
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 19 October 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1036231
 Min Xu1
Min Xu1 Pengzhen Jin2Yingzhi Huang2
Pengzhen Jin2Yingzhi Huang2 Yeqing Qian2Miaochun Lin1Juan Zuo1Jin Zhu1Zhaohui Li1*
Yeqing Qian2Miaochun Lin1Juan Zuo1Jin Zhu1Zhaohui Li1* Minyue Dong2,3*
Minyue Dong2,3*Intracranial hemorrhage is a common complication in preterm infants but occasionally occurs in fetuses. Disruptions of the genes, such as the COL4A1 and COL4A2 genes, are common genetic causes identified in fetal intracranial hemorrhage; however, the disruptions of the JAM3 gene are rarely reported. In the current investigation, fetal intracranial hemorrhage and dilated lateral ventricles were observed in three consecutive siblings in a pedigree. The pregnancies were terminated, and whole-exome sequencing, followed by Sanger sequencing, was performed on the affected fetuses. Pre-implantation genetic testing for monogenic diseases was performed to avoid the recurrence. The compound heterozygous variants of c.712 + 2T > A and c.813C > G p.Tyr271* in the JAM3 gene (NM_032801.4) were identified in the proband and its affected brother, which were predicted to be pathogenic. The variant of c.813C > G p.Tyr271* but not c.712 + 2T > A was identified in the fourth fetus, implying a good prognosis. Our findings expanded the spectrum of the pathogenic mutations in the JAM3 gene and revealed an important application of fetal whole-exome sequencing in idiopathic fetal intracranial hemorrhage.
Intracranial hemorrhage (ICH) is defined as non-traumatic bleeding within the cranium (de Oliveira et al., 2016; Kutuk et al., 2014). It is a complication in preterm neonates and occasionally occurs in preterm fetuses with an incidence ranging from 1/1,00,000 to 1/1,000 (Cavaliere et al., 2021; Kutuk et al., 2014). Fetal ICH is usually associated with cerebral palsy, neurodevelopmental delay, and other adverse impacts (Cavaliere et al., 2021; Kutuk et al., 2014), increasing the risk of perinatal mortality and morbidity (Sileo et al., 2022). Advanced maternal age, hypertension, preeclampsia or eclampsia, coagulopathy, alloimmune thrombocytopenia, twin-to-twin transfusion syndrome, and infectious diseases are known causes leading to fetal ICH (Hausman-Kedem et al., 2021; Meeks et al., 2020). However, nearly 75% of the cases have no identifiable risk factors (Armstrong-Wells et al., 2009).
Recently, a number of pathogenic mutations were detected in idiopathic ICH, including the COL4A1, COL4A2, F11, F7, FGA, VWF, and GP1BA genes (Cavaliere et al., 2021; Hausman-Kedem et al., 2021; Lichtenbelt et al., 2012; Vermeulen et al., 2011). Observations about the disruption of the JAM3 gene leading to ICH are currently available. ICH, caused by pathogenic mutations in the JAM3 gene, has been reported in three publications (Akawi et al., 2013; De Rose et al., 2021; Mochida et al., 2010), few of which were diagnosed prenatally.
Patients with ICH caused by JAM3 mutations usually die in infancy (Akawi et al., 2013; De Rose et al., 2021; Mochida et al., 2010). Therefore, the prenatal identification of genetic causes is necessary for therapeutic decision-making. Whole-exome sequencing (WES) is recommended as a routine test when severe structure abnormalities are newly diagnosed (Jin et al., 2020).
Herein, we described a pedigree with three consecutive fetal ICH and dilated lateral ventricles. WES, followed by Sanger sequencing, was performed on the proband (II 2) and his affected brother (II 3). The compound heterozygous variants of c.712 + 2T > A and c.813C > G were identified in the JAM3 gene, which were predicted to be pathogenic. Then, pre-implantation genetic testing for monogenic diseases (PGT-M) was performed. The variant of c.813C > G p.Tyr271* but not c.712 + 2T > A was identified in the fourth fetus, implying a good pregnancy outcome. The current investigation expands the mutation spectrum of the JAM3 gene, and the combination of ultrasound and fetal whole-exome sequencing is an effective method for the prenatal diagnosis of fetal ICH.
The 33-year-old non-consanguineous healthy couple conceived three fetuses with ICH. No obvious risk factors such as hypertension, coagulopathy, alloimmune thrombocytopenia, and infection were found during the pregnancy.
Their first female child (II 1) was delivered by emergency cesarean section at the 34th week of gestation (Figure 1A). Severe dilated, bilateral ventricles were noticed, and she died in the neonatal period. Unfortunately, no genetic tests were performed and no biological samples were available.
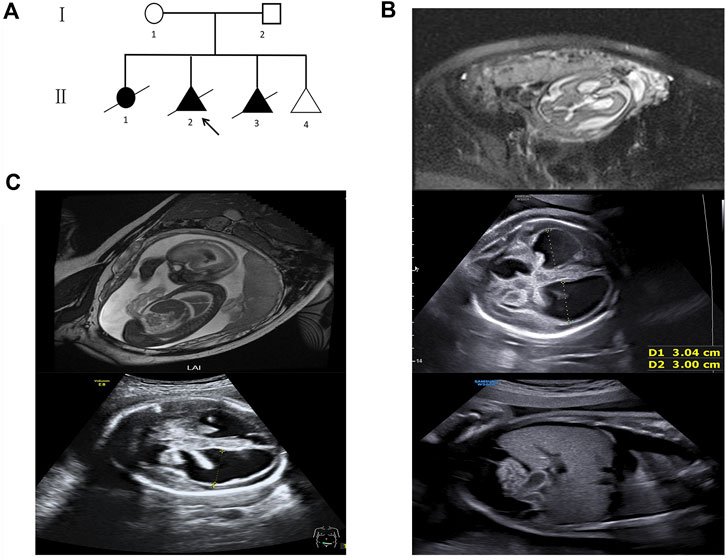
FIGURE 1. Pedigree and fetal ultrasound scan and MRI findings. (A) Pedigree of the family. The arrow refers to the proband. The solid circle (female) and triangle (fetus) represent the affected siblings; (B) ultrasound and MR imaging in the fetus (II 2): the enlarged bilateral ventricles, intraventricular hemorrhage and severe hydrocephalus, the infantile edema of the skin, bilateral pleural effusion, and ascites in the fetus; (C) ultrasound and MR imaging in the fetus (II 3): the enlarged bilateral ventricles, intraventricular hemorrhage, and severe hydrocephalus in the fetus.
The second fetus (II 2) was conceived naturally. Fetal cranial magnetic resonance imaging (MRI) at the 29th week revealed hydrocephalus and dilated ventricles (Figure 1B). The ultrasound scan at the 31st week showed severe cerebral ventriculomegaly measuring 30 mm, edema of the skin, bilateral pleural effusion, and ascites in the fetus (Figure 1B). The pregnancy was terminated after genetic counseling. Then, a chromosomal microarray assay (CMA) was provided, but no duplication or deletion was identified (Supplementary Material).
The third fetus (II 3) was also naturally conceived. Fetal cranial MRI was performed at the 24th week, and dilated ventricles and intraventricular hemorrhage were observed (Figure 1C). The ultrasound scan at the 29th week showed severe cerebral ventriculomegaly measuring 28 mm. Bright echogenic linings were described in the bilateral choroid plexus (Figure 1C). The pregnancy was terminated after genetic counseling, and samples were taken for genetic testing.
The fourth conception was conceived via assisted reproductive technology (ART) with PGT-M. The mother came to our clinic for genetic counseling and received prenatal diagnosis at the 18th week of gestation.
The use of data and medical record was approved by the Ethics Committee of Women’s Hospital, School of Medicine Zhejiang University, and conformed to the Declaration of Helsinki (IRB-20220222-R). All participants provided their written informed consent.
Amniocentesis was performed at the 18th week as usual. A measure of 20 ml of the amniotic fluid, after discarding the first 2 ml, was taken for karyotyping or DNA extraction under real-time sonographic guidance as a routine. Amniotic fluid cells were cultured, and G-banding karyotyping was conducted on metaphase preparations with a targeted 400-band level using the GSL-120 CytoVision platform (Leica, Germany). The karyotypes were described according to the International System for Human Cytogenetic Nomenclature (Stevens-Kroef et al., 2017).
Genomic DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN, Duesseldorf, Germany), and then, chromosomal microarray assay (CMA) was performed using CytoScanTM HD arrays (Affymetrix, Santa Clara, CA, United States). Chromosome Analysis Suite (Thermo Fisher Scientific, ChAS) software was used to analyze the molecular karyotype. Copy number variations were interpreted according to the standards and guidelines of the American College of Medical Genetics (ACMG) (South et al., 2013).
Whole-exome sequencing (WES) was performed on the Illumina HiSeq2000 platform (Illumina, San Diego, CA, United States), with variants filtered through online population databases. The variants were subsequently annotated by multiple databases, including ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), gnomAD (http://gnomad.broadinstitute.org/), OMIM (https://www.omim.org/), ExAC (http://exac.broadinstitute.org/), HGMD (http://www.hgmd.cf.ac.uk/ac/index.php), and Leiden Open Variation Database (LOVD: http://www.dmd.nl/), and only variants with an allele frequency ≤1% were selected. To predict the pathogenic potential of the variant, several software programs including SpliceAI (https://spliceailookup.broadinstitute.org/), NetGene2 (https://services.healthtech.dtu.dk/service.php?NetGene2-2.42), and NNSplice (http://www.fruitfly.org/seq_tools/splice.html) were used. Finally, these variants were interpreted and referred to the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015).
Sanger sequencing was carried out to validate the variants with an ABI 3130 DNA analyzer (Applied Biosystems™). The forward primer (5′-GTCAGGGAGGAACATGCACAGT-3′) and reverse primer (5′-CGGAAGAGTTCTCTAAGCTGATG-3′), and forward primer (5′-GTTCTAGGCTAGAAGGATTGTAAG-3′) and reverse primer (5′-CTCAGGAGCTGCACAATCACTC-3′) were used to amplify the PCR products in the JAM3 gene. The procedure of the PCR was as follows: 95°C for 10 min, then followed by 35 cycles of 60°C for 30 s and 72°C for 1 min, and then 72°C for 10 min. The reaction was kept at 16°C.
The compound variants of c.712 + 2T > A and c.813C > G p.Tyr271* were identified in the JAM3 (NM_032801.4) gene, both in the proband (II 2) and his brother (II 3), neither of which were recorded in the gnomAD exome database, the Clinvar database, or the HGMD database (PM2). Segregation was observed in the family (PP1). The variant of c.712 + 2T > A, with a single nucleotide change at the canonical splicing sites, was predicted to impair the protein function (PSV1). It was predicted to cause abnormal splicing by SpliceAI software (0.98) and NetGene2 (PP3). The variant of c.813C > G p.Tyr271* generated non-sense-mediated premature termination codons, producing dominant-negative truncated proteins, leading to the deficiency of the protein (PSV1).According to the ACMG guidelines (Richards et al., 2015), both of the mutations were classified as pathogenic.
Two primer sets were designed, and Sanger sequencing was performed on all the subjects except the first child (II 1). As shown in Figure 2, the mutations of c.813C > G p.Tyr271* and c.712 + 2T > A were confirmed both in the proband and his affected brother, which were inherited from his mother (I 1) and father (I 2), respectively.
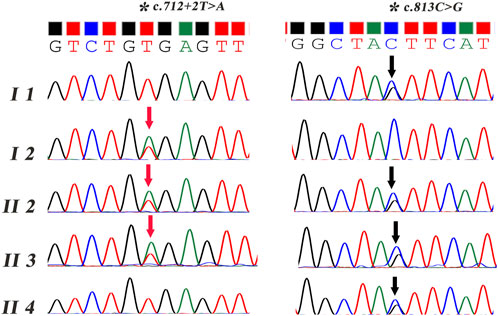
FIGURE 2. Sanger sequencing of the compound heterozygous variants in the JAM3 gene. The red arrow refers to the variant of c.712 + 2T > A. The black arrow refers to the variant of c.813C > G.
Karyotyping was normal, and no duplication or deletion was detected in the fourth fetus. The variant of c.813C > G p.Tyr271*, but not c.712 + 2T > A, was identified, which was consistent with the results of PGT-M. No structural abnormalities were observed during the pregnancy.
In the current investigation, WES, followed by Sanger sequencing, was performed to identify the pathogenic variants in a pedigree with the recurrent occurrence of fetal ICH and dilated lateral ventricles, and compound heterozygous variants of c.712 + 2T > A and c.813C > G were detected in the JAM3 gene. Pre-implantation genetic testing for monogenic disease (PGT-M) was applied to avoid the inheritance of the disease.
Generally, ICH can be classified into five types, according to its anatomical location: intraventricular hemorrhage (IVH), subarachnoid hemorrhage (SAH), cerebellar hemorrhage, subdural hemorrhage, and intraparenchymal hemorrhage (Elchalal et al., 2005). IVH and cerebellar hemorrhage are more common in fetuses than in the other individuals (Hausman-Kedem et al., 2021). Advanced maternal age, hypertension, preeclampsia or eclampsia, coagulopathy, alloimmune thrombocytopenia, twin-to-twin transfusion syndrome, and infectious disease are independently associated with fetal ICH (Hausman-Kedem et al., 2021; Meeks et al., 2020). However, up to 75% of the cases have no identifiable risk factors (Armstrong-Wells et al., 2009) and are therefore classified as idiopathic (Cavaliere et al., 2021).
Recently, an increasing number of publications have disclosed the genetic causes identified in idiopathic ICH, and the known pathogenic variants include the following: COL4A1, COL4A2, F11, F7, FGA, VWF, GP1BA, and X-linked GATA1 gene mutations (Cavaliere et al., 2021; Hausman-Kedem et al., 2021). Apparently, genetic causes are greatly underestimated for the most part, and the identification of the genetic causes is essential to determine prognosis, estimate the recurrence risk, and guide therapeutic decision-making. Recently, observations about disruptions of the JAM3 gene leading to ICH have attracted the attention of researchers. To date, mutations in the JAM3 gene have been identified in five unrelated families (Table 1).
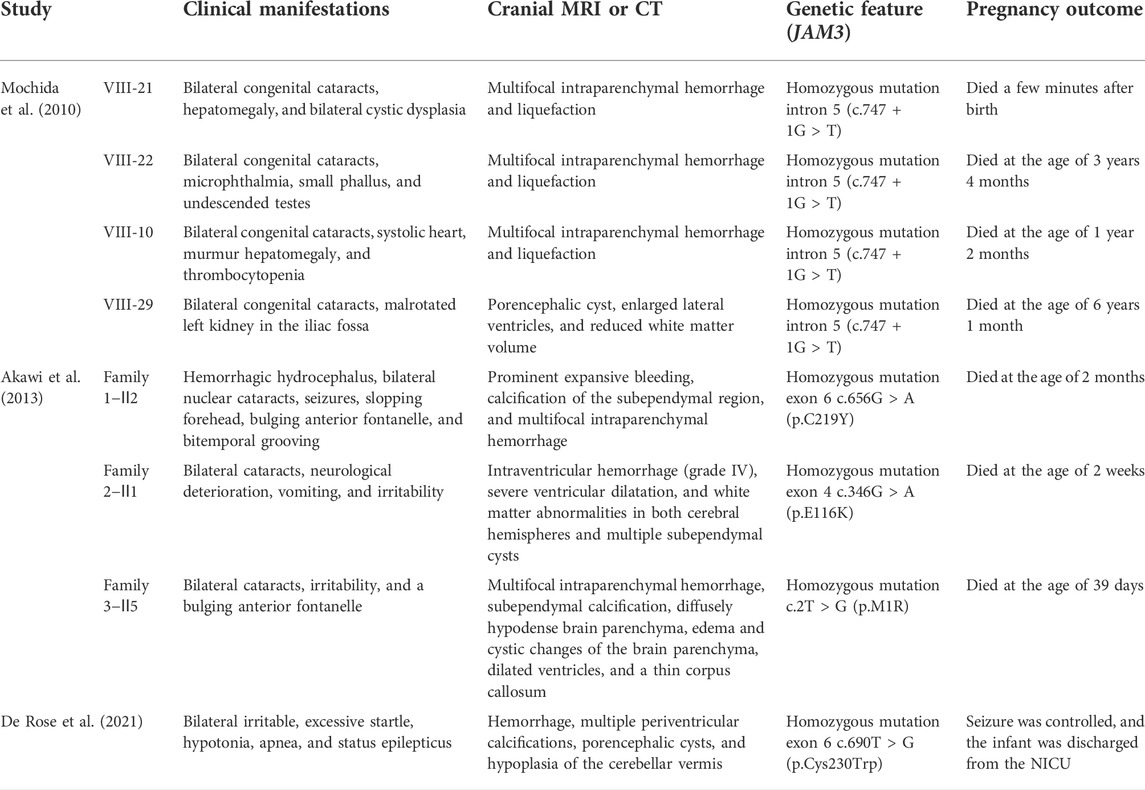
TABLE 1. Summary of clinical and genetic features of mutations in the JAM3 gene leading to fetal ICH.
The JAM3 gene is located on chromosome 11q25, with 930 bases in the open-reading frame, revealing 53% identity with JAM2 or JAM1 (Arrate et al., 2001; Luissint et al., 2014). It encodes junctional adhesion molecule 3 (JAM3), the third member of the JAM family, with three closely related proteins including JAM1 (JAM-A), JAM2 (JAM-B), and JAM3 (JAM-C) (Arrate et al., 2001; Langer et al., 2011; Mochida et al., 2010). JAM3 is widely distributed in the brain, bone marrow, heart, lung, liver, kidney, spleen, and testis nerve (Mochida et al., 2010), which functions through maintaining vascular homeostasis and barrier permeability, regulating cell adhesion and polarization (Gliki et al., 2004), and inducing self-renewal (Akawi et al., 2013; Luissint et al., 2014; Mandell and Parkos., 2005). Therefore, the absence of JAM3 is usually accompanied with growth retardation (Imhof et al., 2007), nuclear cataracts (De Rose et al., 2021), seizures (Mochida et al., 2010), and defects in multiple organ systems. Occasionally, hemorrhagic destruction of the brain and hydrocephalus is observed in the patients with the absence of JAM3 (Akawi et al., 2013; De Rose et al., 2021; Mochida et al., 2010).
Generally, JAM3 is distributed diversely between species. In humans, JAM3 is expressed extensively in the brain (Arrate et al., 2001), whereas the expression of Jam3 in the brain of adult mice remains at a low level (Mochida et al., 2010). Therefore, Jam3 was described to be absent in the mouse brain in some studies (Gliki et al., 2004; Mochida et al., 2010). Wyss et al. (2012) backcrossed JAM-C−/− mice into the C57BL/6 background. They found that JAM-C−/− C57BL/6 mice manifested hemorrhage, hydrocephalus, and enlarged lateral ventricles, providing a valuable model for the human-specific manifestations caused by the absence of JAM3.
In a consanguineous pedigree from the United Arab Emirates, eight individuals were characterized by intracranial hemorrhage, subependymal calcification, and congenital cataracts, four of who died in infancy. Among the survivors, seizures, generalized spasticity, and renal abnormalities were observed. Finally, a homozygous variant of c.747 + 1G > T was identified in the JAM3 gene in this pedigree, uncovering their pathogenicity. It is heartbreaking to witness such an enormous number of sufferers in a family; therefore, the early detection and diagnosis is valuable in a pedigree when the proband is noticed.
In this investigation, intracranial hemorrhage and hydrocephalus were observed in consecutive siblings. The compound heterozygous variants of c.712 + 2T > A and c.813C > G were identified in the JAM3 gene both in the proband and his affected brother, which were predicted to be pathogenic. To avoid the recurrence of the identical symptoms in the next conception, PGT-M was carried out after the genetic counseling.
PGT-M means identifying monogenic diseases by performing a genetic test before the embryo is implanted (Cimadomo et al., 2020). Only those unaffected embryos can be transferred (Traeger-Synodinos, 2006), which avoid the misery of affected pregnancies (He et al., 2019; Natsuaki and Dimler, 2018). It is an ideal method to improve pregnancy outcomes when monogenic diseases are confirmed in the family. In addition, severe fetal ICH, especially occurring in consecutive siblings, usually accompanies with a specific genetic cause. Therefore, the prenatal diagnosis is of great significance to guide therapeutic decision-making, which needs to be performed as early as possible.
In conclusion, our investigation identified the compound heterozygous variants of c.712 + 2T > A and c.813C > G in the JAM3 gene, expanding its spectrum. When recurrent occurrence of fetal ICH or other severe structure abnormalities are observed in a pedigree, fetal WES should be considered as routine genetic testing, and the combination of ultrasound and genetic testing might favor the prenatal diagnosis. In addition, the application of PGT-M provides an effective way to terminate the recurrent occurrence of monogenic diseases.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Ethics Committee of Women’s Hospital, School of Medicine Zhejiang University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
MD conceived of the study; MD and ZL participated in its design; MX and PJ drafted the manuscript; ML and JuZ collected the samples; JiZ collected clinical data; YH and YQ carried out the CMA; and ZL helped revise the manuscript. All authors have read and approved the final manuscript.
This work was supported by the Natural Science Foundation of Fujian Province (2021J011454), the Key Research and Development Program of Zhejiang Province (2019C03025), the National Key Research and Development Program (2018YFC1004903), the Key Projects Jointly Constructed by the Ministry and the Province of Zhejiang Medical and Health Science and Technology Project (WKJ-ZJ-2127), and the National Natural Science Foundation of China (81901382).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1036231/full#supplementary-material
Akawi, N. A., Canpolat, F. E., White, S. M., Quilis-Esquerra, J., Morales, S. M., Gamundi, M. J., et al. (2013). Delineation of the clinical, molecular and cellular aspects of novel JAM3 mutations underlying the autosomal recessive hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts. Hum. Mutat. 34 (3), 498–505. doi:10.1002/humu.22263
Armstrong-Wells, J., Johnston, S. C., Wu, Y. W., Sidney, S., and Fullerton, H. J. (2009). Prevalence and predictors of perinatal hemorrhagic stroke: Results from the kaiser pediatric stroke study. Pediatrics 123 (3), 823–828. doi:10.1542/peds.2008-0874
Arrate, M. P., Rodriguez, J. M., Tran, T. M., Brock, T. A., and Cunningham, S. A. (2001). Cloning of human junctional adhesion molecule 3 (JAM3) and its identification as the JAM2 counter-receptor. J. Biol. Chem. 276 (49), 45826–45832. doi:10.1074/jbc.M105972200
Cavaliere, A. F., Turrini, I., Pallottini, M., Vidiri, A., Marchi, L., Perelli, F., et al. (2021). Genetic profiling of idiopathic antenatal intracranial haemorrhage: What we know? Genes (Basel) 12 (4), 573. doi:10.3390/genes12040573
Cimadomo, D., Rienzi, L., Capalbo, A., Rubio, C., Innocenti, F., García-Pascual, C. M., et al. (2020). The dawn of the future: 30 years from the first biopsy of a human embryo. The detailed history of an ongoing revolution. Hum. Reprod. Update 26 (4), 453–473. doi:10.1093/humupd/dmaa019
de Oliveira, M. A., Goffi, A., Zampieri, F. G., Turkel-Parrella, D., Duggal, A., Marotta, T. R., et al. (2016). The critical care management of spontaneous intracranial hemorrhage: A contemporary review. Crit. Care 20, 272. doi:10.1186/s13054-016-1432-0
De Rose, D. U., Gallini, F., Battaglia, D. I., Tiberi, E., Gaudino, S., Contaldo, I., et al. (2021). A novel homozygous variant in JAM3 gene causing hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts (HDBSCC) with neonatal onset. Neurol. Sci. 42 (11), 4759–4765. doi:10.1007/s10072-021-05480-z
Elchalal, U., Yagel, S., Gomori, J. M., Porat, S., Beni-Adani, L., Yanai, N., et al. (2005). Fetal intracranial hemorrhage (fetal stroke): Does grade matter? Ultrasound Obstet. Gynecol. 26 (3), 233–243. doi:10.1002/uog.1969
Ghi, T., Simonazzi, G., Perolo, A., Savelli, L., Sandri, F., Bernardi, B., et al. (2003). Outcome of antenatally diagnosed intracranial hemorrhage: Case series and review of the literature. Ultrasound Obstet. Gynecol. 22 (2), 121–130. doi:10.1002/uog.191
Gliki, G., Ebnet, K., Aurrand-Lions, M., Imhof, B. A., and Adams, R. H. (2004). Spermatid differentiation requires the assembly of a cell polarity complex downstream of junctional adhesion molecule-C. Nature 431 (7006), 320–324. doi:10.1038/nature02877
Hausman-Kedem, M., Malinger, G., Modai, S., Kushner, S. A., Shiran, S. I., Ben-Sira, L., et al. (2021). Monogenic causes of apparently idiopathic perinatal intracranial hemorrhage. Ann. Neurol. 89 (4), 813–822. doi:10.1002/ana.26033
He, H., Jing, S., Lu, C. F., Tan, Y. Q., Luo, K. L., Zhang, S. P., et al. (2019). Neonatal outcomes of live births after blastocyst biopsy in preimplantation genetic testing cycles: A follow-up of 1, 721 children. Fertil. Steril. 112 (1), 82–88. doi:10.1016/j.fertnstert.2019.03.006
Imhof, B. A., Zimmerli, C., Gliki, G., Ducrest-Gay, D., Juillard, P., Hammel, P., et al. (2007). Pulmonary dysfunction and impaired granulocyte homeostasis result in poor survival of Jam-C-deficient mice. J. Pathol. 212 (2), 198–208. doi:10.1002/path.2163
Jin, S. C., Dong, W., Kundishora, A. J., Panchagnula, S., Moreno-De-Luca, A., Furey, C. G., et al. (2020). Exome sequencing implicates genetic disruption of prenatal neuro-gliogenesis in sporadic congenital hydrocephalus. Nat. Med. 26 (11), 1754–1765. doi:10.1038/s41591-020-1090-2
Kutuk, M. S., Yikilmaz, A., Ozgun, M. T., Dolanbay, M., Canpolat, M., Uludag, S., et al. (2014). Prenatal diagnosis and postnatal outcome of fetal intracranial hemorrhage. Childs Nerv. Syst. 30 (3), 411–418. doi:10.1007/s00381-013-2243-0
Langer, H. F., Orlova, V. V., Xie, C., Kaul, S., Schneider, D., Lonsdorf, A. S., et al. (2011). A novel function of junctional adhesion molecule-C in mediating melanoma cell metastasis. Cancer Res. 71 (12), 4096–4105. doi:10.1158/0008-5472.CAN-10-2794
Lichtenbelt, K. D., Pistorius, L. R., De Tollenaer, S. M., Mancini, G. M., and De Vries, L. S. (2012). Prenatal genetic confirmation of a COL4A1 mutation presenting with sonographic fetal intracranial hemorrhage. Ultrasound Obstet. Gynecol. 39 (6), 726–727. doi:10.1002/uog.11070
Luissint, A. C., Nusrat, A., and Parkos, C. A. (2014). JAM-related proteins in mucosal homeostasis and inflammation. Semin. Immunopathol. 36 (2), 211–226. doi:10.1007/s00281-014-0421-0
Mandell, K. J., and Parkos, C. A. (2005). The JAM family of proteins. Adv. Drug Deliv. Rev. 57 (6), 857–867. doi:10.1016/j.addr.2005.01.005
Meeks, J. R., Bambhroliya, A. B., Alex, K. M., Sheth, S. A., Savitz, S. I., Miller, E. C., et al. (2020). Association of primary intracerebral hemorrhage with pregnancy and the postpartum period. JAMA Netw. Open 3 (4), e202769. doi:10.1001/jamanetworkopen.2020.2769
Mochida, G. H., Ganesh, V. S., Felie, J. M., Gleason, D., Hill, R. S., Clapham, K. R., et al. (2010). A homozygous mutation in the tight-junction protein JAM3 causes hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts. Am. J. Hum. Genet. 87 (6), 882–889. doi:10.1016/j.ajhg.2010.10.026
Natsuaki, M. N., and Dimler, L. M. (2018). Pregnancy and child developmental outcomes after preimplantation genetic screening: A meta-analytic and systematic review. World J. Pediatr. 14 (6), 555–569. doi:10.1007/s12519-018-0172-4
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Sileo, F. G., Zöllner, J., D'Antonio, F., Islam, S., Papageorghiou, A. T., and Khalil, A. (2022). Perinatal and long-term outcome of fetal intracranial hemorrhage: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 59 (5), 585–595. doi:10.1002/uog.24766
South, S. T., Lee, C., Lamb, A. N., Higgins, A. W., and Kearney, H. M. (2013). ACMG Standards and Guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: Revision 2013. Genet. Med. 15 (11), 901–909. doi:10.1038/gim.2013.129
Stevens-Kroef, M., Simons, A., Rack, K., and Hastings, R. J. (2017). Cytogenetic nomenclature and reporting. Methods Mol. Biol. 1541, 303–309. doi:10.1007/978-1-4939-6703-2_24
Traeger-Synodinos, J. (2006). Real-time PCR for prenatal and preimplantation genetic diagnosis of monogenic diseases. Mol. Asp. Med. 27 (2-3), 176–191. doi:10.1016/j.mam.2005.12.004
Vermeulen, R. J., Peeters-Scholte, C., Van Vugt, J. J., Barkhof, F., Rizzu, P., van der Schoor, S. R., et al. (2011). Fetal origin of brain damage in 2 infants with a COL4A1 mutation: Fetal and neonatal MRI. Neuropediatrics 42 (1), 1–3. doi:10.1055/s-0031-1275343
Keywords: intracranial hemorrhage, whole-exome sequencing, JAM3, prenatal diagnosis, genetic counseling
Citation: Xu M, Jin P, Huang Y, Qian Y, Lin M, Zuo J, Zhu J, Li Z and Dong M (2022) Case report: Prenatal diagnosis of fetal intracranial hemorrhage due to compound mutations in the JAM3 gene. Front. Genet. 13:1036231. doi: 10.3389/fgene.2022.1036231
Received: 04 September 2022; Accepted: 03 October 2022;
Published: 19 October 2022.
Edited by:
Muhammad Naeem, Hebei Normal University, ChinaReviewed by:
Tayyab Ali, University of Agriculture, Faisalabad, PakistanCopyright © 2022 Xu, Jin, Huang, Qian, Lin, Zuo, Zhu, Li and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaohui Li, bGk2MzM2QDE2My5jb20=; Minyue Dong, ZG9uZ215QHpqdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.