 Dan Lian
Dan Lian Jiufeng Wei
Jiufeng Wei Chao Chen1
Chao Chen1 Qing Zhao
Qing Zhao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 11 November 2022
Sec. Computational Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1045193
The Phyllocephalini is a group of herbivorous insects in Pentatomidae, which lack distinctive morphological characteristics and systematic studies. Up to now, there are only two complete mitochondrial genomes of Phyllocephalini have been reported. In this study, we sequenced and analyzed the complete mitochondrial genomes of three Phyllocephalini species, Gonopsis coccinea, Gonopsimorpha nigrosignata, and Chalcopis glandulosus, which were 16,534, 16,531, and 16,534 bp in length, respectively. The mitochondrial genomes contained 37 genes, including 13 protein-coding genes, two rRNA genes, 22 tRNA genes, and a control region. The gene arrangement was consistent with that of the putative ancestral insect, with no rearrangement. The cox1 gene of Pentatomidae showed the lowest evolutionary rate among the protein-coding genes, the mean genetic distance of species, genera, and subfamilies of Pentatomidae increased hierarchically based on cox1 gene. The 16S rRNA of Pentatomidae was more conserved than 12S rRNA in sequence and secondary structure. All tRNAs could be folded into a typical cloverleaf structure except trnS1. The stem region was more conserved than the loop region in the secondary structure of tRNAs within Pentatomidae. Gonopsis coccinea and Gonopsimorpha nigrosignata had one type of tandem repetition unit in the control region, while C. glandulosus had two types. The heterogeneity analysis of Pentatomidae showed that Phyllocephalinae was the most heterogeneous. Phylogenetic trees based on the newly obtain mitochondrial genomes along with other 50 mitochondrial genomes of Pentatomidae using Bayesian Inference and Maximum Likelihood strongly supported the following three relationships: (((Anaxilaus + (Plautia + Glaucias)) + (Nezara + Palomena)) + (Eysarcorini + Carpocorini)), (Hoplistoderini + (Menidini + Asopinae)), and ((Sephelini + Halyini) + (Caystrini + (Cappaeini + (Placosternum + Phyllocephalini)))). The relationships within Phyllocephalini were (Chalcopis + (Dalsira + (Gonopsimorpha + Gonopsis))). Our results provide valuable molecular data for further phylogenetic analyses of Pentatomidae.
Pentatomidae is one of the largest and most diverse families in Heteroptera with respect to morphology and behavior (Rider et al., 2018). Numerous species are herbivorous, harming several important crops and causing significant economic losses around the world. For example, Erthesina fullo (Thunberg, 1783) is widely distributed in Asia and feeds on over 57 host plants in approximately 30 families in China (Mi et al., 2020). In addition, most species of Asopinae (Heteroptera: Pentatomidae) are predatory stink bugs that feed on the larvae of various Lepidoptera, Coleoptera, and Hemiptera, such as Zicrona caerulea (Linnaeus, 1758) and Picromerus griseus (Dallas, 1851) (Zhao Q. et al., 2017; Zhao et al., 2020).
Recent phylogenetic studies based on multigene fragments have rejected the monophyly of Pentatomidae, whereas earlier studies based on morphology and molecules supported the monophy of Pentatomidae (Grazia et al., 2008; Roca-Cusachs et al., 2021). The early classification of Pentatomidae subfamilies and tribes was mostly based on morphological data, Rider et al. (2018) divided the family into ten subfamilies and provided an extensive description and discussion of the morphology of each subfamily and tribe within Pentatomidae. However, phenotypic plasticity was a major obstacle to morphological taxonomy (Barao et al., 2013; Sheikh, 2017; Sharif et al., 2020; Genevcius et al., 2021). With the widespread application of molecular technology, molecular data are more and more used in classification. Genevcius et al. (2021) made a preliminary discussion on the main phylogenetic relationships within Pentatomidae based on a combination of morphological and molecular data. In the most detailed analysis to date, which examined the phylogenetic relationships of the whole family of Pentatomidae based on mitochondrial (cox1 and 16S rRNA) and nuclear gene fragments (18S and 28S rRNA), Cyrtocorinae was excluded from ten subfamilies. (Roca-Cusachs et al., 2021). Nonetheless, because only Gonopsis Amyot & Serville, 1843 and Minchamia Gross, 1976 were included, the gene fragment-based analysis did not recover the phylogenetic relationships within Phyllocephalini (Pentatomidae: Phyllocephalinae) (Roca-Cusachs et al., 2021). Obviously, additional data are needed to resolve the relationships among some genera, tribes, and subfamilies of Pentatomidae.
Phyllocephalini is the largest tribe in Phyllocephalinae, with 167 species in 32 genera that feed on gramineous plants, but only 11 species in six genera have been reported in China (Kamaluddin and Ahmad, 1988; David and Zheng, 2005; Rider et al., 2018). Unlike other tribes, the tribe lacks unique and remarkable diagnostic features. Members of Phyllocephalini can be distinguished by a somewhat ovate body, short shovel-shaped head, broadly ligulate scutellum, and quadrangular pygophore; however, these characteristics may also apply to Cressonini (Linnavuori, 1982; Kamaluddin and Ahmad, 1988; Rider et al., 2018). Since the 1980s or early 1990s, systematic analyses of Phyllocephalini are rare (Linnavuori, 1982; Ahmad and Kamaluddin, 1988; Kamaluddin and Ahmad, 1988; Ahmad, 1990). The taxonomic status of the Chalcopis glandulosus (Wolff, 1811) has not been determined, with some Chinese scholars suggesting that it should be placed in the Metonymia Kirkaldy, 1909, which is a junior synonym of the Dalsira Amyot & Serville, 1843, while Linnavuori suggested that it should be placed in the Chalcopis (Kirkaldy, 1909), which is quite different from Metonymia in terms of morphological characteristics (Yang, 1962; David and Zheng, 2005). Accordingly, intensive sampling and genome sequencing of species belonging to Phyllocephalini are needed (Zheng et al., 2022).
Insect mitochondrial genomes (mitogenomes) are usually double-stranded circular DNA molecules, consisting of 37 genes: 13 protein-coding genes (PCGs), two ribosomal RNA genes (rRNAs), 22 transfer RNA genes (tRNAs), and a control region (CR) of variable length at the origin of replication and transcription (Wolstenholme, 1992; Boore, 1999; Stephen, 2014). Mitogenomes contain information that is crucial to molecular evolution, such as base compositional bias, codon usage, and substitution rate (Yuan et al., 2022). In addition, mitogenomes are widely used in phylogenetic studies owing to their highly conserved gene arrangement and relatively high rate of evolution (Chris et al., 2006; Zhu et al., 2017; Liu et al., 2019). Furthermore, removing the third codon of PCGs can reduce the adverse effects of heterogeneity and saturation on phylogenetic analyses (Xu et al., 2021). To date, only 50 complete or near-complete mitogenomes of Pentatomidae have been reported in GenBank, just only two of which are from Phyllocephalini. Therefore, limited sampling and molecular markers may hinder phylogenetic studies of Pentatomidae at various taxonomic levels.
In the present study, we sequenced and annotated the mitogenomes of Gonopsis coccinea (Walker, 1868), Gonopsimorpha nigrosignata (Yang, 1934), and C. glandulosus (Wolff, 1811), representing three genera of Phyllocephalini. The features of these mitogenomes, such as the base composition and genetic structure, were also analyzed. We mapped the secondary structures of tRNAs and rRNAs of the three mitogenomes as well as the structure of the control region. The sequence and secondary structure of the RNAs of Pentatomidae were compared and analyzed. We also calculated the evolutionary rate, codon usage, and heterogeneity of PCGs in Pentatomidae. In addition, we analyzed the genetic distances among taxa at different levels in the Pentatomidae based on the cox1 sequence and reconstructed the phylogenetic relationships based on PCGs using BI and ML. This study not only revealed the phylogenetic positions of the three genera but also provided more mitogenome data on phylogenetic relationship of Pentatomidae.
The specimens of Gonopsis coccinea, Gonopsimorpha nigrosignata, and C. glandulosus used in this study were collected in May 2020 from Chongzuo (107°23′6.72″ E, 22°22′48.16″ N), Jingxi (106°25′21.72″ E, 23°8′30.84″ N) and Fangchenggang (108°21′24.84″ E, 21°41′40.56″ N) in Guangxi Province, China, respectively, then immediately immersed in 100% ethanol, and stored at −20°C. Species were identified by Qing Zhao based on adult morphology. Subfamily and tribe classification followed the nine-subfamily classification system (Rider et al., 2018; Roca-Cusachs et al., 2021). These three newly sequenced mitogenomes were submitted to GenBank with accession numbers ON991492, ON991493, and ON991494.
Genomic DNA was extracted from the thoracic muscle of each specimen using the HiPure universal DNA Kitt (Jisi Huiyuan biotechnology, Nanjing, China). The mitogenomes were sequenced on the Illumina NovaSeq platform. Paired-end 150-bp (PE150) sequencing yielded approximately 5.7 Gb (Gonopsis coccinea), 4.8 Gb (Gonopsimorpha nigrosignata), and 7.3 Gb (C. glandulosus) data. Fastp v.0.20.0 (https://github.com/OpenGene/fastp) was used to filter the original data, and high-quality clean data were obtained by removing adaptor sequences and low-quality reads (Chen et al., 2018).
The mitogenomes were assembled using SPAdes v.3.10.1 with default parameters (Bankevich et al., 2012). The three new mitogenomes were annotated using Geneious Prime 2022.1 (Kearse et al., 2012) with a Dalsira scabrata Distant, 1901 obtained by BLAST in the NCBI database as a reference. The PCG boundaries were manually adjusted according to the reference sequence. To ensure the accuracy of the sequences, all PCG sequences were translated to verify amino acid sequences using Muscle as implemented in MEGA v.11.0 (Tamura et al., 2021). The locations and secondary structure of tRNA genes were confirmed using the MITOS web server (Bernt et al., 2013) with the invertebrate mitochondrial code. The rRNA gene boundaries were determined by aligning with available mitogenomes of related species. The locations of the control region were identified by the boundary of neighboring genes.
The base composition and relative synonymous codon usage (RSCU) were analyzed using MEGA v.11.0. The skew of the nucleotide composition was calculated as follows: AT skew = (A− T)/(A+ T); GC skew = (G − C)/(G + C) (Perna and Kocher, 1995). The number of non-synonymous substitutions per nonsynonymous site (Ka) and synonymous substitutions per synonymous site (Ks) for each PCG was calculated using DnaSP v.6.12.03 (Rozas et al., 2017), with the exclusion of stop codons. Under the Kimura 2-parameter model, genetic distances between 53 Pentatomidae species were calculated for the cox1 gene. Tandem repeats in control regions were identified using the Tandem Repeats Finder web server (Benson, 1999). The tRNA genes were also aligned using the MUSCLE algorithm in MEGA v.11.0. The G-INS-i algorithm was selected for rRNA alignment in MAFFT v.7.490 (Katoh and Standley, 2013). Conserved sites of tRNA and rRNA were calculated using MEGA v.11.0 and annotated in their secondary structures.
Phylogenies were constructed using three newly sequenced mitogenomes and 50 species representing four subfamilies (Asopinae, Pentatominae, Phyllocephalinae, and Podopinae) of Pentatomidae, with two Scutelleridae species as outgroups (Table 1). The nucleotide sequences of each gene were extracted using Geneious Prime 2022.1. All PCGs were translated into amino acid sequences with the invertebrate mitochondrial code and aligned using MUSCLE with default parameters in MEGA v.11.0. All alignments were concatenated into a single data matrix using SequenceMatrix v.1.7.8 (Vaidya et al., 2011). To determine whether the sequences contained sufficient phylogenetic information, we tested nucleotide substitution saturation and plotted transition and transversion rates against the TN93 distances for all datasets using DAMBE v.7.3.11 (Xia, 2018). Heterogeneity in sequence divergences within the two datasets was analyzed by using AliGROOVE with the default sliding window size (Kück et al., 2014). Before reconstructing the BI trees, PartitionFinder v.2.1.1 (Lanfear et al., 2017) was used to select the optimal partitioning models, and phylogenetic analyses were conducted in MrBayes v.3.2.7 (Ronquist et al., 2012) with four independent Markov chains (three heated and one cold) run for 10,000,000 generations and sampled every 1,000 cycles. The initial 25% of trees were discarded as burn-in when the average standard deviation value was below 0.01. ML trees were reconstructed using IQ-Tree v.1.6.12 (Nguyen et al., 2015). The ModelFinder (Kalyaanamoorthy et al., 2017) implemented in the IQ-Tree was used to estimate substitution models under the Bayesian information criterion (BIC), and node support was calculated through 100,000 ultrafast bootstraps. The phylogenetic trees were constructed using two datasets: 1) all codon positions of PCGs (P123), 2) first and second codon positions of PCGs (P12). The final tree was drawn using FigTree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
TABLE 1. The species used in phylogenetic analyses.
The total lengths of the mitogenomes of Gonopsis coccinea, Gonopsimorpha nigrosignata, and C. glandulosus were 16,534 bp, 16,531 bp, and 16,534 bp, respectively (Figure 1). The mitochondrial genomes of all three species were closed circular double-stranded molecules composed of 37 coding genes (13 PCGs, two rRNAs, and 22 tRNAs) and a non-coding control region (Figure 1, Supplementary Table S1). The gene arrangements in the three mitogenomes were identical, with 23 genes (nine PCGs and 14 tRNAs) located on the majority strand (J-strand) and 14 genes (four PCGs, eight tRNAs and two rRNAs) on the minority strand (N-strand) (Supplementary Table S1). The nucleotide composition of whole mitogenomes showed a significant bias toward A and T bases, with A + T contents of 78.93% (Gonopsis coccinea), 77.13% (Gonopsimorpha nigrosignata), and 78.94% (C. glandulosus). The AT skew was positive in all cases, while the GC skew was negative (Supplementary Table S2).
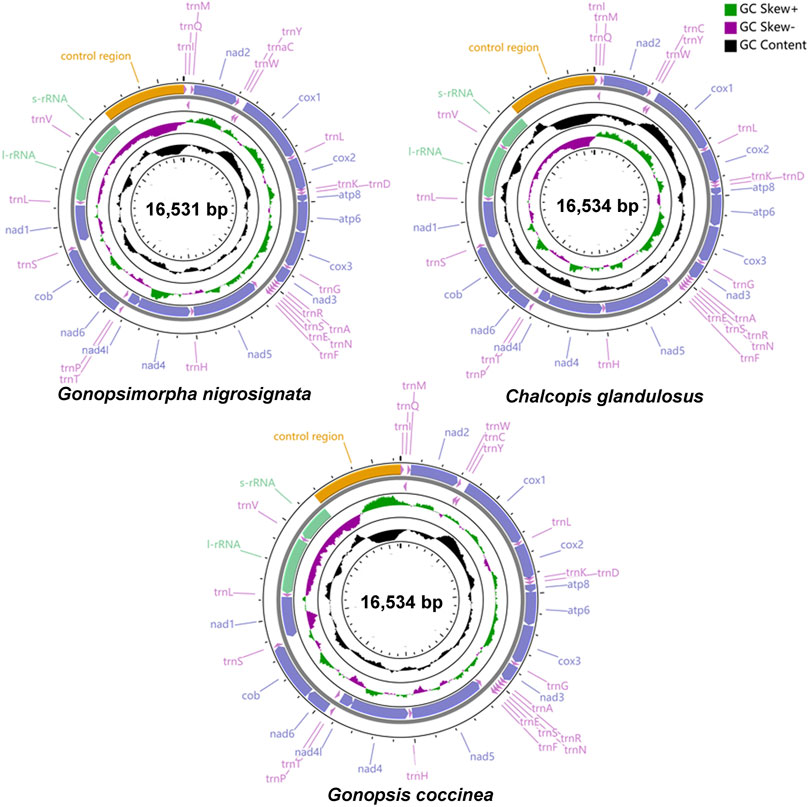
FIGURE 1. Gene structure of three complete mitochondrial genomes.
The longest intergenic spacers were detected between nad1 and trnS1, ranging from 22 to 28 bp in length. There was a conserved 2 bp gene spacer between nad4L and trnT, and a conserved 1 bp interval between trnH and nad5. The longest gene overlap was located between trnC and trnW, with a length of 8 bp. Several regions of conserved gene overlap were also observed in the three mitogenomes, including atp8/atp6 (7 bp), trnN/trnS1 (1 bp), trnE/trnS1 (1 bp), and nad4/nad4L (7 bp) (Supplementary Table S1).
The total lengths of the 13 PCGs of Gonopsis coccinea, Gonopsimorpha nigrosignata, and C. glandulosus were 11,040 bp, 10,992 bp, and 11,022 bp. The nucleotide composition of the 13 PCGs showed high A+ T contents, i.e., 77.73% (Gonopsis coccinea), 75.81% (Gonopsimorpha nigrosignata), and 77.90% (C. glandulosus). A moderate negative AT skew and negligible positive GC skew was observed in 13 PCGs in the three mitogenomes (except in Gonopsimorpha nigrosignata) (Supplementary Table S2). Of all 13 PCGs in the three mitogenomes, nine (nad2, cox1, cox2, atp8, atp6, cox3, nad3, nad6, and cob) were located on the J-strand, and the other four (nad5, nad4, nad4L, and nad1) were found on the N-strand. Among the 13 PCGs, atp8 (159–162 bp) was the shortest, while nad5 (1708–1717 bp) was the longest. Most PCGs were initiated by ATN (ATT/ATA/ATG/ATC) as the start codon, while TTG was the second most common start codon, detected in cox1, nad6, (C. glandulosus), and atp8. Most PCGs ended with the complete termination codon TTA or TAG, except for cox1, (except in Gonopsimorpha nigrosignata), cox2, and nad5, which ended with the incomplete stop codon T (Supplementary Table S1).
We calculated RSCU of PCGs for three species of Phyllocephalini, four subfamilies of Pentatomidae, and 53 species of Pentatomidae respectively (Figure 2; Supplementary Figure S1, S2). The similar RSCU pattern was observed in all the pentatomidae. Among all the codons, UUA (L2) was found to be the most frequently used codon, and we observed that most of the codons with high frequency ended in AT, indicating that a preference for AT over GC in the codon composition of Pentatomidae mitogenomes. Among the four subfamilies of Pentatomidae, the predatory Asopinae showed a weaker preference for RSCU than the other three subfamilies (Figure 2; Supplementary Figure S1).
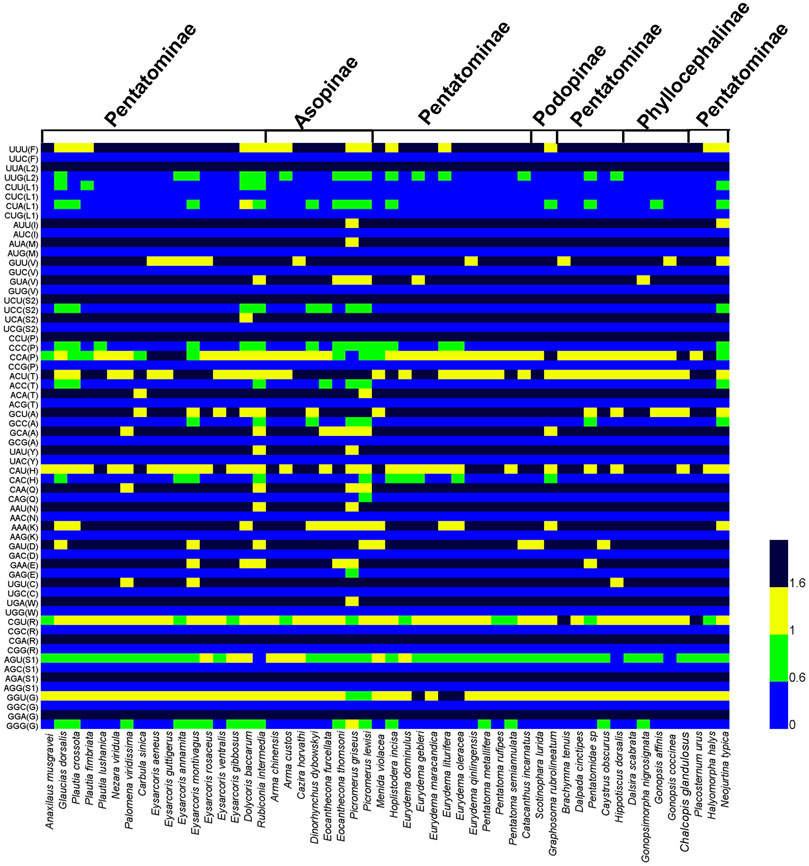
FIGURE 2. Heatmap analysis of relative synonymous codon usage (RSCU) values among the 53 species of Pentatomidae.
In addition, we evaluated the synonymous substitution rate (Ks), non-synonymous substitution rate (Ka), and the Ka/Ks ratio of each PCG to explore patterns of evolution in Pentatomidae (Figure 3). The Ka/Ks ratios for all 13 PCGs were below 0.74, indicating that the genes were under purifying selection. Therefore, all of the PCGs could be used to analyze phylogenetic relationships within Pentatomidae. Among the PCGs, atp8 had the highest Ka/Ks ratio, while cox1 had the lowest value. Owing to its slow rate of evolution, cox1 is commonly used for barcoding analyses and used for genus or species identification (Hebert et al., 2004). We also calculated pairwise genetic distances at different taxonomic levels in Pentatomidae based on cox1 sequences and compared the results with those for Pentatomomorpha and Heteroptera (Park et al., 2011; Tembe et al., 2014) (Table 2). We observed a hierarchical increase in the mean genetic distance across different taxonomic levels. The mean interspecific genetic distance was 15.86% with a range of 2.27%–22.05%. The genetic distance amongst genera averaged 16.30%, with a range of 11.03%–21.02%. The average genetic distance between subfamilies was 16.80%, with a range of 16.06%–17.66%.
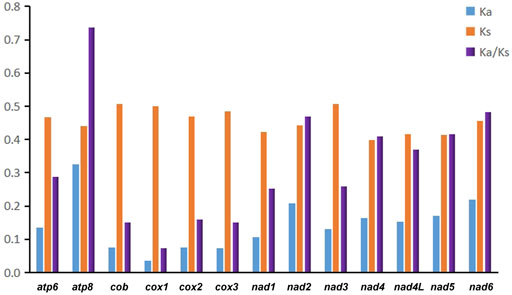
FIGURE 3. The Ka (non-synonymous substitutions), Ks (synonymous substitutions), and Ka/Ks values of each PCGs in Pentatomidae.

TABLE 2. The genetic distances (K2P) at various taxonomic levels in Pentatomidae.
The full lengths of 22 tRNAs of Gonopsis coccinea, Gonopsimorpha nigrosignata, and C. glandulosus were 1,477 bp, 1,478 bp, and 1,473 bp, respectively. The 22 tRNAs showed high A+ T contents of 78.27% (Gonopsis coccinea), 76.59% (Gonopsimorpha nigrosignata), and 78.28% (C. glandulosus), and positive AT and GC skews were also observed (Supplementary Table S2). Fourteen tRNAs (trnI, trnM, trnW, trnL2, trnK, trnD, trnG, trnA, trnR, trnN, trnS1, trnE, trnT, and trnS2) were encoded by the J-strand and the remaining eight (trnQ, trnC, trnY, trnF, trnH, trnP, trnL1, and trnV) were encoded by the N-strand. The lengths of the 22 tRNAs of the three mitogenomes ranged from 63 to 74 bp (Supplementary Table S1). All tRNAs could be folded into typical cloverleaf structures containing of four stems, except for trnS1, whose dihydropyridine (DHU) stem was replaced with a simple loop (Figure 4). In the secondary structures of tRNAs of the Pentatomidae, an anticodon loop (7 nt), anticodon stem (5 bp, except for trnS1), and acceptor stem (7 bp) were conserved in length, while the lengths of the DHU stem (2–4 bp) and TψC stem (3–5 bp, except for trnC) were variable. Among the 22 tRNAs in the Pentatomidae, trnL2 was the most conserved, with the complete conservation of four stems and only a small number of unconserved bases in the loop, while trnV was the least conserved. All tRNAs of the Pentatomidae use the standard anticodon, except for trnV of Gonopsis affinis (Uhler, 1860). Additionally, we observed mismatched base pairs (GT), which play an important role in maintaining the stability of the tRNA secondary structure and can be restored by post-transcriptional editing (Ojala et al., 1981).
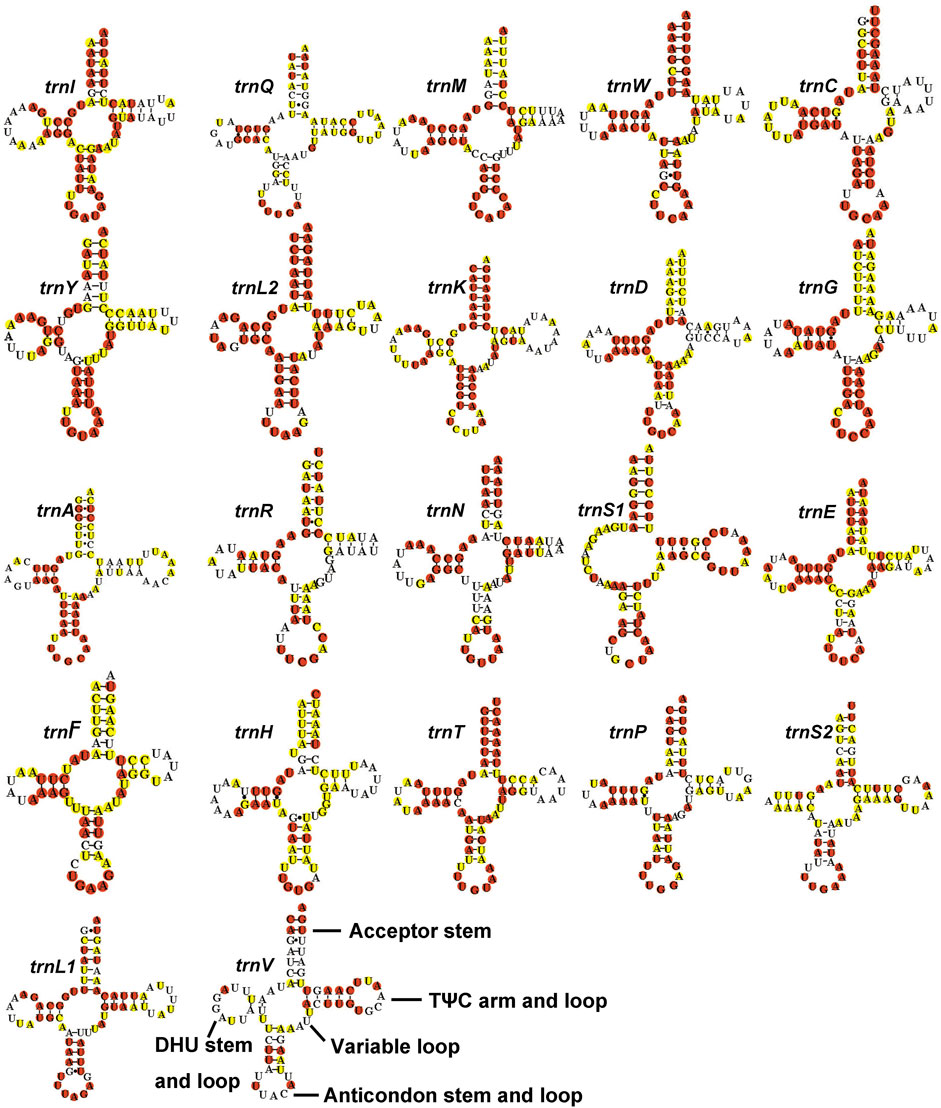
FIGURE 4. The potential secondary structure of the tRNA in Gonopsis coccinea. Conserved sites are marked red in Pentatomidae and yellow in Phyllocephalinae.
The total lengths of two rRNAs of Gonopsis coccinea, Gonopsimorpha nigrosignata, and C. glandulosus were 2,104 bp, 2,079 bp, and 2,080 bp. The two rRNAs showed high A+ T contents of 79.42% (Gonopsis coccinea), 78.69% (Gonopsimorpha nigrosignata), and 80.14% (C. glandulosus), and a negative AT skew and positive GC skew were also observed (Supplementary Table S2). The two rRNAs were encoded on the N-strand in the three mitogenomes. The 12S rRNA gene ranged from 797 bp (Gonopsimorpha nigrosignata) to 815 bp (Gonopsis coccinea) in length and was located between trnV and the control region (Supplementary Table S1). In the 12S rRNA secondary structure of Pentatomidae, domain III was more conserved than domains I and II (Figure 5). The 16S rRNA gene was observed between trnL1 and trnV, with lengths ranging from 1,282 bp to 1,289 bp. In the secondary structure of 16S rRNA, domains IV and V were more conserved than domains I, II, and VI within Pentatomidae (Figure 6).
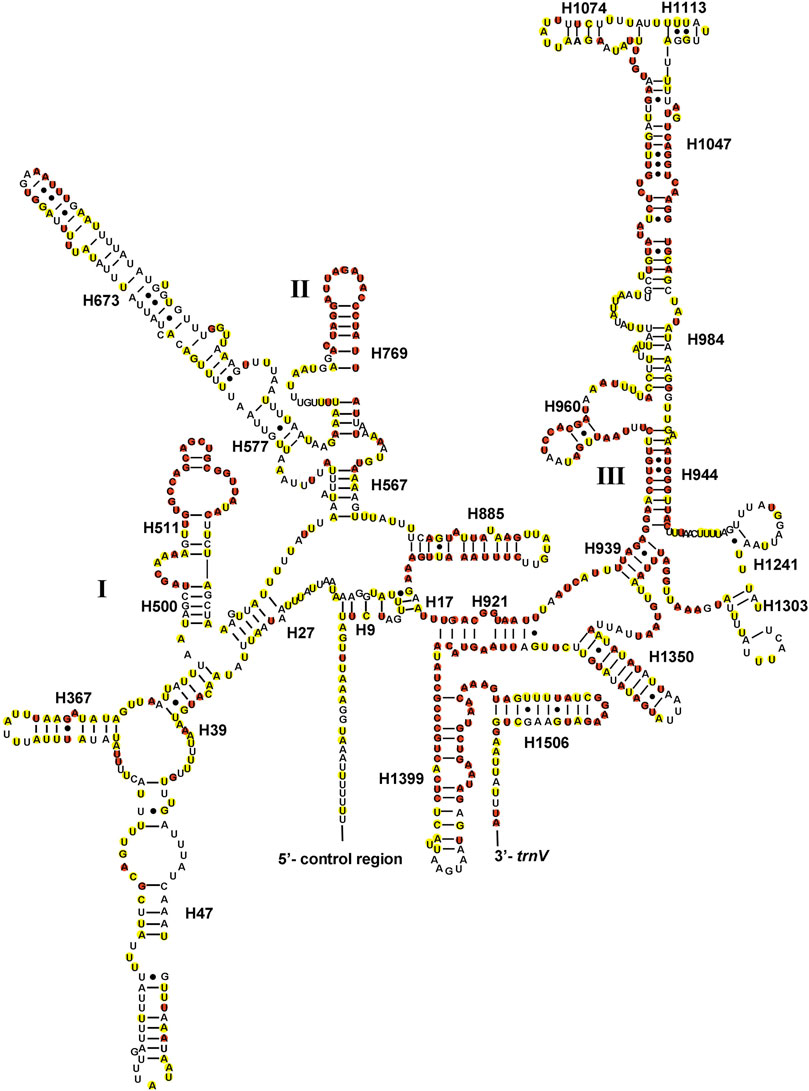
FIGURE 5. The potential secondary structure of the 12S rRNA in Gonopsis coccinea. Conserved sites are marked red in Pentatomidae and yellow in Phyllocephalinae.
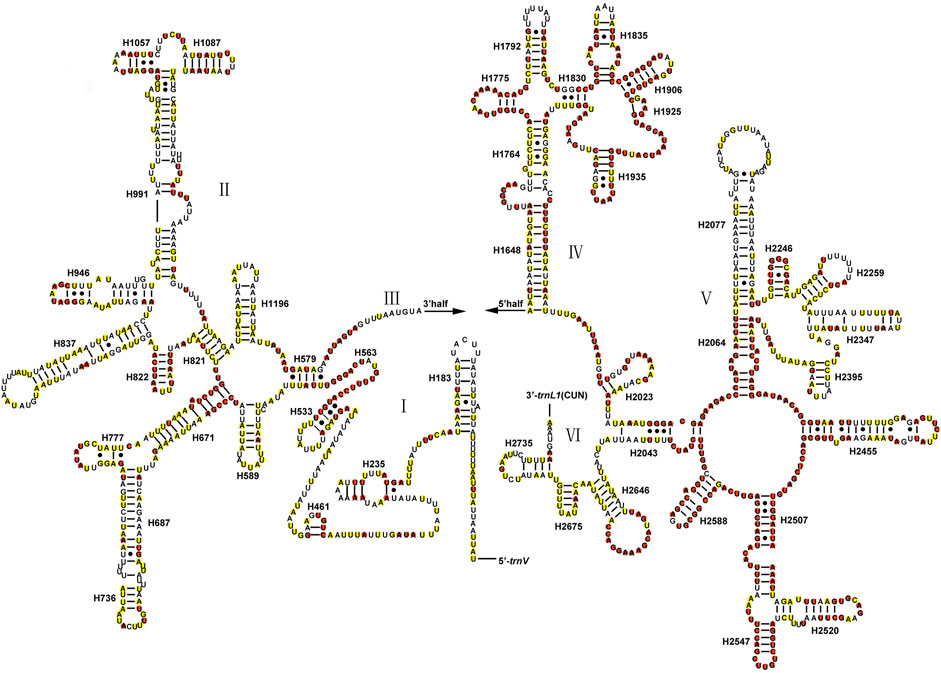
FIGURE 6. The potential secondary structure of the 16S rRNA in Gonopsis coccinea. Conserved sites are marked red in Pentatomidae and yellow in Phyllocephalinae.
The control region is the largest non-coding region. The control regions of the three mitogenomes were located between 12S rRNA and trnI, with lengths of 1,817 bp (Gonopsis coccinea), 1,820 bp (Gonopsimorpha nigrosignata), and 1,881 bp (C. glandulosus), respectively. All three species showed extremely high A+ T contents of 85.25% (Gonopsis coccinea), 82.53% (Gonopsimorpha nigrosignata), and 83.63% (C. glandulosus). Gonopsis coccinea showed moderate positive AT and negative GC skews, while the remaining species showed a slight negative AT skew and strong negative GC skew. Only one type of tandem repeat unit was found in the control region of Gonopsis coccinea and Gonopsimorpha nigrosignata, with lengths of 44 bp and 283 bp, respectively. Two types of tandem repeat units were observed in the control region of C. glandulosus, separated by a 621 bp non-repeat region (Figure 7).
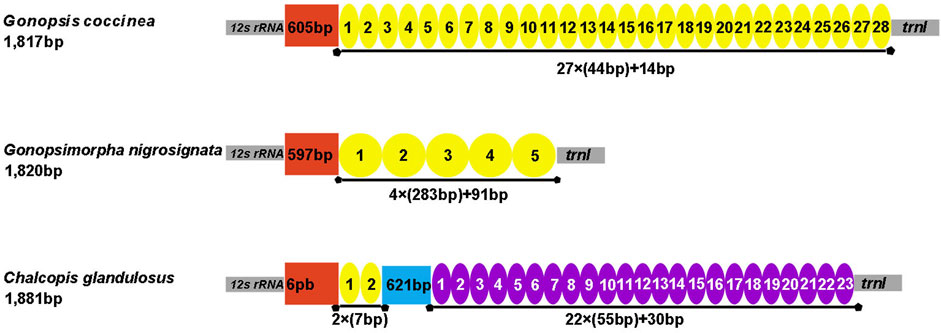
FIGURE 7. Organization of the control region in the three mitochondrial genomes. The location and copy number of tandem repeats are shown by a colored oval with repeat length. Non-repeat regions are shown by the colored box with sequence size inside.
We evaluated substitution saturation in the two PCG datasets (P12 and P123) before the phylogenetic analysis. The saturation index ISS (developed by Xia) for the two datasets was significantly lower than those for a symmetric (Iss.cSym) and asymmetric (Iss.cAsym) topology, indicating that the two data sets were not saturated. The ISS value of P123 was higher than that of P12, indicating that the P123 dataset was more saturated than P12. Furthermore, there was a linear correlation between the base transition and transversion rates and the modified genetic distance, which indicated that the nucleotide sequences of PCGs were generally unsaturated and suitable for phylogenetic analyses of Pentatomidae (Figure 8).
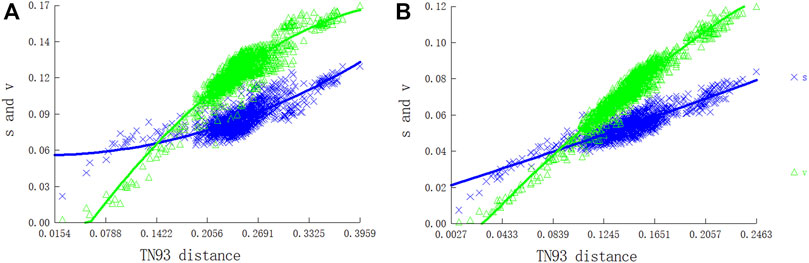
FIGURE 8. Nucleotide substitution saturation plots of all mitochondrial protein-coding genes: (A) All codon positions; (B) First and second codon positions.
An AliGROOVE analysis of the two datasets showed that the degree of heterogeneity of the P123 dataset was higher than that of the P12 dataset. The third codon position had higher heterogeneity, as expected. Among the four subfamilies of Pentatomidae, Phyllocephalinae showed the highest heterogeneity. Neojurtina typica Distant, 1921 showed high heterogeneity within Pentatominae (Figure 9).
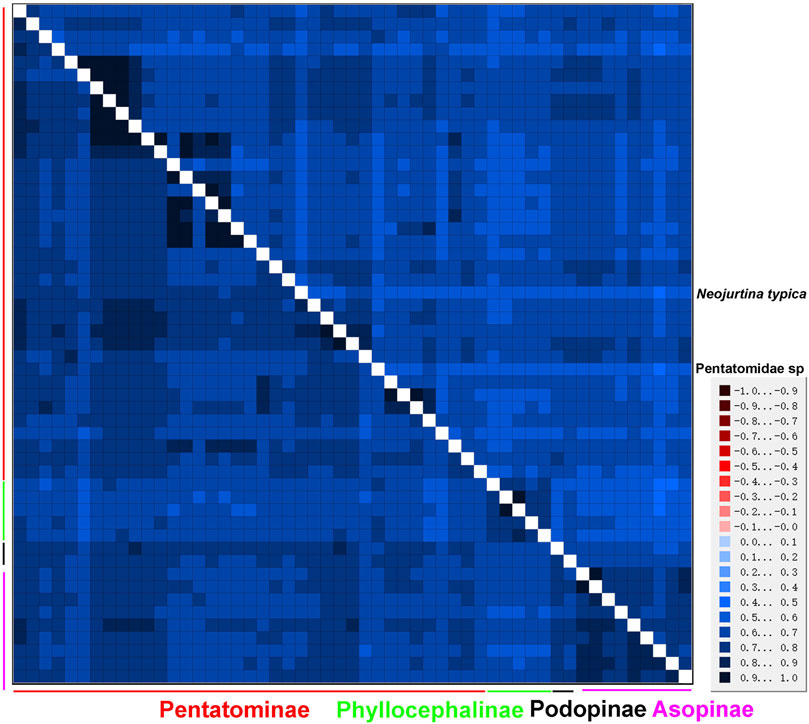
FIGURE 9. AliGROOVE analysis of 53 Pentatomidae species based on PCG12 (lower triangle) and PCG123 (upper triangle). The mean similarity score between sequences is represented by colored squares, based on AliGROOVE scores ranging from −1, which indicates a great difference in rates from the remainder of the data set, that is, heterogeneity (red coloring), to +1, which indicates rates that matched all other comparisons (blue coloring).
We constructed phylogenetic trees of Pentatomidae based on the two data sets using BI and ML (Figure 10, 11, Supplementary Figure S3, S4). The topological structure of the four trees was highly consistent, and most clades had high posterior probabilities. All analyses showed that N. typica was the earliest diverging lineage within Pentatomidae. Asopinae and Phyllocephalinae were recovered as monophyletic groups with strong support values and high internal node support values. Asopinae cannot be subdivided into tribes, and the topology of the internal genera in the P123 tree was ((Arma + Zicrona) + ((Cazira + Dinorhynchus) + (Eocanthecona + Picromerus))). However, the sister group relationship of Cazira and Dinorhynchus was not recovered in the remaining three phylogenetic trees. The species used for the phylogenetic analysis of the Phyllocephalinae all belonged to Phyllocephalini. The internal topology of Phyllocephalini was consistently (Chalcopis + (Dalsira + (Gonopsimorpha + Gonopsis))) with high internal node support. The monophyly of both Pentatominae and Podopinae was rejected, and tribal-level relationships were partially recovered in our analyses. Eysarcorini and Strachiini were recovered as monophyletic with strong support, while the monophyly of Antestiini, Nezarini, and Pentatomini was rejected. The monophyly of the remaining tribes was not clear owing to the limited availability of mitogenomes in the NCBI database.
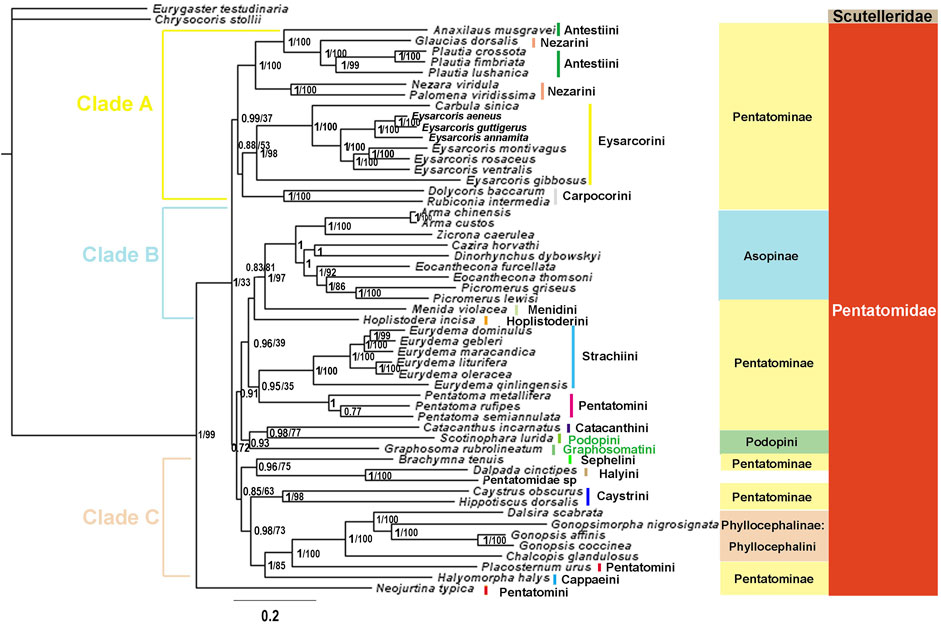
FIGURE 10. The phylogenetic trees of tribes within Pentatomidae were constructed from DNA sequences of 13 PCGs using BI and ML methods. Numbers on branches are posterior probabilities (PP, left) and bootstrap (BS, right). Note: Branches without BS values indicate that the topology is only supported by BI.

FIGURE 11. The phylogenetic trees of tribes within Pentatomidae were constructed from DNA sequences of 13 PCGs excluding the third site using BI and ML methods. Numbers on branches are posterior probabilities (PP, left) and bootstrap (BS, right). Note: Branches without BS values indicate that the topology is only supported by BI.
Most PCGs in the three mitogenomes used the common triplet codon ATN as the start codon, while cox1 and atp8 used TTG as the start codon, which was also found in the mitochondrial genomes of many species within Pentatomidae (Zhao W. et al., 2017; Wang et al., 2017). In addition, nad6 of C. glandulosus used TTG as the start codon, whereas ATN was used in the other two species. Most PCGs utilized TAA as the stop codon, while three PCGs (cox1, cox2, and nad5) used a single T, as observed in most Pentatomidae mitogenomes (Zhao W. et al., 2017). Nevertheless, nad1, nad4, and cob of C. glandulosus used TAG as the stop codon. Our results suggest that some PCGs in C. glandulosus exhibit distinct start and stop codons from homologues in the other two species of Phyllocephalini. In addition, the genetic distances calculated based on the cox1 gene showed that, among the four subfamilies, Asopinae and Phyllocephalinae were the most distantly related, as reflected in the rostrum structures. However, Pentatominae and Podopinae were closely related. This may be one reason why the species of Podopinae in the phylogenetic tree were mixed in Pentatominae. Glaucias Kirkaldy, 1908 and Picromerus were the most distantly related genera, while Arma Hahn, 1832 and Zicrona was the most closely related. Among species, the genetic distance between Arma custos (Fabricius, 1794) and Arma chinensis Fallou, 1881 was the smallest, they have been synonymized (Zhao et al., 2018b).
Most tRNAs of the Pentatomidae have a typical cloverleaf secondary structure, except for the lack of the DHU stem in trnS1, which has also been reported in previous studies of Pentatomidae insects (Zhao et al., 2018a; Mu et al., 2022). The stem region is more conserved than the loop region, suggesting a difference in selective pressure between the two regions. The stem region sequence may be under slower selection to maintain its secondary structure, while the loop region evolves more rapidly (Zhang and Ryder, 1993). The secondary structures of the two rRNAs are conserved within Pentatomidae, and sequence conservation of 16S rRNA (37.75%) was higher than that of 12S rRNA (28.42%). H47 and H673 have a high degree of variation in sequence and secondary structure in 12S rRNA of Pentatomidae. In particular, H47 shows a highly variable structure, suggesting that it shows taxonomic specificity and can be used for classification within Pentatomidae (Yuan et al., 2015). The H921–960, H1047, and H1399 helices at the 3′ end of 12S rRNA were conserved within Pentatomidae. The H2077 helix of 16S rRNA is highly variable in terms of its sequence and secondary structure within Pentatomidae, while six helices (H563, H1775, H2064, H2507, H2547, and H2588) were conserved. Previous studies have shown that the variable length and differentiated tandem repeat units in the control region provide a basis for use as molecular markers to study the evolution and population genetics of pentatomoid species (Wang et al., 2015; Yuan et al., 2015). The control regions of Gonopsis coccinea and Gonopsimorpha nigrosignata were similar in length and each had only one type of tandem repeat unit. The control region of C. glandulosus was much longer than those of the other two species, and two types of repeat units were observed.
The phylogenetic trees based on two datasets using BI and ML methods were completely consistent except for the position instability of Graphosoma rubrolineatum (Westwood, 1837), and were completely consistent with those obtained from PCGs and rRNAs (P123RNA) by BI and ML method (Li et al., 2021). In our study, the third codon position of PCG gene showed higher saturation and heterogeneity compared with the first and second codon positions, which may adversely affect the phylogenetic results. Removing the third site and Bayesian analysis under mixed heterogeneous model is a good solution (Yuan et al., 2015; Xu et al., 2021). However, removing the third site of PCG decreased the nodal support of phylogenetic trees. In the previous heterogeneity analysis of Pentatomoidea, Pentatomidae showed the lowest heterogeneity, and our phylogenetic results under the homogeneous model were completely consistent with those under the heterogeneous model (Liu et al., 2019; Xu et al., 2021). This suggests that the third site of PCGs contains information critical to solving the phylogenetic relationships of the Pentatomidae, and that the Bayesian approach under both heterogeneous and homogeneous models is equally expressive for this group (Zhao W. et al., 2019). In our phylogenetic tree, Neojurtina was the first lineage to diverge and formed a sister group with high node support with the remaining species of Pentatomidae, which was also observed in the phylogenetic tree of Pentatomoidea constructed based on P123RNA and P12RNA datasets under heterogeneous mixed model (Xu et al., 2021).
All phylogenetic analyses strongly supported the following relationship within Phyllocephalini: (Chalcopis + (Dalsira + (Gonopsimorpha + Gonopsis))). These results, combined with the structure of the control region and the initiation and termination codons, suggest that Gonopsimorpha and Gonopsis are more closely related to each other than to Chalcopis. C. glandulosus was the earliest separate clade within Phyllocephalini and did not form a sister group with Dalsira (junior synonym of the Metonymia), which supported morphologic idea that it belongs to Chalcopis (David and Zheng, 2005). Phyllocephalini did not form an independent clade. Phyllocephalini formed a sister group with Placosternum Amyot & Serville, 1843 with high support and then converged with Halyomorpha (Mayr, 1864). The closer relationship between the Phyllocephalini and Halyomorpha was also supported by BI and ML methods based on P123, P12, P12RNA and P123RNA datasets (Chen et al., 2017; Zhao et al., 2020; Zhao et al., 2021).
The species assigned to clade A all belonged to Pentatominae and its topological structure was (((Anaxilaus + (Plautia + Glaucias)) + (Nezara + Palomena)) + (Eysarcorini + Carpocorini)). Neither Antestiini nor Nezarini formed a monophyletic group; however, they were closely related. Rider temporarily placed Plautia (Stål, 1865) in Antestiini, and our phylogenetic results supported this morphology-based view that Plautia may bridge the gap between Antestiini and the Nezarini (Rider et al., 2018). E. gibbosus (Jakovlev, 1904) is the first diverging clade within Eysarcorini, which did not cluster with other species of Eysarcoris Hahn, 1834. This result is consistent with previous results based on molecular and morphological evidence; accordingly, we support the previous proposal that E. gibbosus should be transferred to Stagonomus Gorski, 1852 (Roca-Cusachs and Jung, 2019; Li et al., 2021). In this study, Dolycoris baccarum (Linnaeus, 1758) formed a sister group with Rubiconia intermedia (Wolff, 1811), which has previously been placed in Eysarcorini, supporting the most recent morphological attribution of them to Carpocorini (Puton, 1869; Rider et al., 2018). Eysarcorini and Carpocorini are similar with respect to morphological characteristics and were sister groups in our phylogenetic results, indicating that the species in the two tribes are closely related (Rider et al., 2018; Li et al., 2021).
The topology of clade B was (Hoplistoderini + (Menidini + Asopinae)). In this study, the relationships among the genera in the Asopinae obtained by using the mixed homogeneity model are completely consistent with the results of the mixed heterogeneity model (Xu et al., 2021). The sister group relationship between Menidini and Asopinae has also been found in phylogenetic analyses based on nuclear and mitochondrial genes, consistent with the previously proposed hypothesis, and there were similar pseudoclaspers structures between the two groups (Gapud, 1981; Roca-Cusachs et al., 2021). The topology of clade C was ((Sephelini + Halyini) + (Caystrini + (Cappaeini + (Placosternum + Phyllocephalini)))). This was consistent with the previous phylogenetic relationships of Pentatomomorpha under the site-heterogeneous mixture model and Pentatominae under the homogenous model based on the sequences of the 13 PCGs and two rRNAs (Liu et al., 2019; Li et al., 2021). In this study, the three genera (Pentatoma, Placosternum, Neojurtina) of Pentatomini did not converge, and only the three species of Pentatoma converged. The sternal structure of the Old World genus Placosternum is somewhat more reminiscent of the New World South American genus Evoplitus, and sternal structure of genus Evoplitus strikingly similar to the Old World tribe Rhynchocorini (Rider et al., 2018). Neojurtina was only temporarily placed in Pentatomini, while Pentatoma has always been well placed in Pentatomini (Rider et al., 2018). Therefore, more evidence is needed to determine the phylogenetic position of Placosternum and Neojurtina in Pentatomidae.
Owing to the small number of mitogenomes available, the phylogenetic relationships of Podopinae are unresolved, and stable clustering was not obtained in the four phylogenetic trees. In the phylogenetic BI tree based on the P123 data set, Graphosoma was clustered with Scotinophara, which was a sister group of clade A in the remaining three trees. In terms of morphology, G. rubrolineatum belongs to the Podopinae (Yang, 1962; Rider et al., 2018). In the previous studies based on mitochondrial genome, most of them selected only G. rubrolineatum as the representative species of the Podopinae, so the Podopinae is mixed in the Pentatominae (Chen et al., 2017; Mu et al., 2022; Yuan et al., 2015; Zhao et al., 2021; Zhao et al., 2020; Zhao Q. et al., 2019; Wang et al., 2017; Wang et al., 2019). However, in the study based on the P123RNA and P12RNA datasets, the two representative species of the Podopinae were selected, and the G. rubrolineatum did not converge with other species of the Podopinae (Xu et al., 2021). These results combined with our phylogenetic results indicate that the reason for the lack of stable position of G. rubrolineatum lies in the limited mitogenome and the limitation of molecular marker selection. We think the way to solve this problem is to sequence more mitochondrial genomes of the Podopinae and more molecular markers such as nuclear genes. Although the position of G. rubrolineatum differed between the four phylogenetic trees, Scotinophara lurida (Burmeister, 1834) and Catacanthus incarnatus (Drury, 1773) were sister groups in all trees, which are also observed in the molecular clock analysis (Liu et al., 2019). In addition, the phylogenetic relationship of Eurydema Laporte, 1833 was completely consistent with the results of morphological studies (Zhao W. et al., 2019).
Since the 1990s, few phylogenetic studies have focused on Phyllocephalini. Owing to the limited number of mitogenomes, the internal phylogenetic relationships are not clear. Earlier studies based on morphological characteristics are expected to pay more attention to the molecular phylogenetic study of this group (Rider et al., 2018). In our study, three mitogenomes from Phyllocephalini were sequenced, representing a substantial addition to the two existing mitogenomes of Phyllocephalini. In addition, our results clarified partially the phylogenetic relationships within Pentatomidae at the subfamily, tribe, and genus levels. It is necessary to sequence more mitochondrial genomes, including the other subfamilies and genus groups, to better understand the molecular evolution and phylogenetic relationships of this group.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Conceptualization, DL, JW, and QZ; specimen collection and identification, DL, MN, and QZ; Experiments, DL and CC.; Data analysis, DL.; writing—original draft preparation, DL.; writing—review and editing, DL, JW, and QZ.; funding acquisition, HZ, JW, and QZ. All authors have read and agreed to the published version of the manuscript.
This research was funded by the National Science Foundation Project of China (Nos.31872272, 32100370); the Research Project Supported by Shanxi Scholarship Council of China (Nos. 2020-064, 2020-065), Natural Science Research General project of Shanxi Province (Nos.202103021224331, 202103021224132), Science and Technology Innovation Fund of Shanxi Agricultural University (2020BQ79), the Excellent Doctoral Award of Shanxi Province for Scientific Research Project (SXBYKY2021024) and Science and Technology Innovation Projects of Universities in Shanxi Province (2021L097).
We are grateful to the reserve staff for the collection of samples.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1045193/full#supplementary-material
Ahmad, I., and Kamaluddin, S. (1988). A new tribe and a new species of the subfamily Phyllocephalinae (Hemiptera: Pentatomidae) from the Indo-Pakistan subcontinent. Orient. Insects 22, 241–258. doi:10.1080/00305316.1988.11835490
Ahmad, I. (1990). S. aegyptiaca (Lefebvre) and a new genus for S. orientalis Kamaluddin and Ahmad (Hemiptera: Pentatomidae: Phyllocephalinae) with a key to the worl species and their cladistic analysis. Redescription type species Schysops Spinola 10, 195–208.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi:10.1089/cmb.2012.0021
Barao, K. R., Ferrari, A., and Grazia, J. (2013). Comparative morphology of selected characters of the Pentatomidae foreleg (Hemiptera, Heteroptera). Arthropod Struct. Dev. 42, 425–435. doi:10.1016/j.asd.2013.04.004
Benson, G. (1999). Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi:10.1093/nar/27.2.573
Bernt, M., Donath, A., Jühling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). Mitos: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi:10.1016/j.ympev.2012.08.023
Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780. doi:10.1093/nar/27.8.1767
Chen, C., Wei, J., Ji, W., and Zhao, Q. (2017). The first complete mitochondrial genome from the subfamily Phyllocephalinae (Heteroptera: Pentatomidae) and its phylogenetic analysis. Mitochondrial DNA. B Resour. 2, 938–939. doi:10.1080/23802359.2017.1413313
Chen, Q., Niu, X., Fang, Z., and Weng, Q. (2020). The complete mitochondrial genome of Eysarcoris guttigerus (Hemiptera: Pentatomidae). Mitochondrial DNA. B Resour. 5, 687–688. doi:10.1080/23802359.2020.1714498
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi:10.1093/bioinformatics/bty560
Chris, S., Thomas, R. B., Francesco, F., James, B. S., and Andrew, T. B. (2006). Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 37, 545–579. doi:10.1146/annurev.ecolsys.37.091305.110018
David, A., and Zheng, L. (2005). Checklist and nomenclatural notes on the Chinese Pentatomidae (Heteroptera). III. Phyllocephalinae, Podopinae. J. Proc. Entomol. Soc. WASH 107, 90–98.
Gapud, V. D. L. P. (1981). A generic revision of the subfamily Asopinae, with consideration of its phylogenetic position in the family Pentatomidae and superfamily Pentatomoidea (Hemiptera-Heteroptera). Lawrence, United States: University of Kansas.
Genevcius, B. C., Greve, C., Koehler, S., Simmons, R. B., Rider, D. A., Grazia, J., et al. (2021). Phylogeny of the stink bug tribe chlorocorini (Heteroptera, Pentatomidae) based on DNA and morphological data: The evolution of key phenotypic traits. Syst. Entomol. 46, 327–338. doi:10.1111/syen.12464
Grazia, J., Schuh, R. T., and Wheeler, W. C. (2008). Phylogenetic relationships of family groups in Pentatomoidea based on morphology and DNA sequences (Insecta: Heteroptera). Cladistics. 24, 932–976. doi:10.1111/j.1096-0031.2008.00224.x
Hebert, P. D., Stoeckle, M. Y., Zemlak, T. S., and Francis, C. M. (2004). Identification of birds through DNA barcodes. PLoS Biol. 2, e312. doi:10.1371/journal.pbio.0020312
Hua, J., Li, M., Dong, P., Cui, Y., Xie, Q., and Bu, W. (2008). Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (insecta: Hemiptera: Heteroptera). BMC Genomics 9, 610–625. doi:10.1186/1471-2164-9-610
Jiang, P. (2017). Studies on the comparative mitochondrial genomics and phylogeny of Heteroptera (insecta: Hemiptera). China, Beijing, Haidian District: China Agricultural University.
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi:10.1038/nmeth.4285
Kamaluddin, S., and Ahmad, I. (1988). A revision of the tribe Phyllocephalini (Hemiptera: Pentatomidae: Phyllocephalinae) from Indo-Pakistan subcontinent with description of five new species. Orient. Insects 22, 185–240. doi:10.1080/00305316.1988.11835489
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi:10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi:10.1093/bioinformatics/bts199
Kück, P., Meid, S. A., Groß, C., Wägele, J. W., and Misof, B. (2014). AliGROOVE–visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinforma. 15, 294–315. doi:10.1186/1471-2105-15-294
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2017). PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773. doi:10.1093/molbev/msw260
Lee, W., Kang, J., Jung, C., Hoelmer, K., Lee, S. H., and Lee, S. (2009). Complete mitochondrial genome of Brown marmorated stink bug Halyomorpha halys (Hemiptera: Pentatomidae), and phylogenetic relationships of hemipteran suborders. Mol. Cells 28, 155–165. doi:10.1007/s10059-009-0125-9
Li, R., Li, M., Yan, J., Bai, M., and Zhang, H. (2021). Five mitochondrial genomes of the genus Eysarcoris Hahn, 1834 with phylogenetic implications for the Pentatominae (Hemiptera: Pentatomidae). Insects 12, 597. doi:10.3390/insects12070597
Linnavuori, R. E. (1982). Pentatomidae and Acanthosomatidae (Heteroptera) of Nigeria and the Ivory Coast, with remarks on species of the adjacent countries in West and Central Africa. Acta Zoologica Fennica 163, 176.
Liu, Y., Li, H., Song, F., Zhao, Y., Wilson, J. J., and Cai, W. (2019). Higher-level phylogeny and evolutionary history of Pentatomomorpha (Hemiptera: Heteroptera) inferred from mitochondrial genome sequences. Syst. Entomol. 44, 810–819. doi:10.1111/syen.12357
Mi, Q., Zhang, J., Gould, E., Chen, J., Sun, Z., and Zhang, F. (2020). Biology, ecology, and management of Erthesina fullo (Hemiptera: Pentatomidae): A review. Insects 11, 346. doi:10.3390/insects11060346
Mu, Y. L., Zhang, C. H., Zhang, Y. J., Yang, L., and Chen, X. S. (2022). Characterizing the complete mitochondrial genome of Arma custos and Picromerus lewisi (Hemiptera: Pentatomidae: Asopinae) and conducting phylogenetic analysis. J. Insect Sci. 22, 6. doi:10.1093/jisesa/ieab105
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi:10.1093/molbev/msu300
Ojala, D., Montoya, J., and Attardi, G. (1981). tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474. doi:10.1038/290470a0
Park, D. S., Foottit, R., Maw, E., and Hebert, P. D. (2011). Barcoding bugs: DNA-based identification of the true bugs (insecta: Hemiptera: Heteroptera). PLoS One 6, e18749. doi:10.1371/journal.pone.0018749
Perna, N. T., and Kocher, T. D. (1995). Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358. doi:10.1007/BF00186547
Rider, D. A., Schwertner, C. F., Vilimova, J., Rédei, D., and Thomas, D. B. (2018). Higher systematics of the Pentatomoidea.Invasive stink bugs and related species (Pentatomoidea): Biology, higher systematics, semiochemistry, and management. Boca Raton, FL, USA: CRC Press.
Roca-Cusachs, M., and Jung, S. (2019). Redefining Stagonomus Gorski based on morphological and molecular data (Pentatomidae: Eysarcorini). Zootaxa 4658, 210–374. doi:10.11646/zootaxa.4658.2.10
Roca-Cusachs, M., Schwertner, C. F., Kim, J., Eger, J., Grazia, J., and Jung, S. (2021). Opening pandora's box: Molecular phylogeny of the stink bugs (Hemiptera: Heteroptera: Pentatomidae) reveals great incongruences in the current classification. Syst. Entomol. 47, 36–51. doi:10.1111/syen.12514
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi:10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sánchez-Delbarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi:10.1093/molbev/msx248
Sharif, T., Waheed, I., Bashir, A., Saleem, A., Aftab, M., and Ahmed, S. (2020). Taxonomic studies of family Pentatomidae (Hemiptera) four genera from district Faisalabad Punjab Pakistan with taxonomic keys. J. Entomol. Zool. Stud. 8, 1338–1344.
Sheikh, A. (2017). Contribution to the study of pentatominae (Pentatomidae: Hemiptera) bugs from dumna nature park, jabalpur, India. J. Zoology Stud. 2, 24–27.
Stephen, L. C. (2014). Insect mitochondrial Genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117. doi:10.1146/annurev-ento-011613-162007
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi:10.1093/molbev/msab120
Tang, M., Tan, M., Meng, G., Yang, S., Su, X., Liu, S., et al. (2014). Multiplex sequencing of pooled mitochondrial genomes-a crucial step toward biodiversity analysis using mito-metagenomics. Nucleic Acids Res. 42, e166. doi:10.1093/nar/gku917
Tembe, S., Shouche, Y., and Ghate, H. V. (2014). DNA barcoding of Pentatomomorpha bugs (Hemiptera: Heteroptera) from western ghats of India. Meta Gene 2, 737–745. doi:10.1016/j.mgene.2014.09.006
Vaidya, G., Lohman, D. J., and Meier, R. (2011). SequenceMatrix: Concatenation software for the fast assembly of multi‐gene datasets with character set and codon information. Cladistics. 27, 171–180. doi:10.1111/j.1096-0031.2010.00329.x
Wang, J., Zhang, L., Yang, X. Z., Zhou, M. Q., and Yuan, M. L. (2017). The first mitochondrial genome for the subfamily Podopinae (Hemiptera: Pentatomidae) and its phylogenetic implications. Mitochondrial DNA. B Resour. 2, 219–220. doi:10.1080/23802359.2017.1310605
Wang, Y., Chen, J., Jiang, L. Y., and Qiao, G. X. (2015). Hemipteran mitochondrial genomes: Features, structures and implications for phylogeny. Int. J. Mol. Sci. 16, 12382–12404. doi:10.3390/ijms160612382
Wang, Y., Duan, Y., and Yang, X. (2019). The complete mitochondrial genome of Plautia crossota (Hemiptera: Pentatomidae). Mitochondrial DNA. B Resour. 4, 2281–2282. doi:10.1080/23802359.2019.1627924
Wolstenholme, D R (1992). Genetic novelties in mitochondrial genomes of multicellular animals. Curr. Opin. Genet. Dev. 2, 918–925. doi:10.1016/s0959-437x(05)80116-9
Xia, X. (2018). DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 35, 1550–1552. doi:10.1093/molbev/msy073
Xu, S., Wu, Y., Liu, Y., Zhao, P., Chen, Z., Song, F., et al. (2021). Comparative mitogenomics and phylogenetic analyses of Pentatomoidea (Hemiptera: Heteroptera). Genes. (Basel) 12, 1306. doi:10.3390/genes12091306
Yang, W. I. (1962). Economic insect fauna of China: Fasc. 2. Hemiptera, Pentatomidae. Academia sinica. Beijing: Science Press.
Yuan, L., Liu, H., Ge, X., Yang, G., Xie, G., and Yang, Y. (2022). A mitochondrial genome phylogeny of cleridae (Coleoptera, cleroidea). Insects 13, 118. doi:10.3390/insects13020118
Yuan, M. L., Zhang, Q. L., Guo, Z. L., Wang, J., and Shen, Y. Y. (2015). Comparative mitogenomic analysis of the superfamily Pentatomoidea (insecta: Hemiptera: Heteroptera) and phylogenetic implications. BMC Genomics 16, 460. doi:10.1186/s12864-015-1679-x
Zhang, Q. L., Yuan, M. L., and Shen, Y. Y. (2013). The complete mitochondrial genome of Dolycoris baccarum (insecta: Hemiptera: Pentatomidae). Mitochondrial DNA 24, 469–471. doi:10.3109/19401736.2013.766182
Zhang, Y. P., and Ryder, O. A. (1993). Mitochondrial DNA sequence evolution in the Arctoidea. Proc. Natl. Acad. Sci. U. S. A. 90, 9557–9561. doi:10.1073/pnas.90.20.9557
Zhao, L., Wei, J., Zhao, W., Chen, C., Gao, X., and Zhao, Q. (2021). The complete mitochondrial genome of Pentatoma rufipes (Hemiptera, Pentatomidae) and its phylogenetic implications. Zookeys 1042, 51–72. doi:10.3897/zookeys.1042.62302
Zhao, Q., Cassis, G., Zhao, L., He, Y., Zhang, H., and Wei, J. (2020). The complete mitochondrial genome of Zicrona caerulea (Linnaeus) (Hemiptera: Pentatomidae: Asopinae) and its phylogenetic implications. Zootaxa 4747, 547–561. doi:10.11646/zootaxa.4747.3.8
Zhao, Q., Chen, C., Liu, J., and Wei, J. (2019a). Characterization of the complete mitochondrial genome of Eysarcoris aeneus (Heteroptera: Pentatomidae), with its phylogenetic analysis. Mitochondrial DNA. B Resour. 4, 2096–2097. doi:10.1080/23802359.2019.1622465
Zhao, Q., Wang, J., Wang, M. Q., Cai, B., Zhang, H. F., and Wei, J. F. (2018a). Complete mitochondrial genome of Dinorhynchus dybowskyi (Hemiptera: Pentatomidae: Asopinae) and phylogenetic analysis of Pentatomomorpha species. J. Insect Sci. 18, 44. doi:10.1093/jisesa/iey031
Zhao, Q., Wei, J., Bu, W., Liu, G., and Zhang, H. (2018b). Synonymize Arma chinensis as Arma custos based on morphological, molecular and geographical data. Zootaxa 4455, 161–176. doi:10.11646/zootaxa.4455.1.7
Zhao, Q., Wei, J., Zhao, W., Cai, B., Du, X., and Zhang, H. (2017a). The first mitochondrial genome for the subfamily Asopinae (Heteroptera: Pentatomidae) and its phylogenetic implications. Mitochondrial DNA. B Resour. 2, 804–805. doi:10.1080/23802359.2017.1398599
Zhao, W., Zhao, Q., Li, M., Wei, J., Zhang, X., and Zhang, H. (2017b). Characterization of the complete mitochondrial genome and phylogenetic implications for Eurydema maracandica (Hemiptera: Pentatomidae). Mitochondrial DNA. B Resour. 2, 550–551. doi:10.1080/23802359.2017.1365649
Zhao, W., Zhao, Q., Li, M., Wei, J., Zhang, X., and Zhang, H. (2019b). Comparative mitogenomic analysis of the Eurydema genus in the context of representative Pentatomidae (Hemiptera: Heteroptera) taxa. J. Insect Sci. 19, 20. doi:10.1093/jisesa/iez122
Zheng, B., Han, Y., Yuan, R., Liu, J., Van Achterberg, C., Tang, P., et al. (2022). Comparative mitochondrial Genomics of 104 Darwin wasps (hymenoptera: Ichneumonidae) and its implication for phylogeny. Insects 13, 124. doi:10.3390/insects13020124
Keywords: Pentatomidae, mitochondrial genomes, Phyllocephalini, phylogenetic analysis, Chalcopis, Gonopsimorpha, Gonopsis
Citation: Lian D, Wei J, Chen C, Niu M, Zhang H and Zhao Q (2022) Comparative analysis and phylogeny of mitochondrial genomes of Pentatomidae (Hemiptera: Pentatomoidea). Front. Genet. 13:1045193. doi: 10.3389/fgene.2022.1045193
Received: 21 September 2022; Accepted: 31 October 2022;
Published: 11 November 2022.
Edited by:
Max A. Alekseyev, George Washington University, United StatesReviewed by:
Xianzhao Kan, Anhui Normal University, ChinaCopyright © 2022 Lian, Wei, Chen, Niu, Zhang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Zhao, emhhb3Fpbmc4NjYyM0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.