 Corrado Pancotti
Corrado Pancotti Cesare Rollo
Cesare Rollo Giovanni Birolo
Giovanni Birolo Silvia Benevenuta
Silvia Benevenuta Piero Fariselli
Piero Fariselli Tiziana Sanavia
Tiziana Sanavia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 04 January 2023
Sec. Computational Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1049501
This article is part of the Research TopicStructural Understanding of the Functional Consequences of Missense MutationView all 6 articles
The high cosine similarity between some single-base substitution mutational signatures and their characteristic flat profiles could suggest the presence of overfitting and mathematical artefacts. The newest version (v3.3) of the signature database available in the Catalogue Of Somatic Mutations In Cancer (COSMIC) provides a collection of 79 mutational signatures, which has more than doubled with respect to previous version (30 profiles available in COSMIC signatures v2), making more critical the associations between signatures and specific mutagenic processes. This study both provides a systematic assessment of the de novo extraction task through simulation scenarios based on the latest version of the COSMIC signatures and highlights, through a novel approach using archetypal analysis, which COSMIC signatures are redundant and more likely to be considered as mathematical artefacts. 29 archetypes were able to reconstruct the profile of all the COSMIC signatures with cosine similarity
Somatic mutations in cancer are caused by a wide range of endogenous (i.e. genome instability or deficiency in a DNA repair mechanism) or exogenous (environmental exposures such as ultraviolet radiation or tobacco smoking) mutagenic agents, which stratify over time. It has been hypothesised that a mutational pattern in the genome can be deconvolved considering different generative processes, each of them associated with a specific mutational signature represented by 96 somatic mutation frequencies of six single nucleotide variants (C > A: G > T, C > G: G > C, C > T: G > A, T > A: A > T, T > C: A > G, T > G: A > C) flanked by one nucleotide on each side (Alexandrov et al., 2013). The latest version of the Catalogue Of Somatic Mutations In Cancer (COSMIC) (Bamford et al., 2004) hosts 79 single-base substitution (SBS) signatures extracted from 2,780 genomes of the Pan Cancer Analysis of Whole Genomes (PCAWG) (as described in https://cancer.sanger.ac.uk/signatures/sbs/) using SigProfilerExtractor (Islam et al., 2022), an updated version of the original method based on Non-Negative Matrix Factorization (NMF) proposed by Alexandrov et al. (2020).
Although signature extraction analysis is becoming a routine, there are some issues that should be further investigated. Recently, some studies have highlighted caveats and warnings in using these representations for clinical applications (Maura et al., 2019; Koh et al., 2021). Omichessan et al. (2019) did an empirical evaluation of the main de novo extraction tools showing that the identification of signatures is more difficult for tumours characterised by multiple signatures having a small contribution, and pointing out that different signatures might have very close cosine similarity, as it was observed between COSMIC signatures (cosine similarity
Lal et al. (2021) pointed out that state-of-the-art NMF-based methods aim to minimise the residual error after fitting the data with the discovered signatures to fit the data perfectly, which may generate overfitting issues by including stochastic noise in the data as part of the signatures, or multiple similar signatures for the same underlying process. Indeed, the goal of the signature discovery is not only to fit the data as well as possible, but also to identify signatures that truly reflect separate biological processes and the current version (3.3) of COSMIC database shows several signatures with no experimentally-validated aetiological causes associated yet, suggesting possible mathematical artefacts due to overfitting rather than distinct mutagenic processes (Koh et al., 2021). These issues become more critical when the signature extraction is highly dependent on the number of samples available, which complicates the correct identification of the true components, jeopardising the stability of the results. In addition, the studies of Maura et al. (2019) and Lal et al. (2021) highlighted that the presence of flat signatures, showing similar frequencies across all the 96 mutational classes, could represent a source of background noise and collinearities, making the de novo signature extraction task difficult and ambiguous.
These issues are expected to become more critical in the newest version of COSMIC catalogue, which considers 79 signatures compared to the 30 signatures of the previous versions investigated in the above studies. Therefore, the proposed study will focus on two main goals: 1) to provide a systematic approach to assess to which extent the extraction of the newest version of COSMIC signatures can be affected by the high similarity among the signatures in the same catalogue, the presence of flat signatures and the number of available samples; 2) to provide a compact representation of the current catalogue by prioritising the identification of those profiles representing extreme patterns in the data so that all the observations can be reproduced as mixtures of their extremes. To this aim, Archetypal Analysis (Cutler and Breiman, 1994) was applied to represent how the information from COSMIC can be projected into a reduced number of dimensions and to explain potential instability issues in specific extraction scenarios. Figure 1 displays a summary of the workflow analysis proposed in this study.
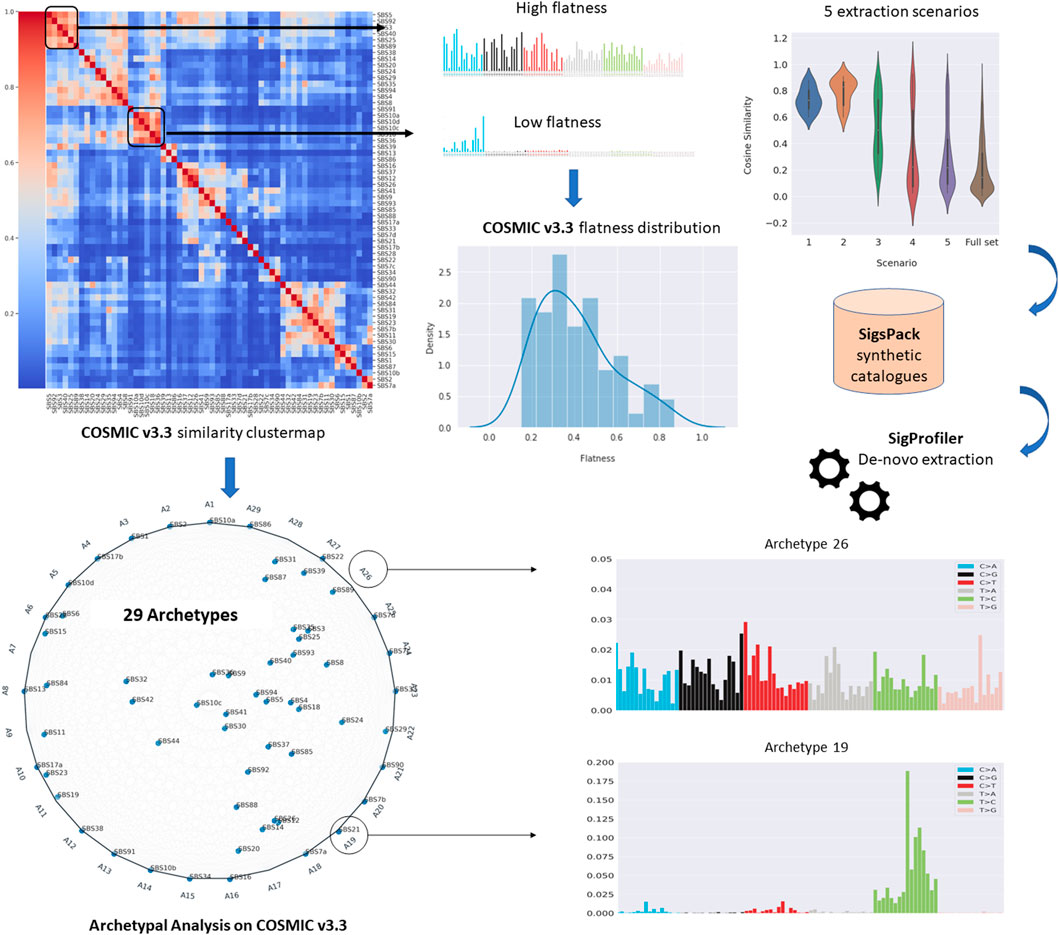
FIGURE 1. Summary of the analyses performed in this study. Firstly, we systematically assessed to which extent the extraction of the newest version of COSMIC signatures (v3.3) can be affected by the high similarity among the signatures in the same catalogue, the level of flatness of the signatures and the number of available samples, assessing the de novo extraction across five different scenarios (upper panel). Then, Archetypal Analyses was applied to provide a compact representation of the current catalogue by prioritising the identification of those profiles representing extreme patterns in the data so that all the observations can be reproduced as mixtures of their extremes (lower panel).
Analyses were performed on COSMIC catalogue v3.3 considering the 79 SBS mutational signature profiles on the reference genome GRCh37 identified by SigProfilerExtractor (Islam et al., 2022). Among these, we removed 19 signatures classified by the catalogue as sequencing artefacts (https://cancer.sanger.ac.uk/signatures/sbs/). Of the remaining 60 signatures considered, 19 neither have a direct association with an experimentally validated mutagenic process nor are they supported by statistical association with a specific process.
We first quantified the pairwise level of similarity in the signature catalogue. Therefore, for each pair of signatures si and sj we calculated the cosine similarity:
obtaining a pairwise cosine distance matrix
SBS3, SBS5, SBS40 and SBS8 are often referred to as flat signatures given their relative featureless profile, almost uniformly distributed across the 96 mutational classes. However, to the best of our knowledge, no quantitative definition of flatness has ever been provided. To fill this gap we formulated a simple definition of signature flatness by calculating the cosine similarity between the signature and the uniform distribution. Therefore the flatness of a signature si can be defined as:
Where su, in the case of SBS mutational signatures, consists of a signal uniformly distributed over the 96 mutational classes. Hence, the flatness is a score ranging from 0 to 1, where 1 represents a perfectly flat profile. Since the presence of flat signatures in a catalogue can complicate the extraction task, a quantitative definition of flatness can be useful to build robust de novo extraction methods and to test their capabilities to correctly extract multiple signatures with different levels of flatness. In this regard, we designed scenarios with different levels of similarity and flatness.
To assess both the reliability and the feasibility of the de novo extraction procedure, several synthetic catalogues were generated considering COSMIC mutational signatures as underlying generative processes. The function create_mut_catalogues from SigsPack R package (https://github.com/bihealth/SigsPack) was used to generate 10 synthetic mutational catalogues for each extraction scenario to take into account statistical fluctuations (Schumann et al., 2019). In particular, create_mut_catalogues takes samples, mutations (per sample) and signatures as input and it generates mutational catalogues with exposures to the specified signatures by sampling the mutations from a distribution of those signature profiles. The contribution of each signature (exposure) is randomly drawn from a uniform distribution for each sample. Different ranges between a minimum of 200 and a maximum of 10,000 samples were set according to the chosen scenario and the number of signatures considered. The number of mutations in each tumour sample was set to 5,000 for each scenario, based on the PCAWG median number of sample mutations. All the simulated scenarios are summarised in Table 1 and the generated catalogues are available at the Github repository https://github.com/compbiomed-unito/archetypal-analysis-cosmic.
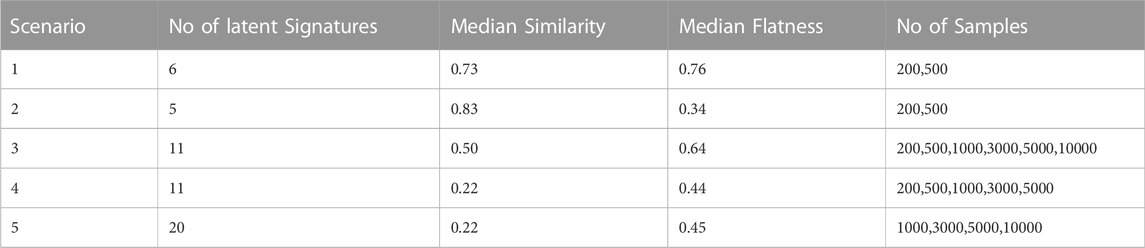
TABLE 1. Summary of the de novo extraction scenario. For each simulated scenario, the number of active signatures, the cosine similarity level. the flatness level and the n° of samples.
De novo extraction analysis was applied to each scenario using the gold-standard approach SigProfilerExtractor (Islam et al., 2022) (https://github.com/AlexandrovLab/SigProfiler), with the aim of evaluating the extraction performance from catalogues with groups of similar latent signatures, varying in number and flatness score. To choose the optimal number of latent signatures, SigProfilerExtractor performs a repeated NMF for a range of k operative signatures. For each value of k, this algorithm applies a custom partition clustering based on the Hungarian algorithm to the signature matrices resulting from the repeated NMF, so that a final consensus signature matrix is obtained. The number of NMF repetitions can be specified by the user. In our experiments this value was set to 30. All the other default parameters were used. To efficiently run SigProfilerExtractor, computational resources from HPC4AI center (Aldinucci et al., 2018) and Occam cluster (Aldinucci et al., 2017) were used, for a total of 64 CPUs and 6 GPUs.
Four metrics were considered for the performance evaluations:
• Frequency (F) of simulation runs where all the signatures are correctly identified:
• Mean square error (MSE) between simulated and reconstructed catalogues:
where xi,j and
• Average stability (Cmean) measured by the mean silhouette coefficient score of the signature clusters generated by SigProfilerExtractor:
cik is the silhouette coefficient of the i−th sample which belongs to cluster k. N is the number of NMF runs performed by SigProfilerExtractor, K is the number of cluster labels, and Ck is the mean silhouette score of the k−th cluster.
cik is given by:
where aik is the mean intra-cluster distance and bik is the mean nearest-cluster distance of the i-th sample which belongs to cluster k.
• Minimum stability (Cmin), represented by the minimum silhouette coefficient score of the signature clusters generated by SigProfilerExtractor:
Archetypal analysis (AA) is an unsupervised learning method that aims to represent data points as sparse convex combinations of their extreme elements. More formally, let
where αik ≥ 0;
In this study, AA was applied directly to the COSMIC signature matrix
The cluster map on COSMIC v3.3 catalogue revealed that there are several groups of signatures showing pairwise cosine similarity
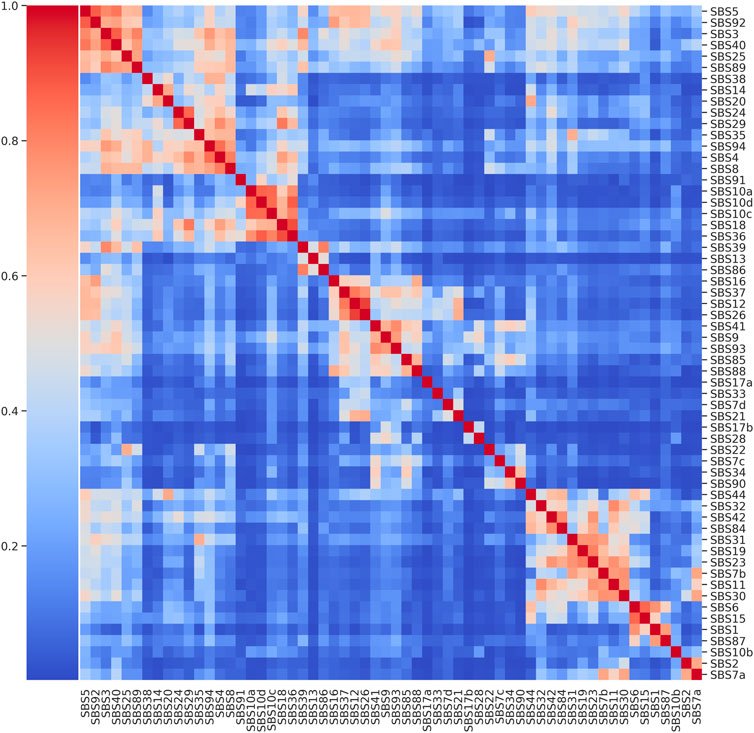
FIGURE 2. Cluster map of COSMIC SBS Mutational Signatures. Pairwise cosine similarity displayed for the 60 SBS signatures from COSMIC catalogue.
A second notable group is characterised by a high pairwise cosine similarity among signatures but with a low level of flatness and it includes SBS36, SBS18 and the three signatures SBS10a, SBS10c and SBS10d associated with an altered activity of polymerase (polymerase epsilon exonuclease domain mutations and defective POLD1 proofreading), which were considered for the second extraction scenario. The median pairwise similarity is 0.83 with a maximum equal to 0.91 between SBS36 and SBS18 while the median flatness is 0.34. In the third extraction scenario the synthetic catalogues were generated from the signatures used in the first and the second scenario together (11 signatures). Finally, in the fourth and fifth extraction scenarios, 11 and 20 signatures with a low flatness score were considered, respectively, where each signature has at least another similar one (cosine similarity
To overcome the qualitative description of flatness, in Eq. (2) (Methods section) we defined a simple way to quantitatively assess the flatness of the signatures, being in line with the qualitative description. Indeed, as shown in Supplementary Table S3, the known flat signatures SBS3, SBS40 and SBS5 show the highest degree of flatness, but a similar level to SBS5 can be found for SBS25 and SBS89. In addition, this definition of flatness appears to be well distributed within COSMIC from a minimum of 0.15 (SBS1) to a maximum of 0.87 (SBS3), showing that this metric can emphasise the differences in shape between the various signatures in COSMIC, as shown in Figure 3. As mentioned in the previous section, the extraction scenarios, built to highlight possible issues in the de novo extraction process, differ in the number of signatures involved, the pairwise similarity between profiles, and the level of flatness. In Supplementary Figure S2, the flatness distribution for each scenario is shown.
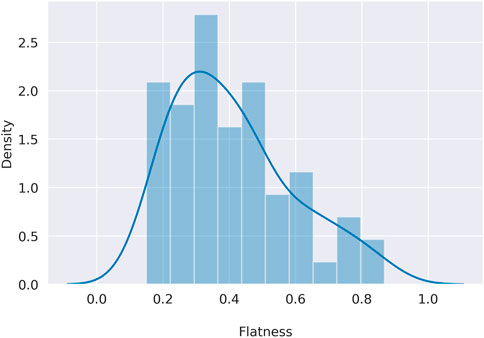
FIGURE 3. Distribution of the COSMIC flatness. On the x axis the flatness defined in Eq. 2, on the y axis the density for each flatness level.
The SigProfilerExtractor performance for each scenario is shown in Table 2. MSE, Cmean and Cmin are reported as their corresponding median values across 10 repetitions, together with their inter-quartile range. When considered separately, signatures involved in scenarios 1 and 2 were almost always correctly extracted at 200 samples (F = 0.9), regardless of the high level of similarity in each group.
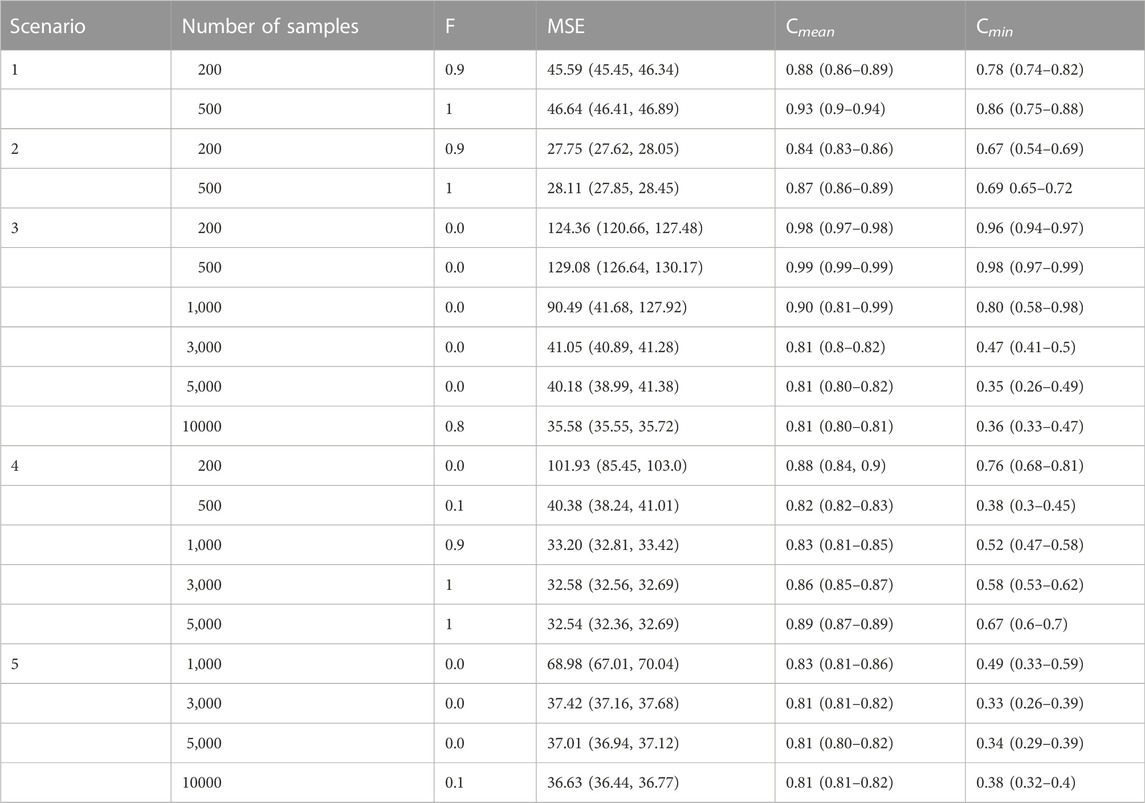
TABLE 2. De novo signature extraction performance. For each simulated scenario, the frequency of runs with all the signatures correctly identified F), the mean square error (MSE) between simulated and reconstructed catalogues, the average Cmean and minimum Cmin stability scores of signature clusters are displayed.
However, when the extraction was performed by combining these two scenarios (scenario 3), SigProfilerExtractor was never able to identify the correct number of signatures up to a high number of samples (5,000) and only at 10,000 samples it succeeded 80% of the times (F = 0.8). In this scenario it is worth noticing that, as the number of available samples increases, while the MSE decreases and the average stability decreases but remaining relatively high, the minimum stability dramatically decreases. As expected, by further increasing the number of samples, both the mean and minimum stability values rise again. However, given that obtaining 10,000 tumour samples is often unfeasible in practice, this scenario highlights well a limitation of the NMF-based extraction process. Indeed, this scenario is particularly complex since it considers two main subgroups, highly similar internally (median pairwise cosine similarity 0.73 and 0.83, respectively) but one at high and the other one at low flatness score (0.76 and 0.34, respectively, as shown in Table 1). Therefore, this difference in the flatness levels makes the extraction process much more difficult if there is not a very large number of samples. As shown in Supplementary Figure S3, the algorithm starts to differentiate similar signatures inside each of these two groups at 3,000 samples, but still failing at differentiating them well even at 5,000 samples.
On the other hand, considering again 11 signatures but with a lower level of similarity and flatness (scenario 4), the algorithm required at least 1,000 samples to identify the signatures with F = 0.9 (Supplementary Figure S3). Finally, when 20 signatures were considered (scenario 5), the algorithm always failed even at 5,000 samples, and it only succeeded 10% of the times at 10,000 samples. It is worth highlighting that the maximum number considered is significantly higher compared to the 2,780 genomes from PCAWG used to build the gold-standard catalogue of mutational signatures available in COSMIC.
The application of AA to the COSMIC SBS mutational signature matrix
The archetypal profiles are summarised in Figure 4. Most of the archetypal profiles coincide almost perfectly with some of the COSMIC signatures. Specifically, 26 out of 29 archetypes correspond to at least one COSMIC signature with cosine similarity of at least 0.97 (Supplementary Figure S6). These results suggest that a subset of signature profiles represents extreme patterns of the catalogue and that a combination of them is capable of reconstructing the entire catalogue with a high level of accuracy. The relationship between signatures and archetypes can be better understood considering the α coefficients of Eq (8). In particular, the coefficients aik represent the weights that each archetype zk has in the reconstruction of the i−th signature xi.
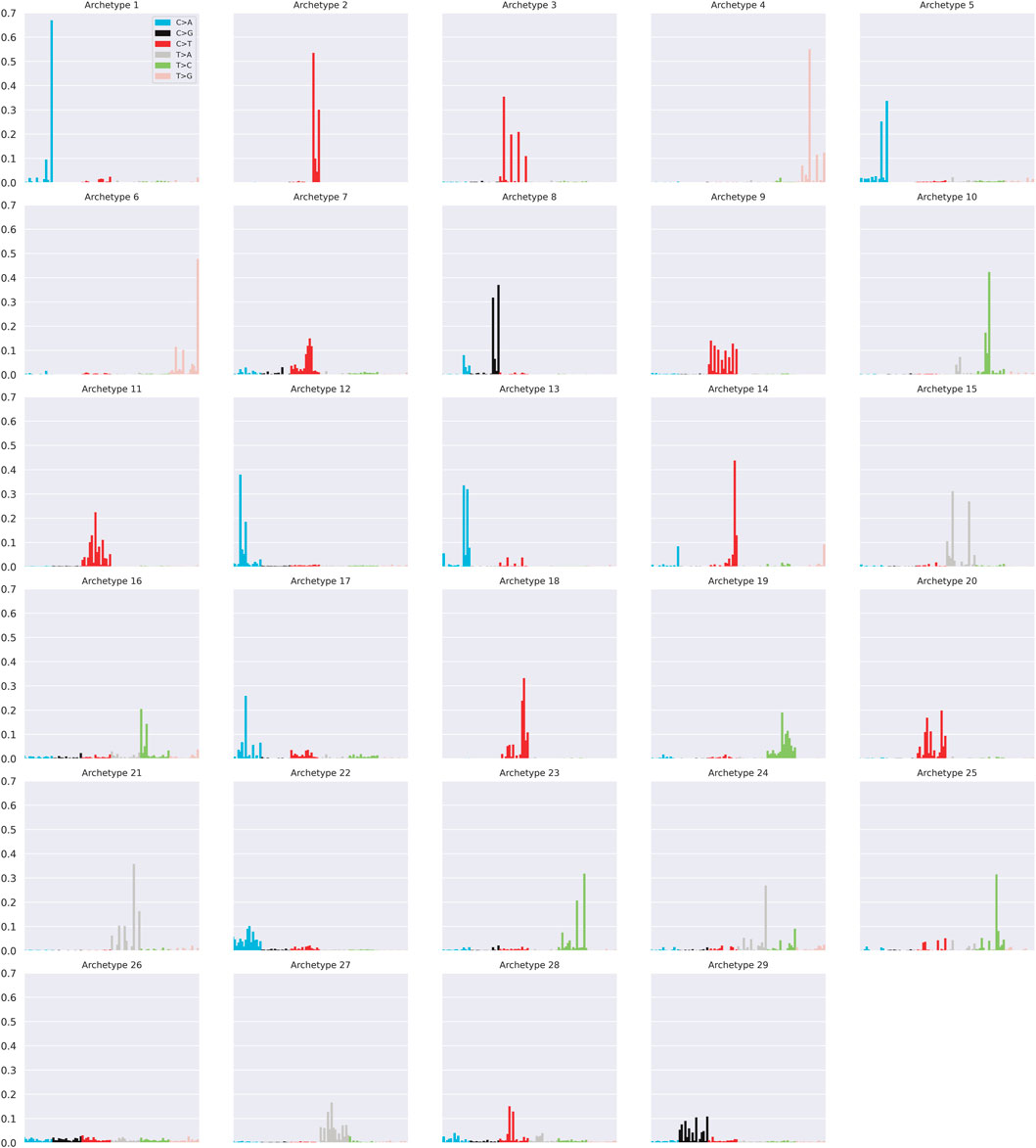
FIGURE 4. Summary of the 29 archetypal profiles.
Figure 5 shows the association between the COSMIC signatures and the archetypes through the α coefficients. The heatmap was consequently clustered to find those signatures which share a common reconstruction pattern through the archetypes. It can be seen that 19 archetypes reconstruct only one signature, indicating a one-to-one relationship between them. Others were found to contribute in more than one signature at different weights, as well as there are groups of reconstructed signatures that are mainly represented by the same archetype, highlighted by different colours in Figure 5.

FIGURE 5. Heatmap highlighting the associations between archetypes and the reconstructed signatures. Different colors highlight groups of reconstructed signatures that are mainly represented by the same archetype. For a better visualisation, α coefficients
Interestingly, AA tends to group similar profiles together fairly well, since the signatures belonging to the same group usually share either the same aetiology or similar biological processes. In Supplementary Figure S7 we further explored the relationship between the mutational signatures and the archetypes by plotting the pairwise cosine similarity distribution of the α coefficients for different categories of pairwise cosine similarity between the original signatures. It is possible to clearly observe that, as the pairwise cosine similarity between the signatures increases, the cosine similarity between α coefficients increases. This confirms that, while providing a more compact representation of the COSMIC signatures, the archetypal analysis is able to maintain a good consistency with the original profiles.
Table 3 summarises some of the qualitative information that can be extracted from the heatmap, showing the relationships between the reconstructed signatures and the archetype that contributed most to them. Each signature was reported with its aetiology, and whether it had been validated experimentally or by statistical association (i.e. unclear evidence for real signature, as reported in COSMIC).
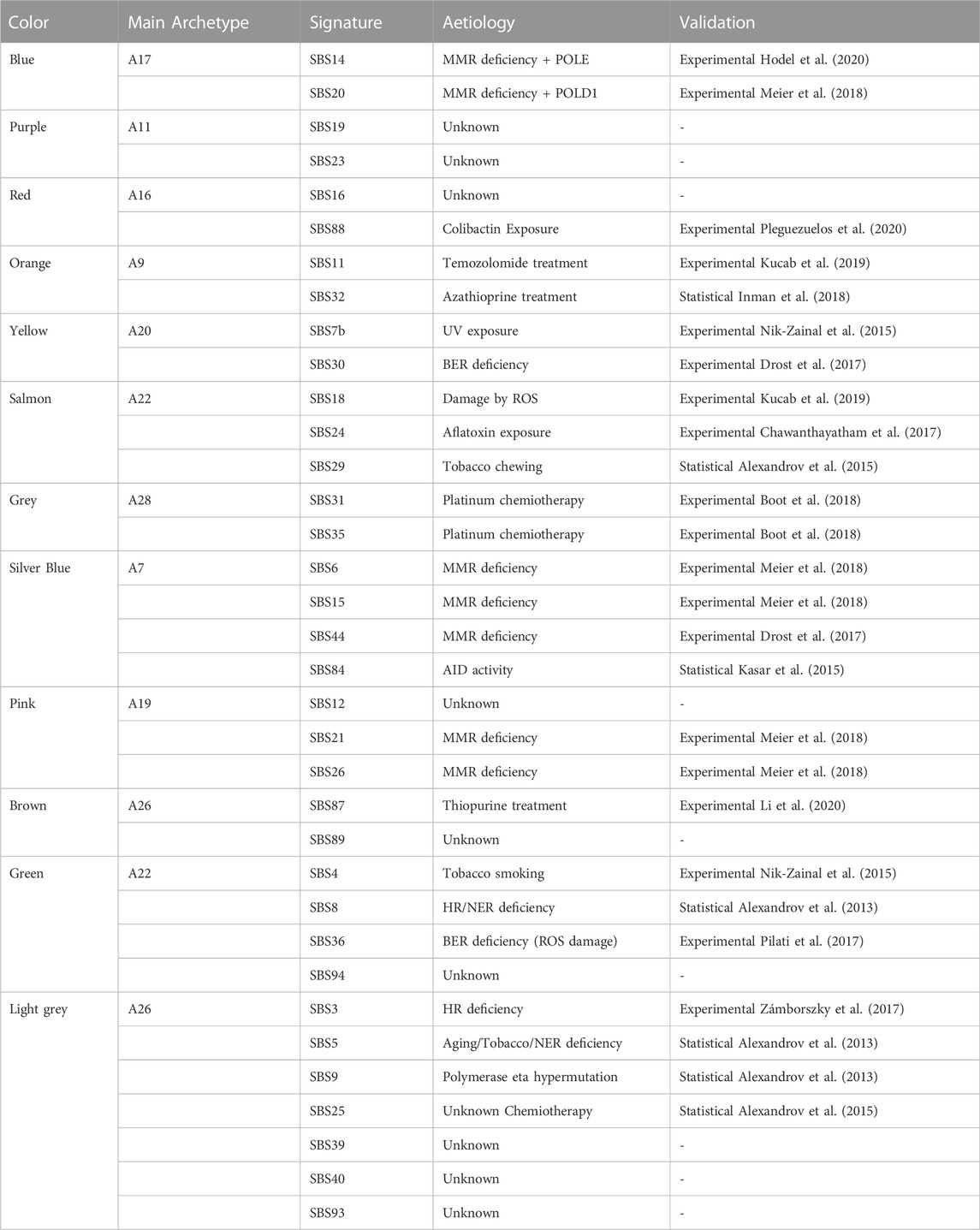
TABLE 3. Aetiological information related to each archetype. For each archetype, the corresponding reconstructed signatures and, when available, their associated aetiologies are reported, indicating the validation studies supporting the biological interpretation.
It is possible to observe that seven signatures (SBS6, SBS14, SBS15, SBS20, SBS21, SBS26, and SBS44), experimentally associated with mismatch repair (MMR) deficiency, are divided into three groups: Blue, Silver Blue and Pink. The Blue group includes two signatures associated with the concurrent effect of MMR deficiency and DNA polymerase (POLD1 and POLE), showing a profile mainly polarised on C
This study investigates the extraction stability issues among the SBS mutational signatures of the most recent version of COSMIC catalogue (3.3). Through a series of simulations considering different scenarios, we showed that high levels of similarity combined with some peculiar (e.g., showing high levels of flatness) signature profiles considerably complicate the de novo extraction. Most of the previous studies evaluated stability issues on COSMIC signatures version 2, which includes 30 signatures. However, here we showed that these issues are becoming more critical in the newest version by evaluating 60 non-artefactual signatures. Although SigProfilerExtractor has been proven to be a robust method for signature extraction, even when the number of samples was high (i.e. up to 5,000), it failed in identifying the correct number of signatures and it succeeded 80% of times with 10,000 samples when we simulated a combined set of six similar signatures at high level of flatness and five similar signatures at a lower level of flatness (scenario 3). Similarly, in scenario 5, considering a higher number of latent signatures (20) with at least each signature highly similar (i.e. pairwise cosine similarity
Although the mutational signatures are not orthogonal by definition, the presence of highly similar signatures, together with the fact that some have a very high level of flatness and there is a lack of an aetiology for many of them, cast some doubts on the real existence of some of these, suggesting that they may be the result of overfitting and hence a mathematical artefact. Several studies already pointed to this issue (Maura et al., 2019; Koh et al., 2021). However, to the best of our knowledge, the most recent assessment of the signature stability observed among COSMIC signatures was performed by Schumann et al. (2019), where they considered the second version of this database, therefore working on half the number of signatures compared to our study and without exploring different scenarios in terms of number of samples, cosine similarity and flat vs non-flat signatures. A limitation of the catalogues used in this work, realised with SigsPack functions, is represented by the random exposure assigned to each latent signature to create the count matrices, subsequently extracted by SigProfilerExtractor. Hence, simulated catalogues may not represent realistic cancer samples. However, this does not affect the technical evaluations of the limitations in the extraction process highlighted by our simulations.
A novelty introduced by this study was the application of AA to investigate whether the information contained in the COSMIC catalogue could be represented more compactly. AA was shown to be an intuitive and straightforward approach to interpret the data like the clustering, but including the flexibility of the matrix factorization (Mørup and Hansen, 2012; Chen et al., 2014). In contrast to the common distance-based approaches, archetypes characterise extremal rather than average properties of the given data and therefore lead to a more compact representation (Abrol and Sharma, 2020). AA is a type of decomposition where convex combinations of extremal points lie on the convex hull of the data and are themselves restricted to being convex combinations of individual observations (Cutler and Breiman, 1994; Mørup and Hansen, 2012). In our study, by applying AA to the COSMIC catalogue, it was possible to identify 29 archetypes able to explain 95% of the variance. Interestingly, it emerged that most of the archetypes correspond almost perfectly (similarity
However, it is worth highlighting that archetypes do not substitute the COSMIC signatures, but emphasise the importance of considering alternative approaches able to reduce redundant information. These observations, together with the lack of known aetiology and experimental validation for many signatures, suggest the need to reformulate the COSMIC catalogue using representations including sparsity constraints in latent vectors during the extraction procedure without loss of information. In the future, archetypal analysis can be also considered to evaluate sparse representations of signatures not only in the context of single base substitutions but also for other types of variants like copy number variations and structural variants (Heller et al., 2020; Steele et al., 2022).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://github.com/compbiomed-unito/archetypal-analysis-cosmic.
Manuscript concept and design: CP and TS; analyses: CP with the support of CR, TS, PF, GB, and SB; manuscript preparation: CP; revision and editing: TS and PF.
This study was supported by the Italian Ministry for Education, University and Research (Ministero dell’Istruzione, dell’Università e della Ricerca- MIUR) under the programmes “Ricerca Locale ex-60%” and “Dipartimenti di Eccellenza 2018–2022” (Project code D15D18000410001).
The authors thank the European Union’s Horizon 2020 projects GenoMed4All (Grant Agreement ID: 101017549) and Brainteaser (Grant Agreement ID: 101017598). We thank PNRR M4C2 HPC – 1.4 “CENTRI NAZIONALI”- Spoke 8 for fellowship support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1049501/full#supplementary-material
Abrol, V., and Sharma, P. (2020). “A geometric approach to archetypal analysis via sparse projections,” in Proceedings of the 37th International Conference on Machine Learning, Vienna, Austria. Editors H. D. III, and A. Singh (PMLR), 42–51.
Aldinucci, M., Bagnasco, S., Lusso, S., Pasteris, P., Rabellino, S., and Vallero, S. (2017). Occam: a flexible, multi-purpose and extendable HPC cluster. J. Phys. Conf. Ser. 898, 082039. IOP Publishing. doi:10.1088/1742-6596/898/8/082039
Aldinucci, M., Rabellino, S., Pironti, M., Spiga, F., Viviani, P., Drocco, M., et al. (2018). “Hpc4ai: an ai-on-demand federated platform endeavour,” in Proceedings of the 15th ACM International Conference on Computing Frontiers, Ischia, Italy, 279–286.
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A., Behjati, S., Biankin, A. V., et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. doi:10.1038/nature12477
Alexandrov, L. B., Jones, P. H., Wedge, D. C., Sale, J. E., Campbell, P. J., Nik-Zainal, S., et al. (2015). Clock-like mutational processes in human somatic cells. Nat. Genet. 47, 1402–1407. doi:10.1038/ng.3441
Alexandrov, L. B., Kim, J., Haradhvala, N. J., Huang, M. N., Tian Ng, A. W., Wu, Y., et al. (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. doi:10.1038/s41586-020-1943-3
An, Y., Shi, X., Tang, X., Wang, Y., Shen, F., Zhang, Q., et al. (2017). Aflatoxin b1 induces reactive oxygen species-mediated autophagy and extracellular trap formation in macrophages. Front. Cell. Infect. Microbiol. 7, 53. doi:10.3389/fcimb.2017.00053
Bagchi, M., Balmoori, J., Bagchi, D., Stohs, S. J., Chakrabarti, J., and Das, D. K. (2002). Role of reactive oxygen species in the development of cytotoxicity with various forms of chewing tobacco and pan masala. Toxicology 179, 247–255. doi:10.1016/s0300-483x(02)00357-8
Bamford, S., Dawson, E., Forbes, S., Clements, J., Pettett, R., Dogan, A., et al. (2004). The cosmic (catalogue of somatic mutations in cancer) database and website. Br. J. Cancer 91, 355–358. doi:10.1038/sj.bjc.6601894
Blokzijl, F., Janssen, R., van Boxtel, R., and Cuppen, E. (2018). Mutationalpatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 10, 33–11. doi:10.1186/s13073-018-0539-0
Boot, A., Huang, M. N., Ng, A. W., Ho, S.-C., Lim, J. Q., Kawakami, Y., et al. (2018). In-depth characterization of the cisplatin mutational signature in human cell lines and in esophageal and liver tumors. Genome Res. 28, 654–665. doi:10.1101/gr.230219.117
Chawanthayatham, S., Valentine, C. C., Fedeles, B. I., Fox, E. J., Loeb, L. A., Levine, S. S., et al. (2017). Mutational spectra of aflatoxin b1 in vivo establish biomarkers of exposure for human hepatocellular carcinoma. Proc. Natl. Acad. Sci. U. S. A. 114, E3101–E3109. doi:10.1073/pnas.1700759114
Chen, Y., Mairal, J., and Harchaoui, Z. (2014). “Fast and robust archetypal analysis for representation learning,” in Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Columbus (Ohio), USA, 1478–1485.
Connell, W. R., Kamm, M. A., Ritchie, J. K., and Lennard-Jones, J. E. (1993). Bone marrow toxicity caused by azathioprine in inflammatory bowel disease: 27 years of experience. Gut 34, 1081–1085. doi:10.1136/gut.34.8.1081
Cutler, A., and Breiman, L. (1994). Archetypal analysis. Technometrics 36, 338–347. doi:10.1080/00401706.1994.10485840
Degasperi, A., Dias Amarante, T., Czarnecki, J., Shooter, S., Zou, X., Glodzik, D., et al. (2020). A practical framework and online tool for mutational signature analyses show inter-tissue variation and driver dependencies. Nat. Cancer 1, 249–263. doi:10.1038/s43018-020-0027-5
Dey, D. K., and Kang, S. C. (2020). Aflatoxin b1 induces reactive oxygen species-dependent caspase-mediated apoptosis in normal human cells, inhibits allium cepa root cell division, and triggers inflammatory response in zebrafish larvae. Sci. Total Environ. 737, 139704. doi:10.1016/j.scitotenv.2020.139704
Drost, J., Van Boxtel, R., Blokzijl, F., Mizutani, T., Sasaki, N., Sasselli, V., et al. (2017). Use of crispr-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 358, 234–238. doi:10.1126/science.aao3130
Heller, D., Vingron, M., Church, G., Li, H., and Garg, S. (2020). Sdip: A novel graph-based approach to haplotype-aware assembly based structural variant calling in targeted segmental duplications sequencing. BioRxiv. doi:10.1101/2020.02.25.964445
Hodel, K. P., Sun, M. J., Ungerleider, N., Park, V. S., Williams, L. G., Bauer, D. L., et al. (2020). Pole mutation spectra are shaped by the mutant allele identity, its abundance, and mismatch repair status. Mol. Cell 78, 1166–1177. doi:10.1016/j.molcel.2020.05.012
Huang, X., Wojtowicz, D., and Przytycka, T. M. (2018). Detecting presence of mutational signatures in cancer with confidence. Bioinformatics 34, 330–337. doi:10.1093/bioinformatics/btx604
Huang, B., Chen, Q., Wang, L., Gao, X., Zhu, W., Mu, P., et al. (2020). Aflatoxin b1 induces neurotoxicity through reactive oxygen species generation, DNA damage, apoptosis, and s-phase cell cycle arrest. Int. J. Mol. Sci. 21, 6517. doi:10.3390/ijms21186517
Inman, G. J., Wang, J., Nagano, A., Alexandrov, L. B., Purdie, K. J., Taylor, R. G., et al. (2018). The genomic landscape of cutaneous scc reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 9, 3667–3714. doi:10.1038/s41467-018-06027-1
Islam, S. M. A., Diaz-Gay, M., Wu, Y., Barnes, M., Vangara, R., Bergstrom, E. N., et al. (2022). Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. Cell Genomics 2, 100179. doi:10.1016/j.xgen.2022.100179
Johnson, S. C. (1967). Hierarchical clustering schemes. Psychometrika 32, 241–254. doi:10.1007/BF02289588
Kasar, S., Kim, J., Improgo, R., Tiao, G., Polak, P., Haradhvala, N., et al. (2015). Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat. Commun. 6, 8866–8912. doi:10.1038/ncomms9866
Koh, G., Degasperi, A., Zou, X., Momen, S., and Nik-Zainal, S. (2021). Mutational signatures: emerging concepts, caveats and clinical applications. Nat. Rev. Cancer 21, 619–637. doi:10.1038/s41568-021-00377-7
Kucab, J. E., Zou, X., Morganella, S., Joel, M., Nanda, A. S., Nagy, E., et al. (2019). A compendium of mutational signatures of environmental agents. Cell 177, 821–836. doi:10.1016/j.cell.2019.03.001
Lal, A., Liu, K., Tibshirani, R., Sidow, A., and Ramazzotti, D. (2021). De novo mutational signature discovery in tumor genomes using sparsesignatures. PLoS Comput. Biol. 17, e1009119. doi:10.1371/journal.pcbi.1009119
Li, B., Brady, S. W., Ma, X., Shen, S., Zhang, Y., Li, Y., et al. (2020). Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 135, 41–55. doi:10.1182/blood.2019002220
Liu, M., and Schatz, D. G. (2009). Balancing AID and DNA repair during somatic hypermutation. Trends Immunol. 30, 173–181. doi:10.1016/j.it.2009.01.007
Martin, A., and Scharff, M. D. (2002). AID and mismatch repair in antibody diversification. Nat. Rev. Immunol. 2, 605–614. doi:10.1038/nri858
Maura, F., Degasperi, A., Nadeu, F., Leongamornlert, D., Davies, H., Moore, L., et al. (2019). A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 10, 2969. doi:10.1038/s41467-019-11037-8
Meier, B., Volkova, N. V., Hong, Y., Schofield, P., Campbell, P. J., Gerstung, M., et al. (2018). Mutational signatures of DNA mismatch repair deficiency in c. elegans and human cancers. Genome Res. 28, 666–675. doi:10.1101/gr.226845.117
Mørup, M., and Hansen, L. K. (2012). Archetypal analysis for machine learning and data mining. Neurocomputing 80, 54–63. doi:10.1016/j.neucom.2011.06.033
Motevalli Soumehsaraei, B., and Barnard, A. (2019). Archetypal analysis package. Canberra, Australia: Commonwealth Scientific and Industrial Research Organisation.
Nik-Zainal, S., Kucab, J. E., Morganella, S., Glodzik, D., Alexandrov, L. B., Arlt, V. M., et al. (2015). The genome as a record of environmental exposure. Mutagenesis 30, 763–770. doi:10.1093/mutage/gev073
Omichessan, H., Severi, G., and Perduca, V. (2019). Computational tools to detect signatures of mutational processes in DNA from tumours: a review and empirical comparison of performance. PloS one 14, e0221235. doi:10.1371/journal.pone.0221235
Pilati, C., Shinde, J., Alexandrov, L. B., Assié, G., André, T., Hélias-Rodzewicz, Z., et al. (2017). Mutational signature analysis identifies mutyh deficiency in colorectal cancers and adrenocortical carcinomas. J. Pathol. 242, 10–15. doi:10.1002/path.4880
Pleguezuelos, C., Puschhof, J., Rosendahl Huber, A., van Hoeck, A., Wood, H. M., Nomburg, J., et al. (2020). Mutational signature in colorectal cancer caused by genotoxic pks+ e. coli. Nature 580, 269–273. doi:10.1038/s41586-020-2080-8
Saha, L. K., Wakasugi, M., Akter, S., Prasad, R., H Wilson, S., Shimizu, N., et al. (2020). Topoisomerase i-driven repair of uv-induced damage in ner-deficient cells. Proc. Natl. Acad. Sci. U. S. A. 117, 14412–14420. doi:10.1073/pnas.1920165117
Schumann, F., Blanc, E., Messerschmidt, C., Blankenstein, T., Busse, A., and Beule, D. (2019). SigsPack, a package for cancer mutational signatures. BMC Bioinforma. 20, 450–459. doi:10.1186/s12859-019-3043-7
Shen, H.-M., Shi, C.-Y., Shen, Y., and Ong, C.-N. (1996). Detection of elevated reactive oxygen species level in cultured rat hepatocytes treated with aflatoxin b1. Free Radic. Biol. Med. 21, 139–146. doi:10.1016/0891-5849(96)00019-6
Steele, C. D., Abbasi, A., Islam, S. M. A., Bowes, A. L., Khandekar, A., Haase, K., et al. (2022). Signatures of copy number alterations in human cancer. Nature 606, 984–991. doi:10.1038/s41586-022-04738-6
Stich, H. F., and Anders, F. (1989). The involvement of reactive oxygen species in oral cancers of betel quid/tobacco chewers. Mutat. Res. 214, 47–61. doi:10.1016/0027-5107(89)90197-8
Sylvester, R. K., Steen, P., M Tate, J., Menta, M., J Petrich, R., Berg, A., et al. (2011). Temozolomide-induced severe myelosuppression: analysis of clinically associated polymorphisms in two patients. Anticancer. Drugs 22, 104–110. doi:10.1097/CAD.0b013e3283407e9f
Zámborszky, J., Szikriszt, B., Gervai, J. Z., Pipek, O., Póti, Á., Krzystanek, M., et al. (2017). Loss of brca1 or brca2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 36, 746–755. doi:10.1038/onc.2016.243
Keywords: archetypal analysis, mutational signatures, matrix factorization, COSMIC, cancer genomics
Citation: Pancotti C, Rollo C, Birolo G, Benevenuta S, Fariselli P and Sanavia T (2023) Unravelling the instability of mutational signatures extraction via archetypal analysis. Front. Genet. 13:1049501. doi: 10.3389/fgene.2022.1049501
Received: 20 September 2022; Accepted: 07 December 2022;
Published: 04 January 2023.
Edited by:
Federico Zambelli, University of Milan, ItalyReviewed by:
Rosario Michael Piro, Politecnico di Milano, ItalyCopyright © 2023 Pancotti, Rollo, Birolo, Benevenuta, Fariselli and Sanavia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiziana Sanavia, dGl6aWFuYS5zYW5hdmlhQHVuaXRvLml0; Piero Fariselli, cGllcm8uZmFyaXNlbGxpQHVuaXRvLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.