus, the phylogenetic position of C. acerifolia remains to be highly controversial.
The chloroplast and nuclear genomes of Clematis are well sequenced and provided valuable information for their phylogeny (Yan et al., 2016; Xiang et al., 2019; Wang et al., 2021), evolution (Sheng et al., 2014), and conservation (Choi et al., 2021). However, it has been reported that the chloroplast genomes provided insufficient phylogenetic information to distinguish between closely related sugarcane cultivars, due to the cultivars’ recent origin and the genome conserved sequences (Evans et al., 2019). In contrast, the mitochondrial genomes (mitogenome) of plants are larger (Sloan et al., 2012), more plastic (Turnel et al., 2003; Rubinstein et al., 2013; Huang et al., 2022), and may contain increased phylogenetic signals (Duminil and Besnard, 2021). For instance, one of the main advantages of the mitogenome as a phylogenetic marker stem from the fact that its 36 genes have different rates of substitution, with some having higher rates than others, allowing relatively recent divisions to be tracked, and those having slower rates of substitution, deemed useful for elucidating deeper relationships (Saccone et al., 1999; Cho et al., 2004). In essence, mitogenomes can provide solutions for a very wide range of phylogenetic depths, from the timing of shallow divergence between populations of individual species to deep divergence within entire clades (Feng et al., 2021). The mitogenome has often been shown to help elucidate previously intractable phylogenies, clarifying the relationships between groups of phylogenetic difficulties that rapid radiation invalidates other markers. Other advantages of the mitogenome include: the wealth of genetic information it contains; its short binding time due to its haploid and matrilineal inheritance (very few species are biparental) (Moore, 1995; Breton et al., 2007); its relatively low recombination rate (Dong et al., 2018); and the clear homology of its coding genes (Stern and Palmer, 1984). In addition to sequence information, the mitogenome gene sequence (which will be discuss later in more details) is sometimes used as corroborating evidence in phylogenetics.
To date, more than 9,500 plant organelle genomes have been submitted to the NCBI’s (National Center for Biotechnology Information) organelle genome resource, but despite this, mitogenomes have rarely been studied in Clematis, with only 499 complete plant mitogenomes, less than one-tenth of those of chloroplasts, which is closely related to the complex structure of mitogenomes and the difficulty of their assembly. The development of genomics and sequencing technologies has provided new opportunities to address this issue, and third-generation sequencing allows access to large reads (Kumar et al., 2019), facilitating the assembly and annotation of plant organelle genomes. Xiang et al. (2019) completed the assembly of three chloroplast genomes of C. acerifolia, Clematis smilacifolia and Clematis uncinata, which included only minimal information on the organelle genome. The chloroplast genomes of the native Korean Clematis brachyura and Clematis trichotoma were later analyzed in more depth by Choi et al. (2021) as well. However, the evolution of mitochondria in Clematis remains unanswered. Deciphering this last unknown genetic material (mitogenome) is crucial to understanding the evolution and genomic resources of Clematis. Unfortunately, very little information is known about the genome of Clematis, and the mitogenome of Clematis has never been reported, which seriously hampers a comprehensive understanding of its genomic evolution.
In this study we characterized the first mitogenome of Clematis, and the complete mitogenome of C. acerifolia was sequenced and assembled using Nanopore and Illumina. The aims of this study were to: 1) determine the molecular characteristics of C. acerifolia mitogenome, 2) contribute to the understanding of the evolution of the C. acerifolia organelle genome through computational analysis of its GC content, codon usage preference, repeats, intracellular gene transfer analysis, RNA editing site prediction, and synteny analysis, 3) explore the evolutionary relationships between the Clematis species based on the protein-coding genes (PCGs) of the mitogenome, compared with the genomes of other angiosperms, and 4) synteny analysis was performed on closely related species of Clematis to investigate the effectiveness of mitogenome analysis in determining the phylogenetic position of clematis plants.
In May 2022, leaves of well-grown living C. acerifolia from 8-year-old plants were collected from the Hongyashan State-owned Forest Farm, Yi County, Baoding City, Hebei Province (N39°20′50″, E115°30′39″), and the collected young fresh leaves were stored in liquid nitrogen for reserve. Total plant genomic DNA and the remaining portion of the plant specimen (barcode number sdf1003248) were stored at the Shandong Forest and Grass Germplasm Resource Centre (code qytxl2022hys09), and total DNA was obtained following the steps of the blood/cell/tissue genomic DNA extraction kit (TIANamp Genoic DNA Kit) from Tiangen.
We used both the Nanopore GridION sequencing platform (Oxford Nanopore Technology, Oxford Science Park) and the Illumina Novaseq 6000 platform for sequencing and library construction, and obtained raw sequence data. Clean data were obtained by using Trimmomatic (Bolger et al., 2014). Here, we removed low-quality sequences, including sequences with a quality value of Q < 19 that accounted for more than 50% of the total bases and sequences in which more than 5% bases were "N."
We used a hybrid Illumina and Nanopore strategy to assemble the C. acerifolia mitogenome. Plant mitochondrial assemblies were first performed using the default parameters of GetOrganelle software (default parameters: v1.7.5) on second-generation DNA sequencing data to obtain a graphical representation of the mitochondrial genome (Jin et al., 2020). The C. acerifolia mitogenome was then visualized using Bandage software and single stretches of the chloroplast and nuclear genomes were manually removed (Wick et al., 2015). Finally, the Nanopore data was compared to the graphical mitogenome fragments using BWA software and the resulting Nanopore data was used to resolve the repetitive sequence regions of the graphical plant mitogenome (Li and Durbin, 2009). The multi-branched C. acerifolia mitogenome was finally obtained by the above method. We used Geseq to annotate the mitogenome of C. acerifolia (Tillich et al., 2017), and Arabidopsis thaliana (NC_037304.1) and Aconitum kusnezoffii (NC_053920.1) were selected as the reference genome for the protein-coding genes (PCGs) of the mitogenome. The tRNA and rRNA of the mitogenome were annotated using tRNAscan-SE and BLASTN (Lowe and Eddy, 1997; Chen et al., 2015), respectively. When errors were made in the annotation of the mitogenome, Apollo was used to manually correct the errors (Lewis et al., 2002). The annotation of the mitogenome of C. acerifolia has been uploaded to GenBank (Acession number: ON674117 and ON674118) and Figshare (doi: https://doi.org/10.6084/m9.figshare.21299757.v1).
The protein-coding sequences of the genome were extracted using Phylosuite (Zhang et al., 2020). The PCGs of the mitogenome were analyzed for codon preference using Mega 7.0 and RSCU values were calculated (Kumar et al., 2016). The neutral plot analysis and ENC-plot analysis were used to perform codon preference analysis. The neutral plot analysis is based on the codon 3 GC content (GC3) as the horizontal coordinate and the average of codon 1 and 2 GC content (GC12) as the vertical coordinate, the correlation between GC12 and GC3 can determine the influencing factors of codon preference. When GC12 is significantly correlated with GC3, codon preference is influenced by mutation, and vice versa by natural selection; ENC-plot analysis was performed by establishing a coordinate system with GC3 as the horizontal coordinate and ENC as the vertical coordinate, and then adding the ENC standard curve to the coordinate system (Wang et al., 2022). If the gene points are distributed along or close to the standard curve, the codon preference of the gene is affected by mutation only, and conversely, if the gene points are far below the standard curve, the codon preference of the gene is affected by selection.
We used (MISA https://webblast.ipk-gatersleben.de/misa/) (Beier et al., 2017), TRF (https://tandem.bu.edu/trf/trf.unix.help.html) and REPuter web server (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) to identify repetitive sequences including microsatellite sequence repeats, tandem repeats and scattered repeats (Benson, 1999; Stefan et al., 2001). The results were visualized using the Circos package (Zhang et al., 2013). In addition, we generated the edited mRNA sequences of PCGs manually, and then calculated the codon usage again. Codon usage before and after RNA editing was used for comparison.
The chloroplast genome was assembled and annotated using GetOrganelle and CPGAVAS2 (Shi et al., 2019; Jin et al., 2020), respectively. Homologous fragments were analyzed using BLASTN (Chen et al., 2015), and the results were visualized using the Circos package (Zhang et al., 2013). Prediction of RNA editing events based on the online website PREP suit (http://prep.unl.edu/) (Mower, 2009).
Based on sequence similarity, the C. acerifolia mitogenome was mapped with multiple synteny of closely related species using MCscanX (Wang et al., 2012). Pairwise comparisons of dot plots were generated and conserved co-linear blocks were plotted (Katoh et al., 2019). Mauve 2.3.1 was used for further comparative analysis of the collinearity of the mitogenomes of two closely related species (Anemone maxima and A. kusnezoffii) (Darling et al., 2004). Mitogenomes of closely related species were selected and downloaded (https://www.ncbi.nlm.nih.gov/) based on affinity, and then shared genes were extracted using PhyloSuite (Zhang et al., 2020), multiple sequence alignment analysis was performed using MAFFT with a bootstrap value of 1,000 (Katoh and Standley, 2013), and phylogenetic analysis using MRBAYES (Huelsenbeck and Ronquist, 2001). The results of the phylogenetic analysis were visualized in ITOL software (Letunic and Bork, 2019).
We sequenced DNA samples of C. acerifolia using Illumina and Nanopore sequencing platforms to obtain basic data on mitogenome assembly. Among them, Nanopore raw data was 10. 14 Gb, N 50 is 20, 564 bp and Illumina raw data was 13.50 Gb (Supplementary Tables S1, S2). First, we obtained a sketch of the C. acerifolia mitogenome assembled from Illumina data, containing a total of 136 nodes that were linked to each other to form overlapping regions, and resolved these repetitive regions in turn by selecting the most appropriate pathway supported by the ONT long read data (Figure 1). This resulted in two independent C. acerifolia mitochondrial DNA molecules containing 7 contigs (numbered according to their length), one of which has a closed-loop structure, while the other has a multi-branching structure with a variety of different potential conformations. In order to include all contigs as much as possible, we unfolded them in the order contig3-contig5-contig7-contig4-contig5-contig7-contig6-contig2. There is a dynamic transformation of mitochondrial DNA conformation in plants due to repeat-mediated reasons, so we emphasized that the pathway unraveled here is not unique and that the pathway represents only one of these cases.
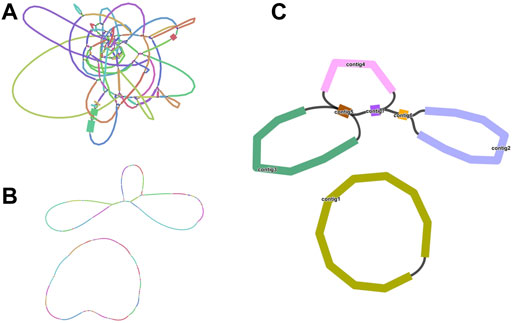
FIGURE 1. Branched conformation of C. acerifolia mitogenome. (A): Sketch of the mitogenome of C. acerifolia, containing 136 nodes. (B): The mitogenome sketch obtained after selecting the best path supported by the long read data of ONT and resolving the individual repeat regions. (C): Mitogenome mapping of C. acerifolia, with circular DNA molecules comprising contig1 and linear DNA molecules consisting of contig2, contig3, contig4, contig5, contig6 and contig7.
C.acerifolia mitogenome showed a multi-branched structure with a total size of 698, 247 bp and a GC content of 46.91%, consisting of a linear molecule (Molecule1, 425, 973 bp) and a circular molecule (Molecule2, 272, 274 bp) with GC contents of 46.82 and 47.06%, respectively (Figure 2). Fifty-five genes were annotated, including 35 protein-coding genes (PCGs), 17 tRNA genes (trnC-GCA, trnE-UUC, trnK-UUU, trnM-CAU, trnP-UGG, and trnQ-UUG are multi-copy genes), and three rRNA genes (Table 1). The PCGs include 24 unique mitochondrial core and 11 non-core genes. There are seven main categories of core genes, including five ATP synthase genes (atp1, atp4, atp6, atp8, and atp9); nine NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9); four ubiquinol cytochrome c reductase genes (ccmB, ccmC, ccmFc, and ccmFn); 3 cytochrome C oxidase genes (cox1, cox2, and cox3); 1 maturation enzyme gene (matR); 1 panthenol-cytochrome C reductase gene (cob), and 1 membrane transport protein gene (mttB). The non-core genes consist of three ribosomal large subunit genes (rpl2, rpl5, and rpl10) and eight ribosomal small subunit genes (rps1, rps3, rps4, rps11, rps12, rps13, rps14, and rps19). In Molecule1, nad2 and nad5 contained 4 introns, ccmFC, rps3, rps4, rps11, trnQ-UUG contained 1 intron, and there is trans splicing in the nad1, nad2 and nad5 genes, while in Molecule2, nad4 and nad7 have 3 and 4 introns, respectively, while cox2, trnE-UUC and trnQ-UUG all have one intron.
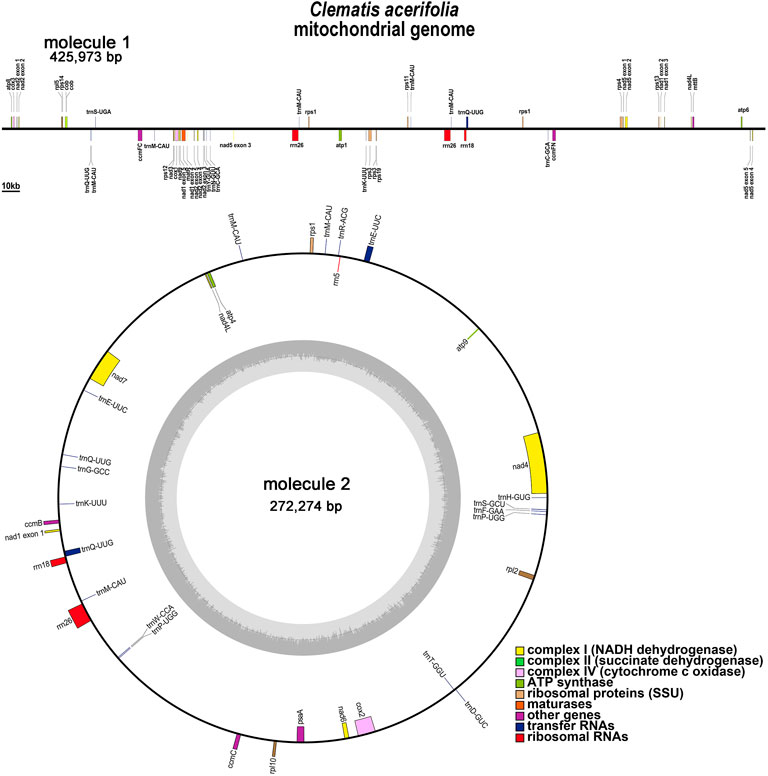
FIGURE 2. C. acerifolia mitogenome gene map. Genes shown on the outside and inside of the circle are transcribed clockwise andcounterclockwise, respectively. The dark gray region in the inner circle depicts GC content.
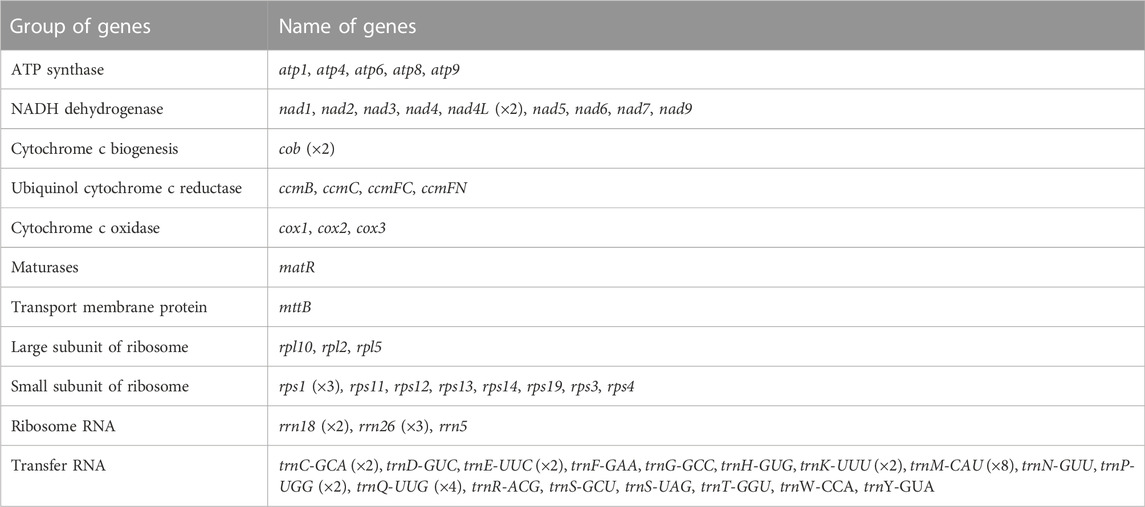
TABLE 1. Gene composition in the C. acerifolia.
Codon usage preference is usually considered to be the result of the tendency to develop relative equilibrium within cells during species evolution, and codons with a relative synonymous codon usage (RSCU) greater than 1 are considered to be used by amino acid preference. As shown in Figure 3A, the codon preference analysis of 35 PCGs revealed that GCU (Ala), UAA (End), CAA (Gln), CAU (His), and UAU (Tyr) were the most common codons, with Ala having the highest RSCU value of 1.58 for the GCU codon in mitochondrial PCGs (Table S3). There were 26 codons with RSCU values greater than 1. Except for two codons, AUG and UGG, other codons ending in A/U (T) bases accounted for 92.3%. Except for the methionine (AUG) and tryptophan (UGG) start codons with RSCU values of 1, there is a broad codon usage preference for PCGs in C. acerifolia mitogenome.
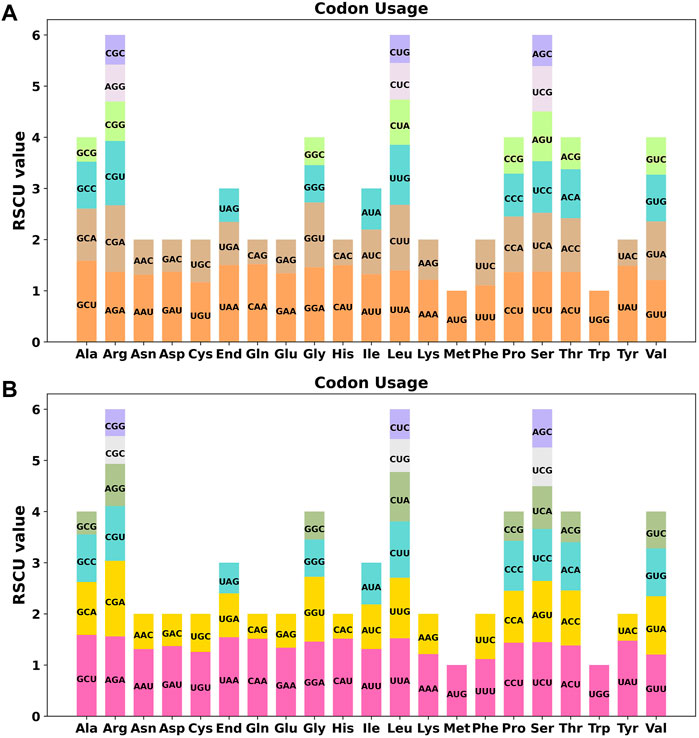
FIGURE 3. C. acerifolia mitogenome relative synonymous codon usage (RSCU). (A) Before RNA editing. (B) After RNA editing. Codon families are shown on the x-axis. RSCU values are the number of times a particular codon is observed relative to the number of times that codon would be expected for a uniform synonymous codon usage.
Neutrality plot analysis allows preliminary determination of the factors affecting codon preference. Supplementary Figure S1A shows that GC12 was between 0.3462–0.5254 and GC3 was between 0.2533–0.5799. Codon GC3 was significantly higher than GC12 for most genes, and GC12 and GC3 correlation analysis was significantly correlated (Supplementary Table S4), with most genes near the diagonal but some genes slightly further away from the diagonal, indicating that C. acerifolia mitogenome codon usage preference, although influenced by natural selection, is more influenced by gene mutations. ENC-plot can further determine the factors affecting codon preference. Most of the gene sites are distributed near the standard curve (Supplementary Figure S1B), which means that the effect of mutational pressure plays a dominant role in the formation of codon preference.
We used PREP (predictive RNA editors for plants) suit to identify RNA edits for the 35 PCGs in the C. acerifolia mitogenome (Figure 4). A total of 676 potential RNA editing sites were identified for the 35 PCGs at a cutoff value = 0.2 criterion, and all were clip C to U edits. Nad4 gene identified 57 RNA editing sites, the highest number of edits among all mitochondrial genes. This was followed by the ccmFN and ccmB genes, each had 40 RNA editing events. The atp9 gene was predicted to have only one RNA editing event, the least of all genes. The comparative analysis of codon usage before and after RNA editing showed that the preference of most codons did not change (Figure 3B and Supplementary Table S3), but we also noticed that some amino acids such as AGA (Arg), CGA (Arg), UAA (End), CCU (Pro), CCC (Pro) and UCU (Ser) were significantly enhanced after the RNA editing event.
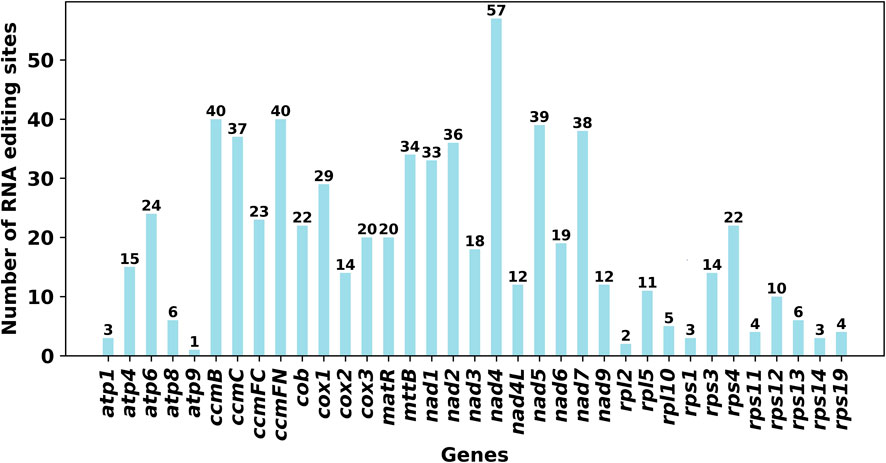
FIGURE 4. Number of RNA editing sites predicted by individual PCGs in mitochondria.
There are a large number of repeats in biological cell DNA sequences including tandem repeats (TRs) and interspersed repeats (TEs), which can be classified by size as large (LR, >500 bp), medium (IntR, 50–500 bp), and short (SR, <50 bp) repeats (Gualberto and Newton, 2017). Microsatellite repeats are a special kind of presence among TRs, which can better reflect species’ genetic structure and genetic diversity changes (Li et al., 2019). We found 126 and 81 SSRs in molecule 1 (linear) and molecule 2 (circular), respectively (Figure 5, SupplementaryTable S7 and Supplementary Table S10). In molecule 1 (linear), most SSRs were in tetrameric form, accounting for 32.54% (41) of the total SSRs, with AAGA and GAAG being the most common types of tetramers. The least number of hexamers (3); among the 28 monomeric SSRs, adenine (A) monomeric repeats were the most abundant (15), accounting for 53.57% of the monomeric SSRs. The tetrameric form of SSRs was also the most abundant in molecule 2 (circular), accounting for 37.04% (30) of the total SSRs, with two of each of the four types CATT, CAAG, TAGA, and AAGG in the tetramer and only one of the other types of tetramers. In contrast the pentamer was the least with only one. SSRs in monomeric and dimeric forms accounted for 44.44% of the total SSRs. Thymine (T) monomeric repeats accounted for 56.25% (9) of the 16 monomeric SSRs. Among the two mitochondrial molecules, TA repeat sequences were the most common type of dimeric SSRs, accounting for 30.77 and 25.00% of the dimeric SSRs, respectively.
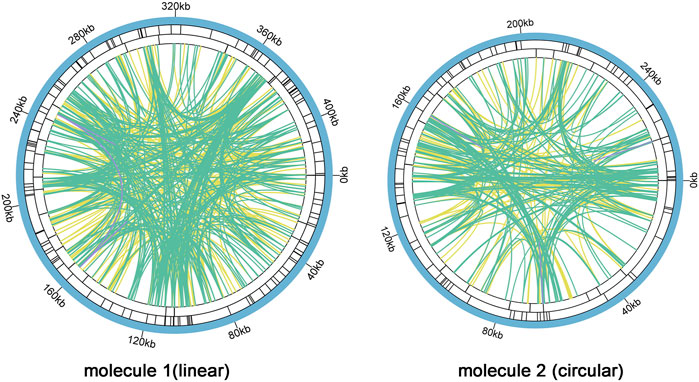
FIGURE 5. C. acerifolia mitogenome repeatedsequence diagram. Two mitochondrial molecules were analyzed separately forrepetitive sequences (one linear and one circular), taking the linear molecule 1 as an example. For example, the colored lines on the innermost circle connect the two repetitive sequences of TEs, the green line represents Palindromic Match (P), the yellow line represents Forward Match (F), and the purple line represents Reverse Match (R). The black line on the second circle represents TRs, and the black line on the outermost circle represents SSRs. molecule 2 and so on.
Analysis of TRs and TEs of the mitogenome revealed 24 TRs with greater than 61% match and length between 9 and 50 bp in M1 (Supplementary Table S5); 680 pairs of TEs with length greater than or equal to 30, including 335 pairs of Forward Match, 342 pairs of Palindromic Match and 3 pairs of Reverse Match, and no Complement Match detected (Supplementary Table S6). The longest Forward Match was 11,827 bp. A total of 16 TRs in M2 with 81% or more match and length between 3 and 39 bp (Supplementary Table S8); 447 pairs of TEs with length greater than or equal to 30, including 201 pairs of Forward Match, 241 pairs of Palindromic Match 241 pairs, Reverse Match 3 pairs, and Complement Match 2 pairs were deteected (Supplementary Table S9). The longest Forward Match is 243 bp.
According to sequence similarity analysis (Figure 6 and Supplementary Table S11), a total of 28 fragments were homologous to the mitochondrial and chloroplast genomes, with a total length of 25, 049 bp. In addition, five of the 28 homologous fragments exceeded 1, 000 bp, with fragments 12 and 13 being the longest at 4, 643 bp. By annotation of these homologous sequences, 13 complete genes were also found on 28 homologous fragments, including 2 PCGs (petL and petG), 9 tRNA genes (trnN-GUU, trnA-UGC, trnI-GAU, trnN-GUU, trnA-UGC, trnI-GAU, trnN-GUU, trnA-UGC, trnI-GAU, and trnN-GAU). TrnR-ACG, trnW-CCA, trnP-UGG, trnD-GUC, trnH-GUG, trnM-CAU) and two rRNA genes (rrn5S and rrn4.5 S).
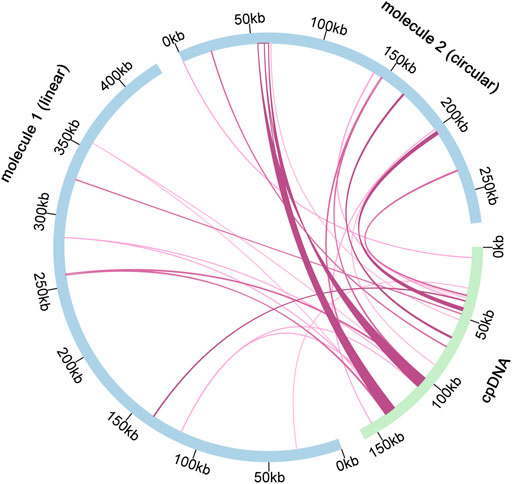
FIGURE 6. Schematic representation of gene transfers between chloroplast and mitogenome in C. acerifolia. The blue arcs in the diagram represent the mitogenome, the green arcs represent the chloroplast genome, and the rosy red lines between the arcs correspond to the genomic fragments that are homologous.
To understand the evolutionary status of C. acerifolia, based on 39 conserved mitochondrial PCGs for seven orders of angiosperms (Ranunculales, Proteales, Trochodendrales, Vitales, Ericales, Caryophyllales, and Zygophyllales), were constructed as phylogenetic trees, and Zygophyllum fabago and Tribulus terrestris of the Zygophyllales were set as outgroups (see Supplementary Table S12, for mitogenome abbreviations and accession numbers). As shown Figure 7 Figure 8, all seven taxa of the studied order showed good clustering, and this phylogenetic tree strongly supports the clustering of C. acerifolia, A. kusnezoffii and A. maxima into one group, with C. acerifolia being more closely related to A. maxima and belonging to the Ranunculaceae of Ranunculales.
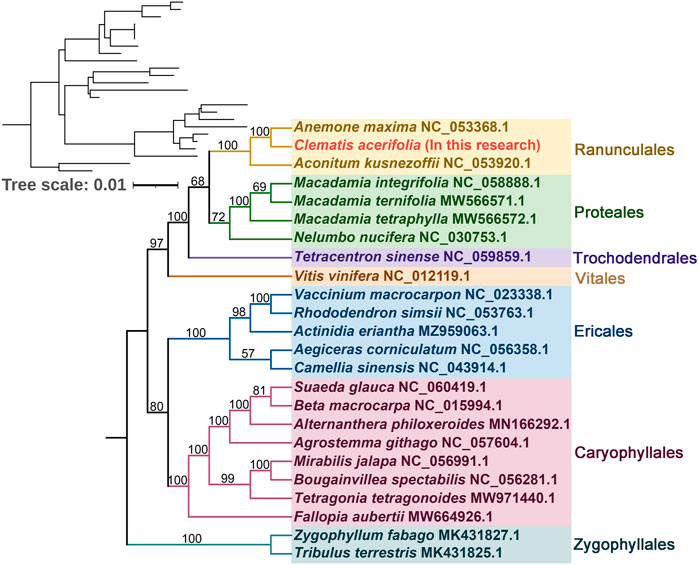
FIGURE 7. Phylogenetic tree of 24 angiosperms based on the sequences of 39 conserved mitochondrial PCGs. Zygophyllum fabago and Tribulus terrestris were selected as outgroups. The number at each node is the bootstrap probability.
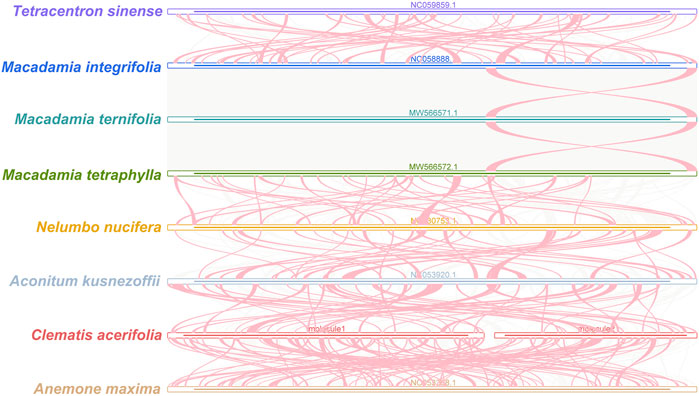
FIGURE 8. C. acerifolia mitogenome synteny. Bars indicated the mitogenomes, and the ribbons showed the homologous sequences between the adjacent species. The red areas indicate where the inversion occurred, the gray areas indicate regions of good homology. Common blocks less than 0.5 kb in length are not retained, and regions that fail to have a common block indicate thatthey are unique to the species.
Plant mitogenomes are extremely conserved in terms of the number and types of functional genes as well as their sequences, but functional genes vary greatly in the location and order of mitogenomes of different species (Tan and Pang, 2013). By comparing the genome sequences of some closely related species, large co-linear blocks can be obtained, and these regions include very rich homology information, so the degree of co-linearity can be used to measure the kinship and evolutionary distance between species, as well as to discover unknown genes and improve the quality of genome annotation (McCouch, 2001). Based on sequence similarity, we mapped Multiple Synteny Plot of C. acerifolia with seven closely related species. A large number of homologous co-linear blocks were detected between C. acerifolia and its closely related species, especially with A. maxima of Anemone. The dot plot analysis showed that there were only sporadic collinear regions between the three mitogenomes of C. acerifolia, A. maxima and A. kusnezoffii, showing poor collinearity (Figures 9A,B). Similar results were obtained by Mauve collinearity analysis (Figure 9C). We found that the co-linear blocks were not arranged in the same order between different mitogenomes, which means that the C. acerifolia mitogenome has undergone extensive genomic rearrangements, and the C. acerifolia mitogenome is extremely unconserved in structure. In addition, it is particularly noteworthy that the three Macadamia species (Macadamia integrifolia, Macadamia ternifolia, and Macadamia tetraphylla) share extremely high sequence homology with each other and no rearrangement events were detected.
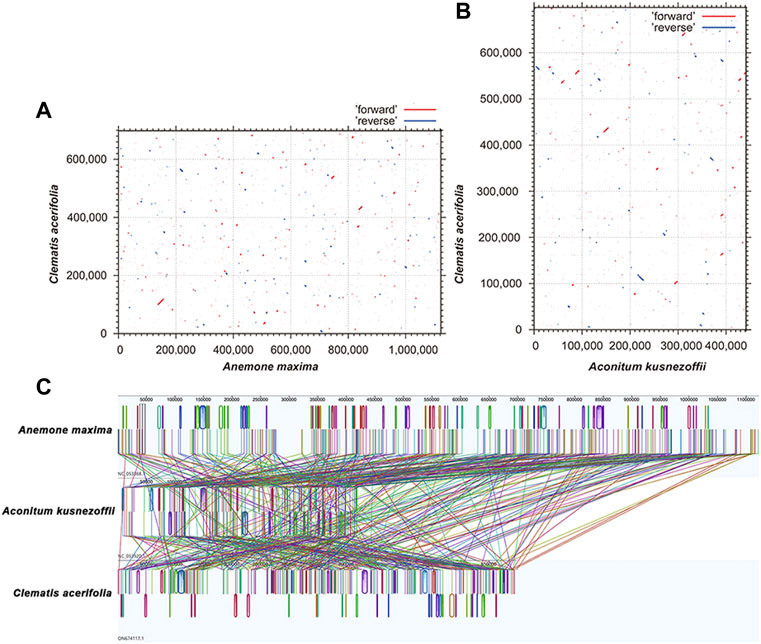
FIGURE 9. Dot plot and collinearity analysis of the mitogenomes of C. acerifolia, A. maxima and A. kusnezoffii. (A, B) are dot plots of A. maxima and A. kusnezoffii with C. acerifolia, respectively. (C) is the collinearity analysis between mitogenomes, with homologous regions represented by the same color blocks and connected by lines.
Plant mitogenomes size are much larger than other species. For example, animal mitogenomes are usually only 15–17 kb, algal mitochondria are slightly larger and can reach 13–96 kb, while angiosperm mitogenome size is usually in the range of 200–700 kb (Gualberto and Newton, 2017), Silene conica (11.3 Mb) is the largest plant mitogenome found so far (Sloan et al., 2012). C. acerifolia mitogenome size is between that of A. kusnezoffii (440, 720bp) and A. maxima (1, 122, 546) of the Ranunculaceae (Park and Park, 2020; Li et al., 2021). Although plant mitogenomes are very large, they do not contain many coding genes, typically 50–60 (Kitazaki and Kubo, 2010). With 55 genes (35 PCGs) annotated in the C. acerifolia mitogenome, which is close to the number of genes annotated in Actinidia chinensis (Wang et al., 2019), Quercus acutissima (Liu et al., 2022), and Populus simonii (Bi et al., 2022). The sequences of the coding regions of angiosperms account for only 7–17% of the total genome, and the rest is occupied by intergenic regions (Kubo and Newton, 2008).
GC content is also one of the important indicators for species evaluation, and C. acerifolia mitogenome GC content was 46.91%, which is similar to the GC content of the mitogenomes of most angiosperms (Q. acutissima, 45.72%; Dalbergia odorifera, 45.1%, Cucumis hystrix, 44.59%; Elymus sibiricus; 44.47%) and higher than that of the clematis chloroplast genome (37.87–38.19%) (Hong et al., 2021; Liu et al., 2022; Xia et al., 2022). The C. acerifolia mitogenome exhibits a multi-branched structure (linear molecule 1 and circular molecule 2), and the vast majority of published plant mitogenomes are presented as circular DNA molecules, and there are also conformational forms such as Y- and H-type linear, but mitochondrial DNA in its real state is often a mixture of different conformational forms (Backert and Börner, 2000; Kitazaki and Kubo, 2010; Bonen, 2014; Christensen, 2019).
Codon usage preference analysis provides a better understanding of the role of the mitogenome in evolution. Although the mitogenomes of different species may differ in size, structure, and sequence, there is a great deal of similarity in the gene products they encode (Li et al., 2011). Analysis of the codon usage preferences of C. acerifolia mitogenome reveals that, like most other plants, Leu, Ser, and Arg are the most common amino acids, compared to Met and Trp, which are much less common (Ma et al., 2022). The preference of genes encoded in C. acerifolia mitogenome for codons ending in A/T is consistent with the codon preferences of A. thaliana and Nicotiana tabacum (Duret and Mouchiroud, 1999), in contrast to monocotyledons (G/C base endings) (Mazumdar et al., 2017). An increasing number of studies have shown that genetic codon usage preference is an important feature of biological evolution, but there is no single influencing factor responsible for this feature; in addition to the effect of mutational pressure (Rao et al., 2011), natural selection (Wang et al., 2022), tRNA abundance (Behura and Severson, 2010), gene length (Wei et al., 2014), and expression level (Benjamini and Speed, 2012) all contribute to codon usage preferences. In this study, codon usage preference in the C. acerifolia mitogenome was influenced by both mutational and selective pressures, with mutational pressure playing a dominant role, but the exact mechanism of influence needs to be further investigated.
RNA editing is one of the required steps for gene expression in plant mitogenomes and is widespread in higher plant mitochondria, occurring mainly at one of the first two positions of the codon, which affects its amino acid alterations and plays a crucial role in plant evolution (Edera et al., 2018). Some amino acids with good codon usage preference in the mitogenome of C. acerifolia are significantly enhanced after RNA editing. The occurrence frequency of editing sites varies widely among plants, with more than 2,000 sites having been reported in bryophytes (Hecht et al., 2011), between 200–700 in angiosperms (Mower, 2009; Sloan et al., 2010; Richardson et al., 2013), and about 500 in gymnosperms (Salmans et al., 2010), while some plants, such as green algae, have not been found to have RNA editing sites. Unlike other plants, angiosperms are experiencing a loss of editing sites at this stage, mainly due to the replacement of editable thymidine by cytosine in the genome (Shields and Wolfe, 1997; Cuenca et al., 2010). In C. acerifolia, 676 RNA editing sites were identified, similar to the number of editing sites in the Ranunculaceae plant A. maxima (687) (Park and Park, 2020). RNA editing includes different forms such as C to U, U to C, and A to I (Small et al., 2020). However, all of the C. acerifolia mitochondria genomes were edited by editing C to U. Notably, the stop codon of the plant mitochondrial atp9 gene, which was generated by RNA editing in 60% of the plants, was predicted only once in C. acerifolia (Handa, 1993). In addition, abnormal RNA editing in organelles affecting embryonic endosperm development, plant growth and abiotic stress resistance has been elaborated on in many studies, which also indicates that RNA editing has a non-negligible role in plant growth and development (Yan et al., 2018).
Containing abundant repeats is one of the distinctive features of mitogenomes in higher plants. The large number of repeats leads to frequent homologous recombination and is more prone to gene rearrangement events, which is closely related to the complexity of mitochondrial gene structure (Stern and Palmer, 1984).
TRs and TEs play an important role in genome evolution, with high mutation rates of TRs accelerating gene coding and regulatory sequence evolution (Gemayel et al., 2010), and TEs being able to alter DNA sequences through a series of events such as cleavage, duplication, and reintegration (Han, 2012). It has been shown that these repeats are active in plant mitogenome reorganization, affecting structural changes in the mitogenome and producing greater mitogenome size (Guo et al., 2016). There are three pairs of LRs in C. acerifolia mitogenome, with the longest TEs exceeding 10 kb (11, 827 bp), and we speculate that intra- and inter-molecular recombination may have occurred through long repeats, leading to the generation of heteromeric forms or subgenomic molecules in the C. acerifolia mitogenome.
We mentioned earlier, non-coding sequences constitute a large part of plant mitogenomes, consisting of repeat fragments, sequences transferred from the chloroplast and nuclear genomes, and even sequences transferred at the gene level to other species. Gene fragments from chloroplasts are commonly present in the mitogenomes of higher plants and represent a high proportion of them (Zhang et al., 2011). The mitogenome of Amborella trichopoda, the oldest angiosperm, contains a large number of sequence fragments from mosses, green algae and other angiosperms (Rice et al., 2013).
Length of homologous fragments between C. acerifolia mitogenome and chloroplast genome accounted for 3.59% of the total mitogenome, while the mitogenome of Vitis vinifera contained 30 fragments from the chloroplast, totaling 68, 237 bp, accounting for 8.8% of the entire mitogenome and 42.4% of its entire chloroplast genome, this value is the largest among sequenced plant mitogenomes (Goremykin et al., 2009). Interestingly, we found that the proportion of chloroplast DNA sequences was much lower in C. acerifolia, but compared with the lower plant Marchantia polymorpha, which has 27 tRNAs but no chloroplast-derived sequences (Oda et al., 1992), the transfer of chloroplast gene sequences to mitochondria may be unique to flowering plants.
In our study, C. acerifolia, A. maxima, and A. kusnezoffii were grouped together in the Ranunculaceae, and the topology of the mitochondrial DNA-based phylogeny coincided with the Angiosperm Phylogeny Group (APG) classification. The order of the 24 species in the phylogenetic tree is consistent with the species evolutionary relationships, which also illustrates the consistency between traditional and molecular taxonomy. Previously, C. acerifolia was traditionally classified in the subsection Montanae Schneider (Grey-Wilson, 2000; Johnson, 2001), while Wang’s recent revision of the genus Clematis placed it in Sect. Cheiropsis as a single species in Subsect (Wang, 2002; Wang and Li, 2005). Xie (2011) also considered C. acerifolia to be a phylogenetically isolated lineage and speculated that this species may represent a relic species of Holarctic Clematis.
The co-linearity between C. acerifolia mitogenomic and A. maxima is high, with large homozygous co-linear blocks, suggesting that the two species have undergone extensive rearrangement phenomena. We speculate that a break fusion occurred between their different chromosomes, that is, C. acerifolia and A. maxima did not diverge simultaneously, but rather as one species evolved from the other. Wang’s analysis of important morphological characters, such as plant trophic and reproductive organs, revealed that Clematis is adjacent to Trollius and Anemone and originated from Anemone (Wang and Li, 2005). Tamura also highlighted the existence of a close kinship between Clematis and Anemone (Tamura, 1955; Tamura, 1967; Tamura, 1970; Tamura, 1987; Tamura, 1995; Tamura, 1997). All these studies confirm the accuracy of the mitogenome of C. acerifolia in determining its phylogenetic position.
Clematis is one of the most widespread genera of flowering plants, and its ornamental value and species diversity have evoked research and exploration. In recent years, C. acerifolia has attracted much attention, but the phylogenetic status of the genus is highly controversial. Here, we have assembled and annotated the complete mitogenome of C. acerifolia, the first mitogenome of Clematis. The C. acerifolia mitogenome is a relatively rare multi-branched structure that including a linear molecule (M1) and a circular molecule (M2), which we speculate may have some association with long repeats. We performed extensive analyses based on the DNA and amino acid sequences of the annotated genes, and we found that mutational pressure was the main factor influencing codon usage preferences. In addition, C. acerifolia has similar affinities to A. maxima and substantial genomic rearrangements occur between it and closely related species. Our study is expected to enrich the information on the organelle genome of C. acerifolia and provide many new insights into Clematis genetics and evolution, but the determination of the position of C. acerifolia in the genus still requires the addition of more genetic information, and we hope that subsequent studies will help better clarification of these relationships.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
DL, KQ, and YY participated in manuscript preparation, data analysis for this study; ZZ, YC, and BH participated in the resource investigation and collection of plant material; YY assembled the sequence and data analysis; DL and KQ wrote this manuscript; WL and YE-K participated in revising and correcting language errors in the manuscript; XX (lead), BT, and HL conceived and designed the study and supervised it; all authors read and approved the final manuscript.
This research was supported by the National Forest Germplasm Resources Sharing Service Platform Construction and Operation Project (2005 - DKA21003), and the State Forestry and Grassland Administration Rare and Endangered Species Investigation and Supervision and Industry Regulation Project (2,020,070,316).
The authors acknowledge support from the laboratory and our collaborators for many useful suggestions. We sincerely thank the experimental personnel and bioinformatics analysts at Wuhan Benagen Tech Solutions Company Limited (www.benagen.com) and MitoRun research group participated in this project.
YY was employed by Wuhan Benagen Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1050040/full#supplementary-material
Backert, S., and Börner, T. (2000). Phage T4-like intermediates of DNA replication and recombination in the mitochondria of the higher plant Chenopodium album (L.). Phage T4-like Intermed. DNA replication Recomb. mitochondria High. plantCurr. Genet. 37 (5), 304–314. doi:10.1007/s002940050532
Behura, S. K., and Severson, D. W. (2010). Coadaptation of isoacceptor tRNA genes and codon usage bias for translation efficiency in Aedes aegypti and. Insect Mol. Biol. 20 (2), 177–187. doi:10.1111/j.1365-2583.2010.01055
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-Web: A web server for microsatellite prediction. Bioinformatics 33 (16), 2583–2585. doi:10.1093/bioinformatics/btx198
Benjamini, Y., and Speed, T. P. (2012). Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 40 (10), e72. doi:10.1093/nar/gks001
Benson, G. (1999). Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27 (2), 573–580. doi:10.1093/nar/27.2.573
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., and Yin, T. (2022). Deciphering the multi-chromosomal mitochondrial genome of Populus simonii. Front. Plant Sci. 13, 914635. doi:10.3389/fpls.2022.914635
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi:10.1093/bioinformatics/btu170
Bonen, L. (2014). “Mitochondrial genomes in land plants,” in Molecular life Sciences. Editor E. Bell, 1–9. doi:10.1007/978-1-4614-6436-5
Breton, S., Beaupré, H. D., Stewart, D. T., Hoeh, W. R., and Blier, P. U. (2007). The unusual system of doubly uniparental inheritance of mtDNA: isn’t one enough? Trends Genet. 23 (9), 465–474. doi:10.1016/j.tig.2007.05.011
Castro, J. A., Picornell, A., and Ramon, M. (1998). Mitochondrial DNA: A tool for populational genetics studies. Int. Microbiol. 1 (4), 327–332.
Chen, Y., Ye, W., Zhang, Y., and Xu, Y. (2015). High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 43 (16), 7762–7768. doi:10.1093/nar/gkv784
Cho, Y., Mower, J. P., Qiu, Y-L., and Palmer, J. D. (2004). Mitochondrial substitution rates are extraordinarily elevated and variable in a genus of flowering plants. Proc. Natl. Acad. Sci. U. S. A. 101 (51), 17741–17746. doi:10.1073/pnas.0408302101
Choi, K-S., and Park, S. (2021). Complete plastid and mitochondrial genomes of aeginetia indica reveal intracellular gene transfer (IGT), horizontal gene transfer (HGT), and cytoplasmic male sterility (CMS). Int. J. Mol. Sci. 22 (11), 6143. doi:10.3390/ijms22116143
Choi, K. S., Ha, Y-H., Gil, H-Y., Choi, K., Kim, D-K., and Oh, S-H. (2021). Two Korean endemic clematis chloroplast genomes:inversion, reposition, expansion of the inverted repeat region, phylogenetic analysis, and nucleotide substitution rates. Plants 10 (2), 397. doi:10.3390/plants10020397
Christensen, A. C., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 15 (8), e1008373. doi:10.1371/journal.pgen.1008373
Cuenca, A., Petersen, G., Seberg, O., Davis, J. I., and Stevenson, D. W. (2010). Are substitution rates and RNA editing correlated? BMC Evol. Biol. 10 (1), 1–15. doi:10.1186/1471-2148-10-349
Daniell, H., Lin, C-S., Yu, M., and Chang, W-J. (2016). Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 17 (1), 134–162. doi:10.1186/s13059-016-1004-2
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14 (7), 1394–1403. doi:10.1101/gr.2289704
Dong, S., Zhao, C., Chen, F., Liu, Y., Zhang, S., Wu, H., et al. (2018). The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genomics 19 (1), 614. doi:10.1186/s12864-018-4991-4
Duminil, J., and Besnard, G. (2021). Utility of the mitochondrial genome in plant taxonomic studies. Methods Mol. Biol. 2222, 107–118. doi:10.1007/978-1-0716-0997-2_6
Duret, L., and Mouchiroud, D. (1999). Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila. Proc. Natl. Acad. Sci. U. S. A. 96 (8), 4482–4487. doi:10.1073/pnas.96.8.4482
Edera, A. A., Gandini, C. L., and Sanchez-Puerta, M. V. (2018). Towards a comprehensive picture of C-to-U RNA editing sites in angiosperm mitochondria. Plant Mol. Biol. 97 (3), 215–231. doi:10.1007/s11103-018-0734-9
Evans, D. L., Hlongwane, T. T., Joshi, S. V., and Pachón, D. M. R. (2019). The sugarcane mitochondrial genome:assembly, phylogenetics and transcriptomics. PeerJ 7, e7558. doi:10.7717/peerj.7558
Feng, Y., Xiang, X., Akhter, D., Pan, R., Fu, Z., and Jin, X. (2021). Mitochondrial phylogenomics of fagales provides insights into plant mitogenome mosaic evolution. Front. Plant Sci. 12, 762195. doi:10.3389/fpls.2021.762195
Gemayel, R., Vinces, M. D., Legendre, M., and Verstrepen, K. J. (2010). Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 44 (1), 445–477. doi:10.1146/annurev-genet-072610-155046
Goremykin, V. V., Salamini, F., Velasco, R., and Viola, R. (2009). Mitochondrial DNA of Vitis vinifera and the issue of rampant horizontal gene transfer. Mol. Biol. Evol. 26 (1), 99–110. doi:10.1093/molbev/msn226
Grey-Wilson, C. (2000). Clematis, the genus: A comprehensive guide for gardeners, horticulturists and botanists. Portland: Timber Press.
Gualberto, J. M., and Newton, K. J. (2017). Plant mitochondrial genomes: Dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 68 (1), 225–252. doi:10.1146/annurev-arplant-043015-112232
Guo, W., Grewe, F., Fan, W., Young, G. J., Knoop, V., Palmer, J. D., et al. (2016). Ginkgo and Welwitschia Mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol. Biol. Evol. 33 (6), 1448–1460. doi:10.1093/molbev/msw024
Han, L., Song, J. H., Yoon, J. H., Park, Y. G., Lee, S. W., Choi, Y. J., et al. (2012). TNF-Α and TNF-β polymorphisms are associated with susceptibility to osteoarthritis in a Korean population. Korean J. Pathol. 28 (12), 30–37. doi:10.4132/KoreanJPathol.2012.46.1.30
Handa, H. (1993). RNA editing of rapeseed mitochondrial atp9 transcripts: RNA editing changes four amino acids, but termination codon is already encoded by genomic sequence. Jpn. J. Genet. 68 (1), 47–54. doi:10.1266/jjg.68.47
Hecht, J., Grewe, F., and Knoop, V. (2011). Extreme RNA editing in coding islands and abundant microsatellites in repeat sequences of Selaginella moellendorffii mitochondria: The root of frequent plant mtDNA recombination in early tracheophytes. Genome Biol. Evol. 3, 344–358. doi:10.1093/gbe/evr027
Hong, Z., Liao, X., Ye, Y., Zhang, N., Yang, Z., Zhu, W., et al. (2021). A complete mitochondrial genome for fragrant Chinese rosewood (Dalbergia odorifera, Fabaceae) with comparative analyses of genome structure and intergenomic sequence transfers. BMC Genomics 22 (1), 1–13. doi:10.1186/s12864-021-07967-7
Huang, Y., Si, X., Shao, M., Teng, X., Xiao, G., and Huang, H. (2022). Rewiring mitochondrial metabolism to counteract exhaustion of CAR-T cells. J. Hematol. Oncol. 15, 38–57. doi:10.1186/s13045-022-01255-x
Huelsenbeck, J. P., and Ronquist, F. (2001). Mrbayes: Bayesian inference of phylogenetic trees. Bioinformatics 17 (8), 754–755. doi:10.1093/bioinformatics/17.8.754
Jin, J-J., Yu, W-B., Yang, J-B., Song, Y., dePamphilis, C. W., Yi, T-S., et al. (2020). GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21 (1), 1–31. doi:10.1186/s13059-020-02154-5
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20 (4), 1160–1166. doi:10.1093/bib/bbx108
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi:10.1093/molbev/mst010
Kitazaki, K., and Kubo, T. (2010). Cost of having the largest mitochondrial genome: Evolutionary mechanism of plant mitochondrial genome. Journal of botany article ID. J. Bot. 620137, 1–12. doi:10.1155/2010/620137
Kubo, T., and Newton, K. J. (2008). Angiosperm mitochondrial genomes and mutations. Mitochondrion 8 (1), 5–14. doi:10.1016/j.mito.2007.10.006
Kumar, K. R., Cowley, M. J., and Davis, R. L. (2019). Next-generation sequencing and emerging technologies. Semin. Thromb. Hemost. 45 (7), 661–673. doi:10.1055/s-0039-1688446
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi:10.1093/molbev/msw054
Lehtonen, S., Christenhusz, M. M., and Falck, D. (2016). Sensitive phylogenetics of clematis and its position in ranunculaceae. Bot. J. Linn. Soc. 182 (4), 825–867. doi:10.1111/boj.12477
Letunic, I., and Bork, P. (2019). Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 47 (1), W256–W259. doi:10.1093/nar/gkz239
Lewis, S. E., Searle, S. M. J., Harris, N., Gibson, M., Iyer, V., Richter, J., et al. (2002). Apollo: A sequence annotation. Genome Biol. 3(12), 0082–114. doi:10.1186/gb-2002-3-12-research0082
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, S., Yang, Y-Y., Xu, L., Xing, Y-P., Zhao, R., Ao, W-L., et al. (2021). The complete mitochondrial genome of Aconitum kusnezoffii Rchb. (Ranales, Ranunculaceae). Mitochondrial DNA. B Resour. 6 (3), 779–781. doi:10.1080/23802359.2021.1882894
Li, X., Guo, Q., Dong, Li., Sun, Y., Niu, D., Liu, J., et al. (2019). EST-SSR analysis of genetic diversity of Robinia pseudoacacia clones in Jixian County. J. Beijing For. Univ. 41 (07), 39–48. doi:10.13332/j.1000-1522.20190108
Li, Y., Zhao, H., Tan, H., Liu, X., Zhang, C., and Dong, Y. (2011). Analysis and comparison on characteristic of mitochondrial genome of eight plants. Biol. Bull-Us., 156–162. doi:10.13560/j.cnki.biotech.bull.1985.2011.10.033
Liu, D., Guo, H., Zhu, J., Qu, K., Cheng, Y., Guo, Y., et al. (2022). Complex physical structure of complete mitochondrial genome of Quercus acutissima (fagaceae): A significant energy plant. Genes 13, 1321. doi:10.3390/genes13081321
Lopez-Pujol, J., Zhang, F. M., and Ge, S. (2003). Population genetics and conservation of the critically endangered. Clematis acerifolia (Ranunculaceae). Can. J. Bot. 83 (10), 1248–1256. doi:10.1139/b05-097
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25 (5), 955–964. doi:10.1093/nar/25.5.955
Ma, Q., Wang, Y., Li, S., Wen, J., Zhu, L., Yan, K., et al. (2022). Assembly and comparative analysis of the first complete mitochondrial genome of acer truncatum bunge: A woody oil-tree species producing nervonic acid. BMC Plant Biol. 22 (1), 29–17. doi:10.1186/s12870-021-03416-5
Maximowicz, C. J. (1879). Ad florae Asiae orientalis cognitionem meliorem fragmenta. Moscow: Russian National Library.
Mazumdar, P., Binti Othman, R., Mebus, K., Ramakrishnan, N., and Ann Harikrishna, J. (2017). Codon usage and codon pair patterns in non-grass monocot genomes. Ann. Bot. 120 (6), 893–909. doi:10.1093/aob/mcx112
McCouch, S. R. (2001). Genomics and synteny. Plant Physiol. 125 (1), 152–155. doi:10.1104/pp.125.1.152
Moore, W. S. (1995). Inferring phylogenies from mtDNA variation: Mitochondrial-gene trees versus nuclear-gene trees. Evolution 49 (4), 718–726. doi:10.1111/j.1558-5646.1995.tb02308.x
Moreira, S., Valach, M., Aoulad-Aissa, M., Otto, C., and Burger, G. (2016). Novel modes of RNA editing in mitochondria. Nucleic Acids Res. 44 (10), 4907–4919. doi:10.1093/nar/gkw188
Mower, J. P. (2009). The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 37, W253–W259. doi:10.1093/nar/gkp337
Niu, L., Zhang, Y., Yang, C., Yang, J., Ren, W., Zhong, X., et al. (2022). Complete mitochondrial genome sequence and comparative analysis of the cultivated yellow nutsedge. Plant Genome 15. doi:10.1002/tpg2.20239
Oda, K., Yamato, K., Ohta, E., Nakamura, Y., Takemura, M., Nozato, N., et al. (1992). Gene organization deduced from the complete sequence of liverwort Marchantia polymorpha mitochondrial DNA: A primitive form of plant mitochondrial genome. J. Mol. Biol. 223 (1), 1–7. doi:10.1016/0022-2836(92)90708-r
Pang, J., Yuan, Y., Zhou, M., Ren, J., Gao, P., and Cheng, K. (2022). Species niche of Clemtis acerifolia Maxim.community. Acta Ecol. Sin. 42 (8), 3449–3457. doi:10.5846/stxb202009142401
Park, S., and Park, S. (2020). Large-scale phylogenomics reveals ancient introgression in Asian Hepatica and new insights into the origin of the insular endemic Hepatica maxima. Sci. Rep. 10 (1), 16288. doi:10.1038/s41598-020-73397-2
Rao, Y., Wu, G., Wang, Z., Chai, X., Nie, Q., and Zhang, X. (2011). Mutation bias is the driving force of codon usage in the Gallus gallus genome. DNA Res. 18 (6), 499–512. doi:10.1093/dnares/dsr035
Rice, D. W., Alverson, A. J., Richardson, A. O., Young, G. J., Sanchez-Puerta, M. V., Munzinger, J., et al. (2013). Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 342 (6165), 1468–1473. doi:10.1126/science.1246275
Richardson, A. O., Rice, D. W., Young, G. J., Alverson, A. J., and Palmer, J. D. (2013). The “fossilized” mitochondrial genome of Liriodendron tulipifera: Ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Bio. 11 (1), 1–17. doi:10.1186/1741-7007-11-29
Rubinstein, N. D., Feldstein, T., Shenkar, Noa., Botero-Castro, F., Griggio, F., Mastrototaro, F., et al. (2013). Deep sequencing of mixed total DNA without barcodes allows efficient assembly of highly plastic ascidian mitochondrial genomes. Genome Biol. Evol. 5 (6), 1185–1199. doi:10.1093/gbe/evt081
Saccone, C., Giorgi, D., Gissi, C., Pesole, G., and Reyes, A. (1999). Evolutionary genomics in metazoa: The mitochondrial DNA as a model system. Gene 238 (1), 195–209. doi:10.1016/s0378-1119(99)00270-x
Salmans, M. L., Chaw, S. M., Lin, C. P., Shih, A. C-C., and Mulligan, R. M. (2010). Editing site analysis in a gymnosperm mitochondrial genome reveals similarities with angiosperm mitochondrial genomes. Curr. Genet. 56 (5), 439–446. doi:10.1007/s00294-010-0312-4
Sheng, L., Ji, K., and Yu, L. (2014). Karyotype analysis on 11 species of the genus Clematis. Braz. J. Bot. 37 (4), 601–608. doi:10.1007/s40415-014-0099-5
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47 (W1), W65–W73. doi:10.1093/nar/gkz345
Shields, D. C., and Wolfe, K. H. (1997). Accelerated evolution of sites undergoing mRNA editing in plant mitochondria and chloroplasts. Mol. Biol. Evol. 14 (3), 344–349. doi:10.1093/oxfordjournals.molbev.a025768
Sloan, D. B., Alverson, A. J., Chuckalovcak, J. P., Wu, M., McCauley, D. E., Palmer, J. D., et al. (2012). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 10 (1), e1001241. doi:10.1371/journal.pbio.1001241
Sloan, D. B., MacQueen, A. H., Alverson, A. J., Palmer, J. D., and Taylor, D. R. (2010). Extensive loss of RNA editing sites in rapidly evolving Silene mitochondrial genomes: Selection vs. retroprocessing as the driving force. Genetics 185 (4), 1369–1380. doi:10.1534/genetics.110.118000
Small, I. D., Schallenberg-Rudinger, M., Takenaka, M., Mireau, H., and Ostersetzer-Biran, O. (2020). Plant organellar RNA editing: What 30 years of research has revealed. Plant J. 101 (5), 1040–1056. doi:10.1111/tpj.14578
Stefan, K., Choudhuri, J. V., Enno, O., Chris, S., Jens, S., and Robert, G. (2001). REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29 (22), 4633–4642. doi:10.1093/nar/29.22.4633
Stern, D. B., and Palmer, J. D. (1984). Recombination sequences in plant mitochondrial genomes: Diversity and homologies to known mitochondrial genes. Nucleic Acids Res. 12 (15), 6141–6157. doi:10.1093/nar/12.15.6141
Tamura, M. (1987). A classification of genus. Clematis. Acta Phytotax. Geobot. 38, 33–44. doi:10.18942/bunruichiri.KJ00002992228
Tamura, M. (1995). Engler’s die natürlichen pflanzenfamilien. Zweite Aufl. Berlin Dunker Humbolt 17 (4), 365–388.
Tamura, M. (1967). Morphology, ecology and phylogeny of the Ranunculaceae. Sci. Rep. (Osaka Univ. 16, 21–43.
Tamura, M. (1997). Taxonomic studies of the ranunculaceae: Retrospect and prospect. Memoirs of the school of B. O. S. Trans. Kinki Univ. 2, 69–85.
Tan, T., and Pang, E. (2013). Identification and application of colinearity in close related species. J. Beijing Normal Univ. Nat. Sci. 49 (01), 38–41.
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45 (1), W6–W11. doi:10.1093/nar/gkx391
Turnel, M., Otis, C., and Lemieux, C. (2003). The mitochondrial genome of Chara vulgaris: Insights into the mitochondrial DNA architecture of the last common ancestor of green algae and land plants. Plant Cell 15 (8), 1888–1903. doi:10.1105/tpc.013169
Wang, R., Luo, Y., Liu, X., Lu, X., Yin, G., and Deng, Z. (2022). Synonymous codon usage in the chloroplast genome of Metasequoia glyptostroboides. J. Hubei Minzu Univ. Nat. Sci. Ed. 40 (2), 121–128. doi:10.13501/j.cnki.42-1908/n.2022.06.001
Wang, S., Li, D., Yao, X., Song, Q., Wang, Z., Zhang, Q., et al. (2019). Evolution and diversification of kiwifruit mitogenomes through extensive whole-genome rearrangement and mosaic loss of intergenic sequences in a highly variable region. Genome Biol. Evol. 11 (4), 1192–1206. doi:10.1093/gbe/evz063
Wang, W., and Li, L. (2005). A new system of classification of the genus. Clematis (Ranunculaceae). Acta Phytotaxon. Sin. 43 (5), 431–488. doi:10.1360/aps040130
Wang, X., Li, M., Tian, L., and Liu, D. (2021). ISSR and rDNA-ITS sequence analysis of the genetic relationship of clematis in Hebei Province. Acta Hortic. Sin. 48 (9), 1755–1767. doi:10.16420/j.issn.0513-353x.2020-0953
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40 (7), e49. doi:10.1093/nar/gkr1293
Wei, L., He, J., Jia, X., Qi, Q., Liang, Z., Zheng, H., et al. (2014). Analysis of codon usage bias of mitochondrial genome in Bombyx mori and its relation to evolution. BMC Evol. Biol. 14 (1), 1–12. doi:10.1186/s12862-014-0262-4
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 31 (20), 3350–3352. doi:10.1093/bioinformatics/btv383
Xia, L., Cheng, C., Zhao, X., He, X., Yu, X., Li, J., et al. (2022). Characterization of the mitochondrial genome of Cucumis hystrix and comparison with other cucurbit crops. Gene 823, 146342. doi:10.1016/j.gene.2022.146342
Xiang, Q-H., He, J., Liu, H-J., Lyu, R-D., Yao, M., Guan, W-B., et al. (2019). The complete chloroplast genome sequences of three Clematis species (Ranunculaceae). Mitochondrial DNA Part B 4 (1), 834–835. doi:10.1080/23802359.2019.1567290
Yan, J., Zhang, Q., and Yin, P. (2018). RNA editing machinery in plant organelles. Sci. China. Life Sci. 61 (2), 162–169. doi:10.1007/s11427-017-9170-3
Yan, S., Liu, H-J., Lin, L-L., Liao, S., Li, J-Y., Pei, L-Y., et al. (2016). Taxonomic status of Clematis acerifolia var. elobata, based on molecular evidence. Phytotaxa 268 (3), 209–219. doi:10.11646/phytotaxa.268.3.5
Yuan, Y., Ren, J., Zhou, M., Pang, J., and Cheng, K. (2021). Study on the individual morphology, growth characteristics and cluster composition of clematis acerifolia. J. West China For. Sci. 50 (1), 35–41. doi:10.16473/j.cnki.xblykx1972.2021.01.005
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20 (1), 348–355. doi:10.1111/1755-0998.13096
Zhang, H., Meltzer, P., and Davis, S. (2013). RCircos: an R package for Circos 2D track plots. BMC Bioinforma. 14 (1), 244–245. doi:10.1186/1471-2105-14-244
Zhang, X., Zhang, R., Hou, S-y., Shi, J., and Guo, S-D. (2011). Research progress on mitochondrial genome of higher plant. J. Agr. Sci. Tech-Iran 13 (4), 23–31. doi:10.3969/j.issn.1008-0864
Keywords: clematis, mitochondrial genome, organelle genome, Clematis acerifolia, phylogenetic relationship, repeats
Citation: Liu D, Qu K, Yuan Y, Zhao Z, Chen Y, Han B, Li W, El-Kassaby YA, Yin Y, Xie X, Tong B and Liu H (2023) Complete sequence and comparative analysis of the mitochondrial genome of the rare and endangered Clematis acerifolia, the first clematis mitogenome to provide new insights into the phylogenetic evolutionary status of the genus. Front. Genet. 13:1050040. doi: 10.3389/fgene.2022.1050040
Received: 21 September 2022; Accepted: 05 December 2022;
Published: 04 January 2023.
Edited by:
Fei Shen, Beijing Academy of Agricultural and Forestry Sciences, ChinaReviewed by:
Zhe Hou, Suzhou Polytechnic Institute of Agriculture, ChinaCopyright © 2023 Liu, Qu, Yuan, Zhao, Chen, Han, Li, El-Kassaby, Yin, Xie, Tong and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoman Xie, eHhtNTI5QDEyNi5jb20=; Boqiang Tong, dGJxMTAwMUBzaGFuZG9uZy5jbg==; Hongshan Liu, bGhzMTM4MzEyMzgzMzVAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.