Abstract
A comprehensive understanding of genetic background for rare species will provide an important theoretical basis for the future species management, monitoring and conservation. Tetraena mongolica is restrictedly distributed in the western Ordos plateau of China and has been listed as a national protected plant. We generated 13 chloroplast (cp) genomes of T. mongolica (size range of 106,062–106,230 bp) and conducted a series of comparative analyses of six Zygophyllaceae cp genomes. T. mongolica cp genome exhibited a quadripartite structure with drastically reduced inverted repeats (IRs, 4,315 bp) and undergone the loss of a suit of ndh genes and a copy of rRNAs. Furthermore, all the T. mongolica populations were divided into two genetic groups based on complete cp phylogenomics. In addition, notably variable genome size, gene order and structural changes had been observed among the six Zygophyllaceae cp genomes. Overall, our findings provide insights into the cp genome evolution mode and intraspecific relationships of T. mongolica, and provide a molecular basis for scientific conservation of this endangered plant.
Introduction
Dramatic global climate change and human activities have triggered the loss of biodiversity and the sixth mass extinction (Barnosky et al., 2011). It is noteworthy that rare species—that is, represented by only a few individuals or restricted to limited areas—act as the cornerstones of the community they support, however, they would be vulnerable to extinction when against the climate change and anthropogenic pressures (Bracken and Low, 2012; Mouillot et al., 2013). Thus, a comprehensive understanding of genetic background for species, especially for rare species, is essential, which will provide an important theoretical basis for the future species management, monitoring and conservation.
Tetraena mongolica Maxim, a super-xerophilous shrub, is a monotypic genus of Zygophyllaceae (Zhao, 1997). T. mongolica is considered as a Tertiary relic species with extant populations are restrictedly distributed in the western Ordos plateau of China (Wang, 2005). In addition, intensifying anthropogenic activities, such as urbanization, grazing, mining and woodcutting, all contributed to the significantly population declined and habitat fragmentation of T. mongolica in recent decades (Wang, 2005). In China, T. mongolica has been listed as a national protected species for its higher extinction risk (Fu, 1992).
Previous molecular studies based on a limited number of markers have provided insights into the genetic diversity, genetic structure and phylogenetic relationships of T. mongolica (Ge et al., 2011; Zhi et al., 2018). However, both of the studies suffered from a lack of resolution for intraspecific relationships, which may be largely related to the insufficient phylogenetic signals. Furthermore, based on numerous SNPs from Genotyping-by-Sequencing, Cheng et al. (2020) revealed that T. mongolica was divided into two groups but with lower support values. To obtain more comprehensively understanding of this endangered species, we will explore the related evolutionary questions with complete chloroplast (cp) genome, which can provide more phylogenetic signals compared with a limited number of markers and reflect the evolutionary history of a single parental lineage.
Sequencing technical feasibility has made cp genome a workhorse of plant phylogeny and genome evolution (e.g., Song et al., 2017; Sullivan et al., 2017; Fu et al., 2019; Zhai et al., 2019; Cauz-Santos et al., 2020; Guo et al., 2021a; Yang et al., 2021). Given that the advantages of cp genomes, such as moderate genomic size, generally recombination free and uniparental inheritance (Birky et al., 1983; Wolfe and Randle, 2004), they act as a cornerstone in plant phylogenetic inferences. Most frequently, cp phylogenomics has been rendered fully resolved phylogenetic relationships, even in enigmatic plant lineages (Ma et al., 2014; Zhang et al., 2017; Fu et al., 2019; Yang et al., 2021). Most cp genomes of land plants show a simple mode of evolution, which is mainly characterized by highly conserved genome structure and organization. It has been well documented that most cp genomes have a typical quadripartite structure, with two copies of inverted repeat (IR) regions separating the small and large single copy (SSC and LSC, respectively) regions (Jansen et al., 2005; Jansen and Ruhlman, 2012). Additionally, most cp genomes encode around 101–118 distinct genes distributed over 120–170 kb (Bock, 2007; Jansen and Ruhlman, 2012; Ruhlman and Jansen, 2014). While exceptions to the simple mode of cp evolution have been observed, exemplifying genomic upheaval resulting from gene loss (e.g., Lauraceae, Song et al., 2017), IR expansion/contraction or loss (e.g., Paphiopedilum, Guo et al., 2021a; Geraniaceae, Weng et al., 2014) and rearrangements (e.g., Passiflora, Cauz-Santos et al., 2020). Moreover, high frequency of hybridization and biparental inheritance give rise to recombination of cp genomes (e.g., Picea, Sullivan et al., 2017).
In this study, we generated 15 cp genomes representing three species of Zygophyllaceae comprising 13 accessions of T. mongolica and one each of Zygophyllum mucronatum, Zygophyllum xanthoxylon. We intend to 1) explore the cp genome evolution mode of T. mongolica by comparing with all genera for which reliable cp genomes have been published in Zygophyllaceae; and 2) estimate the intraspecific relationships of T. mongolica with different data partitions.
Materials and methods
Taxon sampling and plant material
Leaves were collected of T. mongolica, Z. mucronatum, and Z. xanthoxylon. Detailed information of sampling is provided in Table 1. The geographical coordinates of 13 locations were collected from field investigations, which covered most of the known area of T. mongolica. Currently, the best-supported taxonomy of Z. mucronatum and Z. xanthoxylon is that they belong to genus Zygophyllum (e.g., Beier et al., 2003; Wu et al., 2018; Zhang et al., 2021), whereas Z. xanthoxylon has been categorized into genus Sarcozygium by Bunge (1843) and Liou (1998). Thus, we collected Z. mucronatum and Z. xanthoxylon for comparative analyses. Totally, we generated 15 new cp genomes, including 13 T. mongolica cp genomes, one Z. mucronatum cp genome and one Z. xanthoxylon cp genome. Additionally, three previously published cp genomes of Tribulus terrestris (Yan et al., 2019), Larrea tridentata and Guaiacum angustifolium (Gonçalves et al., 2019) are retrieved in this study, representing all genera for which reliable cp genomes have been published in Zygophyllaceae (Table 1).
TABLE 1
| Species | Populations | Collection locality | Latitude (N°) | Longitude (E°) | Elevation (m) | GenBank number |
|---|---|---|---|---|---|---|
| Tetraena mongolica | WD01 | Wudaqu, Wuhai, Inner Mongolia | 39.47 | 106.66 | 1,195 | ON408217 |
| QEG09 | Hainanqu, Wuhai, Inner Mongolia | 39.30 | 106.81 | 1,094 | ON408220 | |
| QPJ11 | Etuokeqi, Ordos, Inner Mongolia | 39.41 | 106.98 | 1,259 | ON408221 | |
| HNHXQ15 | Hainanqu, Wuhai, Inner Mongolia | 39.54 | 106.89 | 1,285 | ON408222 | |
| HNHCQ39 | Hainanqu, Wuhai, Inner Mongolia | 39.45 | 106.77 | 1,091 | ON408229 | |
| SC04 | Haibowanqu, Wuhai, Inner Mongolia | 39.66 | 106.85 | 1,165 | ON408218 | |
| MEG07 | Haibowanqu, Wuhai, Inner Mongolia | 39.68 | 106.88 | 1,239 | ON408219 | |
| QLG21 | Haibowanqu, Wuhai, Inner Mongolia | 39.88 | 106.93 | 1,311 | ON408223 | |
| MX23 | Haibowanqu, Wuhai, Inner Mongolia | 39.85 | 106.87 | 1,231 | ON408224 | |
| QPC25 | Etuokeqi, Ordos, Inner Mongolia | 39.90 | 106.75 | 1,089 | ON408225 | |
| TST30 | Etuokeqi, Ordos, Inner Mongolia | 40.17 | 106.92 | 1,062 | ON408226 | |
| SGK32 | Hangjingqi, Ordos, Inner Mongolia | 40.25 | 107.12 | 1,197 | ON408227 | |
| WJRGC35 | Hangjingqi, Ordos, Inner Mongolia | 40.22 | 107.34 | 1,214 | ON408228 | |
| Tribulus terrestris | — | — | — | — | — | MN164624 |
| Larrea tridentata | — | — | — | — | — | MK726018 |
| Guaiacum angustifolium | — | — | — | — | — | MK726011 |
| Zygophyllum mucronatum | — | Shandan, Zhangye, Gansu | 38.43 | 101.14 | 2,289 | ON408230 |
| Zygophyllum xanthoxylon | — | Hainanqu, Wuhai, Inner Mongolia | 39.54 | 106.89 | 1,285 | ON408231 |
Information of samples in this study.
DNA extraction, sequencing, assembly and annotation
Total genomic DNA was extracted for the 15 accessions from silica-dried leaf material following the modified CTAB method (Doyle, 1987). Sequencing was completed on an Illumina Hiseq platform, yielding at least 2 GB clean data for each accession. All of the above work were conducted by Biomarker Technologies Inc (Beijing, China). Clean reads were assembled using MIRA 4.0.2 (Chevreux et al., 2004) and MITObim v1.7 (Hahn et al., 2013) with default settings. In this process, the published cp genome of T. mongolica (MK331720), Z. xanthoxylon (MZ427318) and Zygophyllum fabago (MK341052) were used as reference genome in cp genome assembly of T. mongolica, Z. xanthoxylon and Z. mucronatum, respectively. Annotation of the cp genomes was performed in GENEIOUS v.11 (Biomatters Ltd., Auckland, New Zealand) coupled with manual adjustments.
Sequence divergence, genome rearrangement and IR/SC boundary analysis
Genetic diversity of T. mongolica was assessed. Sequences were aligned by MAFFT with the default settings (Katoh and Standley, 2013). Variable sites, informative sites, nucleotide diversity (Pi) and indels in the aligned result were detected by DnaSP v 5.0 (Librado and Rozas, 2009). Furthermore, Progressive Mauve v 2.4.0 (Darling et al., 2004) was used to identify the possible rearrangements among the six Zygophyllaceae cp genomes. The six Zygophyllaceae cp genome alignment was visualized using mVISTA with the sequence of T. terrestris as the reference (Frazer et al., 2004). In addition, to detect possible expansion or contraction in junction regions, the IR/SC boundaries were analyzed for both T. mongolica cp genomes and Zygophyllaceae cp genomes.
Codon usage bias analysis
To avoid sampling bias, protein-coding regions with the length greater than 300 bp were extracted for the analysis (Wright, 1990). Finally, the number of selected protein-coding genes of T. terrestris, L. tridentata, G. angustifolium, T. mongolica, Z. mucronatum, and Z. xanthoxylon were 58, 57, 56, 41, 41, and 41, respectively. Relative synonymous codon usage (RSCU) is the ratio of the observed frequency of a codon to the expected frequency and is a good indicator of codon usage bias (Sharp and Li, 1986). RSCU values of the six Zygophyllaceae cp genomes were calculated by MEGA v 5.0 (Tamura et al., 2011). The heatmap from all RSCU values was carried out using Morpheus (https://software.broadinstitute.org/morpheus).
Repeat elements and SSRs analysis
Dispersed repeats, tandem repeats and simple sequence repeats (SSRs) within six Zygophyllaceae cp genomes were analyzed, respectively. Firstly, REPuter (Kurtz et al., 2001) was used to identify dispersed repeats, including forward, reverse, complement and palindromic repeats. We focused on the dispersed repeats having a minimal size of 30 bp and 90% or greater similarity between the two repeat copies. The maximum distance between palindromic repeats is 3 kb. Subsequently, tandem repeats (>10 bp in length) were detected using online program Tandem Repeats Finder (Benson, 1999) with default parameters. The minimum alignment score and maximum period size were set as 50 and 500, respectively. All found repeats were manually verified and the redundant results were removed. Finally, SSRs were detected by msatcommander (Faircloth, 2008). The minimum repeat unit were 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexanucleotides, respectively.
Phylogenetic inference and network analysis
Phylogenetic analyses of different datasets were conducted using Maximum likelihood (ML) and Bayesian inference (BI) methods, which were conducted using RAxML v7.2.8 (Stamatakis, 2006) and MrBayes v3.1.2 (Ronquist and Huelsenbeck, 2003), respectively. To estimate the phylogenetic relationships of T. mongolica, the following datasets: 1) complete cp genome sequences (CCS); 2) noncoding region sequences (NCS); and 3) protein-coding region sequences (PCS) were analyzed. In these inferences, L. tridentata (MK726018) and G. angustifolium (MK726011) were treated as outgroups. For Zygophyllaceae, we used Krameria bicolor (MK726015) and Krameria lanceolata (MK726016) as outgroups. Datasets:1) complete cp genome sequences (CCS); and 2) SSC + single IR sequences (SIS) were conducted for phylogenetic analysis. Sequence alignment was performed using MAFFT (Katoh and Standley, 2013) with the default parameters set. The ML tree was inferred with GTR + G model and 1,000 bootstrap replicates. The best-fitting model for BI analyses was determined using Modeltest 3.7 (Posada and Crandall, 1998) based on the Akaike information criterion. Two independent Markov chain Monte Carlo runs were performed for one million cycles with sampling every 100 generations, and the first 25% of the trees were discarded as burn-in.
A TCS network of intraspecific relationships of T. mongolica was performed by PopArt v 1.7 (Leigh and Bryant, 2015) using the 13 cp genome haplotypes identified by DnaSP v 5.0 (Librado and Rozas, 2009).
Divergence time of T. mongolica
The divergence times among T. mongolica lineages were estimated using BEAST v1.8.0 (Drummond and Rambaut, 2007). Dating analysis were conducted under GTR substitution model and an uncorrelated lognormal relaxed clock. A Yule process was chosen to model speciation. Thirty million generations were run of the two independent Markov chain Monte Carlo, with sampling every 3000th generation. Finally, the stationarity of the tree was checked using Tracer v1.5 by assessing the effective sample size (ESS) values (>200). Final trees were edited using FigTree v1.4.3 (http://beast.community/figtree). Considering the ambiguity of Zygophyllaceae fossil record (Bellstedt et al., 2012), we estimated the divergence time twice. Firstly, the stem age of T. mongolica was set at 9.38 Ma and the split between L. tridentata and G. angustifolium was set at 17.22 Ma, as the results of Wu et al. (2015). Secondly, the split between Zygophyllaceae and Krameriaceae was 92.1 Ma following the results of Li et al. (2019). Hence, K. bicolor and K. lanceolata were retrieved as the outgroups.
Results
Characteristics of T. mongolica cp genomes
To assess intraspecific variation within T. mongolica, we sampled 13 populations of T. mongolica covered most of the known area of this endangered plant. A total of 13 complete cp genomes were newly generated by using the next generation sequencing approach (Table 1). T. mongolica cp genomes were highly conserved and all of them displayed the typical quadripartite structure, including LSC, SSC, and two copies of IR regions. The complete cp genome size ranged from 106,062 bp (WJRGC35) to 106,230 bp (MX23) (Table 2). In the LSC region, the length varied from 80,273 bp (WJRGC35) to 80,351 bp (QPC25), in the SSC region from 17,159 bp (WJRGC35) to 17,268 bp (HNHCQ39). Notably, the length of IR was stable among different accessions (4,315 bp). The IR/SC boundaries of these cp genomes were stable, with the only exception that the distance from the trnL (UAG) end to the junction of IRa/SSC boundary of HNHCQ39 and the other accessions were 555 bp and 553 bp, respectively (Supplementary Figure S1). The GC content of 13 cp genomes ranged from 33.60% to 33.63%, and averaged 33.61% (Table 2).
TABLE 2
| Accessions | LSC (bp) | SSC (bp) | IRa (bp) | IRb (bp) | Size (bp) | GC content (%) | Protein-coding genes | tRNA genes | rRNA genes | Total genes |
|---|---|---|---|---|---|---|---|---|---|---|
| WD01 | 80,340 | 17,200 | 4,315 | 4,315 | 106,170 | 33.61 | 67 | 33 | 4 | 104 |
| QEG09 | 80,326 | 17,200 | 4,315 | 4,315 | 106,156 | 33.62 | ||||
| QPJ11 | 80,334 | 17,199 | 4,315 | 4,315 | 106,163 | 33.60 | ||||
| HNHXQ15 | 80,349 | 17,200 | 4,315 | 4,315 | 106,179 | 33.61 | ||||
| HNHCQ39 | 80,280 | 17,268 | 4,315 | 4,315 | 106,178 | 33.62 | ||||
| SC04 | 80,311 | 17,243 | 4,315 | 4,315 | 106,184 | 33.61 | ||||
| MEG07 | 80,302 | 17,244 | 4,315 | 4,315 | 106,176 | 33.61 | ||||
| QLG21 | 80,340 | 17,255 | 4,315 | 4,315 | 106,225 | 33.60 | ||||
| MX23 | 80,337 | 17,244 | 4,315 | 4,315 | 106,230 | 33.60 | ||||
| QPC25 | 80,351 | 17,200 | 4,315 | 4,315 | 106,181 | 33.61 | ||||
| TST30 | 80,284 | 17,245 | 4,315 | 4,315 | 106,159 | 33.61 | ||||
| SGK32 | 80,284 | 17,263 | 4,315 | 4,315 | 106,177 | 33.61 | ||||
| WJRGC35 | 80,273 | 17,159 | 4,315 | 4,315 | 106,062 | 33.63 |
Summary of the 13 T. mongolica chloroplast genomes.
T. mongolica composed of 104 genes, including 67 protein-coding genes, 33 tRNA genes and four rRNA genes (Table 2; Supplementary Table S1). In addition, a total of 77 genes were located in LSC, including 56 protein-coding genes and 21 tRNAs. SSC harbored five protein-coding genes, six tRNAs and four rRNAs. The remaining genes were located in IRs.
The 13 complete cp genomes had an aligned length of 106,471 bp, while there were only 128 variable sites and 41 parsimony-informative sites. Nucleotide diversity (Pi) across the complete cp genomes of total populations was relatively low (Pi = 0.00032). In contrast, non-coding regions showed the highest sequence diversity (Pi = 0.00054) and protein-coding genes exhibited extremely low sequence diversity (Pi = 0.00012) (Table 3).
TABLE 3
| Complete cp genomes | Protein-coding genes | Non-coding regions | |
|---|---|---|---|
| Number of sites | 106,471 | 46,764 | 52,798 |
| Number of variable/informative sites | 128/41 | 21/7 | 104/35 |
| Nucleotide diversity (Pi) | 0.00032 | 0.00012 | 0.00054 |
| Number of indels | 527 | 0 | 533 |
Genetic diversity of T. mongolica.
Characteristics of Zygophyllaceae cp genomes
To investigate the evolution mode of T. mongolica cp genome, a total of six species of Zygophyllaceae were analyzed, including T. terrestris (subfam. Tribuloideae), L. tridentata (subfam. Larreoideae), G. angustifolium (subfam. Larreoideae), T. mongolica (subfam. Zygophylloideae), Z. mucronatum (subfam. Zygophylloideae) and Z. xanthoxylon (subfam. Zygophylloideae), which covered all genera for which reliable cp genomes have been published in Zygophyllaceae. Due to the highly conserved cp genomes, we used WD01 to represent T. mongolica for the subsequent analyses.
The complete cp genome sequences of six Zygophyllaceae species were 105,251 bp (Z. mucronatum) to 158,184 bp (T. terrestris) (Table 4). All of them exhibited a typical quadripartite structure, including a pair of IR regions (8,576–51,684 bp), one LSC region (79,912–88,878 bp) and one SSC region (13,767–17,622 bp). The GC contents of six cp genomes ranged from 33.61% (T. mongolica) to 35.81% (T. terrestris).
TABLE 4
| T. terrestris | L. tridentata | G. angustifolium | T. mongolica | Z. mucronatum | Z. xanthoxylon | |
|---|---|---|---|---|---|---|
| Subfamily | Tribuloideae | Larreoideae | Larreoideae | Zygophylloideae | Zygophylloideae | Zygophylloideae |
| Size (bp) | 158,184 | 135,988 | 130,809 | 106,170 | 105,251 | 105,802 |
| LSC (bp) | 88,878 | 81,323 | 81,554 | 80,340 | 79,912 | 80,263 |
| SSC (bp) | 17,622 | 15,965 | 13,767 | 17,200 | 16,513 | 16,963 |
| IRs (bp) | 51,684 | 38,700 | 35,488 | 8,630 | 8,826 | 8,576 |
| Overall GC content (%) | 35.81 | 35.08 | 35.57 | 33.61 | 33.90 | 33.98 |
| Total genes | 130 | 129 | 129 | 104 | 104 | 104 |
| Protein-coding genes | 84 | 80 | 80 | 67 | 67 | 67 |
| tRNA genes | 37 | 37 | 37 | 33 | 33 | 33 |
| rRNA genes | 8 | 8 | 8 | 4 | 4 | 4 |
| pseudogene(s) | 1 | 4 | 4 | 0 | 0 | 0 |
Summary of the six Zygophyllaceae chloroplast genomes.
Due to the high conservation of cp genomes, we used WD01 (GenBank number: ON408217) to represent Tetraena mongolica for comparative analyses and phylogenetic inference.
The six cp genomes of Zygophyllaceae exhibited two patterns of gene order and content: the cp genomes of subfam. Zygophylloideae possessed basically identical genes, and the remaining three cp genomes shared similar coding genes. The specific details of gene loss were shown in the Supplementary Table S1. The LSC of the six cp genomes possessed remarkable resemblance in terms of gene order and content, encoding from 77 to 84 genes (Figure 1). While these cp genomes differed greatly in size, gene order and content of the remaining regions (Figure 2)—for instance, gene numbers varied from 12 (subfam. Zygophylloideae) to 35 (G. angustifolium) in IRs, which attributed to the gene loss (e.g., ndh gene family) and the transfer of genes from IRs to SSC region (e.g., rRNA genes). Although most IR boundary shifts are small (up to several hundred bp) among land plants (Zhu et al., 2016), significant IR/SC boundaries shifts occurred in Zygophyllaceae cp genomes, yielding strongly shrinking IRs in subfam. Zygophylloideae (Figure 3). Additionally, different from a broad range of land plants with conserved collinear gene order, and also from several lineages (e.g., Geraniaceae and Campanulaceae) with reconfigurable gene order (Cosner et al., 2004; Guisinger et al., 2011; Weng et al., 2014), the six Zygophyllaceae cp genomes had relatively few rearrangement events as indicated in the Mauve alignment (Supplementary Figure S2). As expected, there were much more highly divergent sequences in non-coding regions than in coding regions (Supplementary Figure S3).
FIGURE 1

Characteristics of Zygophyllaceae chloroplast genomes. (A) Number of different functional categories of genes. (B) Number of genes. (C) Size of cp genomes. (D) Number of genes located in different regions.
FIGURE 2

Genes in IRa and SSC of Zygophyllaceae chloroplast genomes.
FIGURE 3

IR/SC boundaries of six Zygophyllaceae chloroplast genomes. Number above the gene features means the distance between the ends of genes and the borders sites. These features are not to scale.
Codon usage analysis
To avoid sampling bias, protein-coding regions with the length greater than 300 bp were analyzed (Wright, 1990), resulting in a total of 13,936 (Z. xanthoxylon) to 24,733 (T. terrestris) codons. The codon usage and RSCU values of six Zygophyllaceae cp genomes were summarized in Supplementary Table S2 and the heatmap of RSCU values were shown in Figure 4. Although the number of codons varied dramatically for each amino acid among these species, the codon bias of each amino acid was largely consistent. For example, the amino acid Leu was encoded by six codons (UUA, UUG, CUU, CUC, CUA, and CUG), and the codon, UUA, was used on average 0.83 times less frequently in T. terrestris genes than in the remaining species genes, however, UUA was always the most frequently occurring codon (with the highest RSCU value) relative to the other codons encoding Leu in six species. Codons AUG and UGG showed no bias (RSCU = 1), as Methionine (Met) and Tryptophan (Trp) were encoded only by AUG and UGG, respectively. Concurrently, 30 codons were translation-preferred (RSCU>1) and characterized with a bias in favor of purine (A/U) at the third codon position (except UUG).
FIGURE 4

The RSCU values of protein-coding genes for six Zygophyllaceae species. Color key: the red values indicated higher values and the blue values indicated lower values.
Repeat sequences and simple sequence repeats
A total of 226 tandem repeats, 121 dispersed repeats and 452 SSRs were identified in the six cp genomes (Figure 5) and the detailed information was shown in Supplementary Table S3. In regard to tandem repeats, T. terrestris contained the largest number of repeats (50) while Z. xanthoxylon had the fewest (25). We found that 96.9% of tandem repeats had the repeat motif less than five and only three of the 226 repeats had larger repeat unit (>100 bp). We detected 121 dispersed repeats, and the majority were forward repeats (52.07%), followed by palindromic repeats (37.2%). In contrast, reverse repeats and complement repeats were found with much lower frequency or even absent in some species. Moreover, there were 115 (95.0%) smaller dispersed repeats (<50 bp) while Z. mucronatum and Z. xanthoxylon had four and two larger dispersed repeats (>100 bp), respectively. Among the 452 SSRs, mononucleotide repeats accounted for the highest proportion (84.7%) and all consisted of A/T repeats. Penta- and hexanucleotide repeats were absent in all species.
FIGURE 5
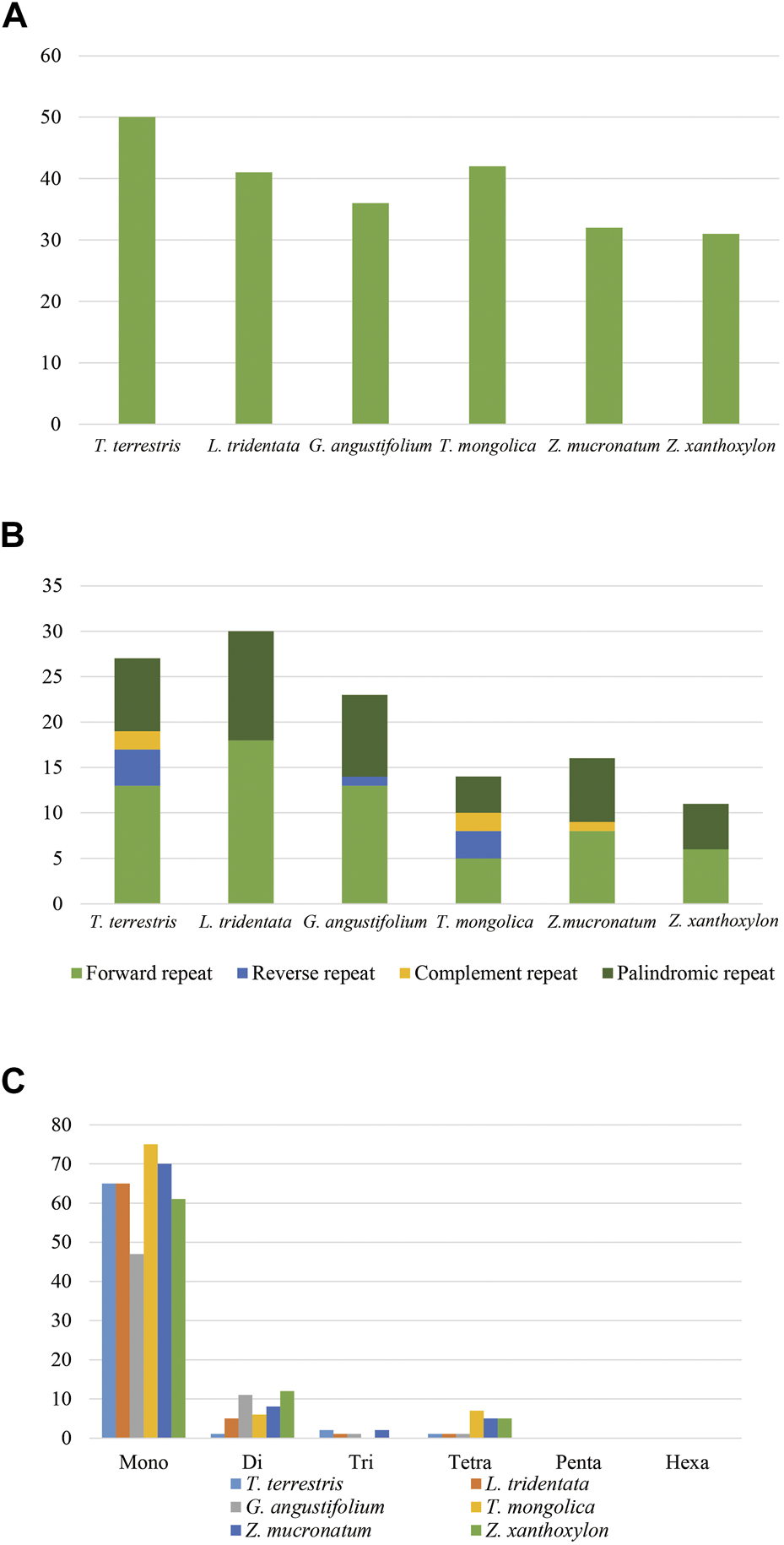
Numbers and types of repeat elements. (A) Tandem repeats. (B) Dispersed repeats. (C) SSRs.
Phylogeny and TCS analysis
Three datasets (CCS, NCS, and PCS) were used to reconstruct the intraspecific relationships of T. mongolica (Figure 6; Supplementary Figures S4,S5). The topologies estimated from ML and BI analyses of the CCS dataset and NCS dataset were broadly similar to each other and received moderate-to-high support. While the ML and BI topologies were generally incongruent inferred from PCS dataset with poor support for most nodes. Here, we reported the relationships from CCS dataset with higher support. Phylogenetic tree resolved two clades comprising: 1) one accession WJRGC35 and 2) the remaining accessions (Figure 6B). With the notable exception of QPC25 and HNHCQ39, accessions respectively belonging to northern populations and southern populations constituted the two subclades of the second clade. Notably, some nodes of the northern subclade received relatively low support values. Similarly, the TCS network result of the 13 cp haplotypes revealed a closer relationship of the most northern populations to southern populations than to WJRGC35 (Figure 6C). Overall, all T. mongolica populations were mainly divided into two groups: the northern group (WJRGC35 and northern populations) and the southern group (the remaining populations).
FIGURE 6

(A) Geographical distributions of sampled T. mongolica populations. (B) The ML tree of 13 T. mongolica inferred from complete chloroplast genomes. Numbers at nodes represented the bootstrap support values and posterior probability values. Dash (–) denoted this relationship was not supported by the BI analysis. (C) The TCS network of intraspecific relationships among the 13 cp haplotypes, the steps among haplotypes showed the degree of genetic variation.
Due to the extensive variation of SSC and IRs, the phylogenetic analyses of Zygophyllaceae were performed based on CCS dataset (Figure 7) and SIS dataset (Supplementary Figure S6). Both ML and BI analyses based on the aforementioned two datasets recovered the same topologies with strong support. Three subfamilies were strongly supported and a closer relationship was exhibited between Larreoideae and Zygophylloideae. Furthermore, our present phylogenetic result showed the closest relationship between Z. mucronatum and Z. xanthoxylon.
FIGURE 7

The ML tree of Zygophyllaceae inferred from complete cp genome dataset. Numbers at nodes represented the bootstrap support values and posterior probability values.
Divergence time estimation of T. mongolica
The BEAST-derived ages of T. mongolica with two calibration points (Figure 8) were largely agreed with those inferred by using one calibration point (Supplementary Figure S7). Thus, we focused on ages mainly from the former result. WJRGC35 diverged from the remainder at ca. 0.304 Ma (node A). The split between the mainly northern group and southern group was ca. 0.225 Ma. The onset of diversification of the mainly northern group and southern group occurred at 0.097 Ma (node B) and 0.082 Ma (node C), respectively.
FIGURE 8

The BEAST-derived chronograms of T. mongolica with two calibration points. The divergence time estimates based on the complete cp genome sequences. Red pentastar represented the calibration points.
Discussion
Unusual structure and gene loss events of the T. mongolica and Zygophyllaceae cp genomes
Despite the high conservation of cp genomes has been observed in most land plants, large-scale variations still have been detected, even within species. For example, extensive population divergences were observed in Monotropa and Hypopitys, suggesting that speciation may be occurring (Braukmann et al., 2017). For T. mongolica, 13 cp genomes possessed identical gene content and order, fairly close GC content, and extremely stable IR/SC boundaries. Additionally, extremely low sequence diversity was exhibited. Consequently, we found that cp genomes of T. mongolica were highly conserved, as observed in most land plants, such as Leonurus Cardiaca (Sun et al., 2021), Cercidiphyllum japonicum (Zhu et al., 2020).
Among the six cp genomes of Zygophyllaceae, genome length, organization and gene content of T. mongolica were more similar to the cp genome of Z. xanthoxylon and Z. mucronatum than to the remaining three species, exhibiting the drastically reduced IRs, the loss of ndh gene family and one copy of rRNAs.
Across the land plants, the significant reduction of cp genome size is mainly due to IR contraction/loss (e.g., IR-lacking clade (IRLC) legumes, some species of Geraniaceae; Duan et al., 2021; Guisinger et al., 2011) and gene loss (e.g., some parasitic plants and mycoheterotrophs; Braukmann et al., 2017; Graham et al., 2017). With the conspicuous exception of T. terrestris (∼158 kb), the remaining species in Zygophyllaceae had smaller cp genomes (c. 106–135 kb) compared with most land plants (an average size of 151 kb) (Guo et al., 2021b). Unlike most angiosperms retaining a 25 kb IR (Ruhlman and Jansen, 2014), variation in the size of the IR, which could range from 4,288 bp (Z. xanthoxylon) and up to 25,842 bp (T. terrestris), accounted for Zygophyllaceae cp genome size variation overall (105,251 bp to 158,184 bp). The phenomenon that IR has undergone expansion and contraction is nearly omnipresent while significant IR boundary shifts are relatively rare (Hansen et al., 2007; Yang et al., 2016; Zhu et al., 2016; Sinn et al., 2018; Wei et al., 2021). In all investigated six species except T. terrestris, the IR was thought to have undergone large-scale contraction relative to the most angiosperms (25 kb), contributing to their smaller IR size (4,288–19,350 bp). Overall GC content is typically in the range of 30–40% (average 37%) in most cp genomes of land plants (Bock, 2007; Civáň et al., 2014), and the average GC content (∼34.7%) of Zygophyllaceae cp genomes is relatively lower than in land plants (37%).
Moreover, the drastically reduced IR, rearrangement events, IR/SC boundary shifts all resulted in reconfiguration of gene content and order. The total gene numbers of the six Zygophyllaceae cp genomes differed greatly, from 104 in subfam. Zygophylloideae, to 130 in T. terrestris. Especially, gene numbers varied from 12 (subfam. Zygophylloideae) to 35 (G. angustifolium) located in IRs. Despite there were different numbers of protein-coding genes (67–84), the codon usage preference of each amino acid showed highly similar in these Zygophyllaceae cp genomes, even more broadly across land plants, as previous research results (e.g., Guisinger et al., 2011; Yang et al., 2018). We observed a similar bias in our analysis in comparison to many other land plants, the frequently used codons (RSCU value > 1) all but UUG ended in A/U (Kim and Lee, 2004). The presence of translation-preferred codons and A/U bias at the third codon position may be the result of both natural selection and mutation biases during the cp genome evolutionary process (Xu et al., 2011; Li et al., 2021).
Most remarkably, the loss of entire family of ndh genes (ndhA–ndhK) occurred in subfam. Zygophylloideae, which had also been detected and verified by PCR product sequencing (Wang et al., 2022). In most cp genomes of land plants, 11 genes (ndhA–ndhK) constitute ndh family and encode subunits of the NDH complex that is involved in photosystem I cyclic electron flow and chlororespiration (Burrows et al., 1998; Peltier and Cournac, 2002; Martín and Sabater, 2010; Peng et al., 2011). However, loss or pseudogenization of ndh gene family are well-known in a number of plant lineages. For example, these genes are commonly lost in non-photosynthetic plants and mycoheterotrophs (Graham et al., 2017; Sabater, 2021). In addition, it has been documented that ndh genes are lost in several fully photosynthetic plant lineages, such as Gnetales, several conifers (Martín and Sabater, 2010), Carnegiea gigantea (Sanderson et al., 2015), Erodium (Chris Blazier et al., 2011) and Kingdonia (Sun et al., 2017). For the loss of ndh genes in these fully photosynthetic plants, one possible reason is that the functional role of the protein products may be simply dispensable, especially for the taxa distributed in mild habitats (Martín and Sabater, 2010; Sabater, 2021). Alternatively, NDH-dependent pathway may be functionally replaced by another pathway (such as antimycin A-sensitive cyclic electron flow pathway), especially for the taxa under light, water and temperature stress (Sanderson et al., 2015). Notably, Balanites aegyptiaca (subfam. Tribuloideae, Zygophyllaceae), an arid and semi-arid plant, possesses all 11 ndh genes in cp genome, possibly indicating the different evolutionary history among the subfamilies of Zygophyllaceae (Al-Juhani et al., 2022). Specifically, considering the role of ndh genes in stress acclimation (Peng et al., 2011), angiosperms lacking ndh genes have a higher risk of extinction in facing variable evolutionary environments (Sabater, 2021) and may be occurring in endangered Kingdonia uniflora (Sun et al., 2017). When considering the loss of the ndh gene family, T. mongolica may have higher extinction risk in facing changing environments.
In most land plants, two copies of rRNAs (5S, 4.5S, 23S, and 16S rRNA) are often present in the IRa and IRb respectively. In subfam. Zygophylloideae, the highly unusual is that there is only one copy of rRNAs with SSC localization, which can be attributed mainly to the transfer of these genes from IRs to SSC rather than the loss of one copy of IR in several plant lineages, as observed for IRLC legumes. It has been detected in many land plants that genes transfer from the SC into the IRs and vice versa (Zhu et al., 2016), and the variation in the number of gene copy due to distribution changes is also present in some plants (e.g., Sinn et al., 2018; Henriquez et al., 2020).
Repeat analyses
Larger and more complex repeat sequences may be correlated with the rearrangement of cp genomes (Timme et al., 2007; Weng et al., 2014). As observed in highly rearranged cp genomes, such as Geraniaceae (Weng et al., 2014) and Passiflora (Cauz-Santos et al., 2020), there are always existing a high frequency of larger repeats. In our study, there were few larger repeats (six of 121 dispersal repeats were longer than 100 bp), meanwhile, a low number of rearrangements were present in Zygophyllaceae. SSRs are ubiquitous throughout genomes and have been widely used as molecular markers (Kumar et al., 2015; Younis et al., 2020). Overall, SSRs present in our analyses could act as markers for genetic diversity and phylogenetic inference in Zygophyllaceae.
Phylogenetic relationships in T. mongolica and Zygophyllaceae
Our phylogenetic result and TCS network result supported T. mongolica was mainly divided into two groups (northern group and southern group) with significantly improved support values, which is generally consistent with previous result inferred from cpDNA marker (Ge et al., 2011), rather than the result based on nuclear genomic data (Cheng et al., 2020). Although two groups were recognized by both cp genomes and nuclear genomic data, the populations located in the central of the distributional range were identified as the northern group and southern group based on cp genomes in our study and nuclear genomic data in Cheng et al. (2020), respectively. The incongruent phylogenetic topologies between nuclear and cp gene trees have been commonly observed, which may be triggered by incomplete lineage sorting (ILS), allopolyploidy, hybridization, introgression and so on (Duan et al., 2021). Interestingly, population WJRGC35 formed a clade that was sister to the remaining populations. The previous study suggested that the northernmost population (WJM population) may be the ancestral population of T. mongolica and spread southward in history (Cheng et al., 2020). In terms of geographical location, both SGK32 and WJRGC35 in our study belonged to the WJM population in the study of Cheng et al. (2020), while WJRGC35 was located in the central area of the WJM population. Accordingly, combined with our divergence time estimates, we supported that the ancestral position of the northernmost population, especially these T. mongolica located in its central area. In addition, the phylogenetic position of QPC25 and HNHCQ39 was not consistent with its geographical location, which may have resulted from ILS. Besides, the collection locations of the two populations were very close to the busy roads (such as beltway and expressway), which may provide opportunities for the long-distance dispersal of seeds from other populations. Despite two groups were well supported, the phylogenetic relationships within group were recovered with low to medium support in all datasets, which may be largely attributed to the low genetic differentiation among the populations rather than the insufficient phylogenetic signals. Such a similar result was also observed in previous study, which inferred from a total of 38,097 SNPs (Cheng et al., 2020).
Although representatives of Morkillioideae and Seetzenioideae were absent, the remaining three of the five subfamilies of Zygophyllaceae were recovered as monophyletic with strong support. In our analyses, Larreoideae was resolved sister to Zygophylloideae, consistent with previous studies (Sheahan and Chase, 2000; Wu et al., 2018). To some extent, the closest phylogenetic relationship coupled with the highly similar cp genomes between Z. xanthoxylon and Z. mucronatum in our study, supported that the two species form a genus (Zygophyllum) rather than two genera (Sarcozygium and Zygophyllum). Such a taxonomy was also suggested in most previous studies (e.g., Beier et al., 2003; Wu et al., 2018; Zhang et al., 2021).
Statements
Data availability statement
The data presented in the study are deposited in the NCBI repository, accession number ON408217–ON408231, BioProject PRJNA866691 and PRJNA866686.
Author contributions
YY, YW, and TZ designed the experiments. YY and YZ collected the plant samples. YY and YJ analyzed the data. YY wrote the paper. All authors read and approved the final manuscript.
Funding
This work was financially supported by the Natural Science Foundation of Inner Mongolia (2021BS03001) (providing open access publication fees), the Scientific Research Project of Higher Education Institutions of Inner Mongolia (NJZZ20360) (providing sequencing fees) and the Initial Scientific Research Fund of Baotou Teachers’ College (BTTCRCQD 2018-01) (providing sampling fees).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1026919/full#supplementary-material
SUPPLEMENTARY FIGURE S1IR/SC boundaries of 13 T. mongolica chloroplast genomes. Number above the gene features means the distance between the ends of genes and the borders sites. These features are not to scale.
SUPPLEMENTARY FIGURE S2Synteny and rearrangements detected in six Zygophyllaceae chloroplast genomes and T. terrestris was set as the reference genome. Colored bars indicated syntenic blocks, connecting lines indicated the correspondence between blocks, and pink boxes indicated the IRs. The numbers indicated the genome length (bp).
SUPPLEMENTARY FIGURE S3Visualization of the comparison of complete six Zygophyllaceae chloroplast genomes. T. terrestris was used as the reference genome.
SUPPLEMENTARY FIGURE S4The ML tree of 13 T. mongolica inferred from the noncoding region sequences. Numbers at nodes represented the bootstrap support values and posterior probability values. Dash (–) denoted this relationship was not supported by the BI analysis.
SUPPLEMENTARY FIGURE S5The ML tree of 13 T. mongolica inferred from the protein-coding region sequences. Numbers at nodes represented the bootstrap support values and posterior probability values. Dash (–) denoted this relationship was not supported by the BI analysis.
SUPPLEMENTARY FIGURE S6The ML tree of Zygophyllaceae inferred from SSC + single IR sequences. Numbers at nodes represented the bootstrap support values and posterior probability values.
SUPPLEMENTARY FIGURE S7The BEAST-derived chronograms of T. mongolica with one calibration point. The divergence time estimates based on the complete cp genome sequences. Red pentastar represented the calibration point.
References
1
Al-Juhani W. S. Alharbi S. A. Al Aboud N. M. Aljohani A. Y. (2022). Complete chloroplast genome of the desert date (Balanites aegyptiaca (L.) Del. comparative analysis, and phylogenetic relationships among the members of Zygophyllaceae. BMC Genomics23, 626–719. 10.1186/s12864-022-08850-9
2
Barnosky A. D. Matzke N. Tomiya S. Wogan G. O. Swartz B. Quental T. B. et al (2011). Has the Earth’s sixth mass extinction already arrived?. Nature471, 51–57. 10.1038/nature09678
3
Beier B. A. Chase M. W. Thulin M. (2003). Phylogenetic relationships and taxonomy of subfamily Zygophylloideae (Zygophyllaceae) based on molecular and morphological data. Plant Syst. Evol.240, 11–39. 10.1007/s00606-003-0007-0
4
Bellstedt D. U. Galley C. Pirie M. D. Linder H. P. (2012). The migration of the palaeotropical arid flora: Zygophylloideae as an example. Syst. Bot.37, 951–959. 10.1600/036364412X656608
5
Benson G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res.27, 573–580. 10.1093/nar/27.2.573
6
Birky C. W. Maruyama T. Fuerst P. (1983). An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics103, 513–527. 10.1093/genetics/103.3.513
7
Bock R. (2007). “Structure, function, and inheritance of plastid genomes,” in Cell and molecular biology of plastids. Editor BockR. (Heidelberg, Berlin: Springer Press), 29–63.
8
Bracken M. E. Low N. H. (2012). Realistic losses of rare species disproportionately impact higher trophic levels. Ecol. Lett.15, 461–467. 10.1111/j.1461-0248.2012.01758.x
9
Braukmann T. W. Broe M. B. Stefanović S. Freudenstein J. V. (2017). On the brink: the highly reduced plastomes of nonphotosynthetic ericaceae. New Phytol.216, 254–266. 10.1111/nph.14681
10
Bunge D. A. (1843). Eine neue gattung aus der familie der Zygophyllaceae. Linnaea17, 7.
11
Burrows P. A. Sazanov L. A. Svab Z. Maliga P. Nixon P. J. (1998). Identification of a functional respiratory complex in chloroplasts through analysis of tobacco mutants containing disrupted plastid ndh genes. EMBO J.17, 868–876. 10.1093/emboj/17.4.868
12
Cauz-Santos L. A. da Costa Z. P. Callot C. Cauet S. Zucchi M. I. Bergès H. et al (2020). A repertory of rearrangements and the loss of an inverted repeat region in Passiflora chloroplast genomes. Genome Biol. Evol.12, 1841–1857. 10.1093/gbe/evaa155
13
Cheng J. Kao H. Dong S. (2020). Population genetic structure and gene flow of rare and endangered Tetraena mongolica Maxim. revealed by reduced representation sequencing. BMC Plant Biol.20, 391–413. 10.1186/s12870-020-02594-y
14
Chevreux B. Pfisterer T. Drescher B. Driesel A. J. Müller W. E. Wetter T. et al (2004). Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res.14, 1147–1159. 10.1101/gr.1917404
15
Chris Blazier J. Guisinger M. M. Jansen R. K. (2011). Recent loss of plastid-encoded ndh genes within Erodium (Geraniaceae). Plant Mol. Biol.76, 263–272. 10.1007/s11103-011-9753-5
16
Civáň P. Foster P. G. Embley M. T. Seneca A. Cox C. J. (2014). Analyses of charophyte chloroplast genomes help characterize the ancestral chloroplast genome of land plants. Genome Biol. Evol.6, 897–911. 10.1093/gbe/evu061
17
Cosner M. E. Raubeson L. A. Jansen R. K. (2004). Chloroplast DNA rearrangements in Campanulaceae: phylogenetic utility of highly rearranged genomes. BMC Evol. Biol.4, 27. 10.1186/1471-2148-4-27
18
Darling A. C. E. Mau B. Blattner F. R. Perna N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res.14, 1394–1403. 10.1101/gr.2289704
19
Doyle J. J. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull.19, 11–15.
20
Drummond A. J. Rambaut A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol.7, 214–218. 10.1186/1471-2148-7-214
21
Duan L. Li S. J. Su C. Sirichamorn Y. Han L. N. Ye W. et al (2021). Phylogenomic framework of the IRLC legumes (Leguminosae subfamily Papilionoideae) and intercontinental biogeography of tribe Wisterieae. Mol. Phylogenet. Evol.163, 107235. 10.1016/j.ympev.2021.107235
22
Faircloth B. C. (2008). MSATCOMMANDER: detection of microsatellite repeat arrays and automated, locus-specific primer design. Mol. Ecol. Resour.8, 92–94. 10.1111/j.1471-8286.2007.01884.x
23
Frazer K. A. Pachter L. Poliakov A. Rubin E. M. Dubchak I. (2004). VISTA: Computational tools for comparative genomics. Nucleic Acids Res.32, W273–W279. 10.1093/nar/gkh458
24
Fu C. N. Mo Z. Q. Yang J. B. Ge X. J. Li D. Z. Xiang Q. Y. J. et al (2019). Plastid phylogenomics and biogeographic analysis support a trans-Tethyan origin and rapid early radiation of Cornales in the Mid-Cretaceous. Mol. Phylogenet. Evol.140, 106601. 10.1016/j.ympev.2019.106601
25
Fu L. G. (1992). The red book of Chinese plants–rare and endangered plants. Beijing: Science Press.
26
Ge X. J. Hwang C. C. Liu Z. H. Huang C. C. Huang W. H. Hung K. H. et al (2011). Conservation genetics and phylogeography of endangered and endemic shrub Tetraena mongolica (Zygophyllaceae) in Inner Mongolia, China. BMC Genet.12, 1–12. 10.1186/1471-2156-12-1
27
Gonçalves D. J. Simpson B. B. Ortiz E. M. Shimizu G. H. Jansen R. K. (2019). Incongruence between gene trees and species trees and phylogenetic signal variation in plastid genes. Mol. Phylogenet. Evol.138, 219–232. 10.1016/j.ympev.2019.05.022
28
Graham S. W. Lam V. K. Merckx V. S. (2017). Plastomes on the edge: the evolutionary breakdown of mycoheterotroph plastid genomes. New Phytol.214, 48–55. 10.1111/nph.14398
29
Guisinger M. M. Kuehl J. V. Boore J. L. Jansen R. K. (2011). Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol. Biol. Evol.28, 583–600. 10.1093/molbev/msq229
30
Guo Y. Y. Yang J. X. Bai M. Z. Zhang G. Q. Liu Z. J. (2021a). The chloroplast genome evolution of venus slipper (Paphiopedilum): IR expansion, SSC contraction, and highly rearranged SSC regions. BMC Plant Biol.21, 248–314. 10.1186/s12870-021-03053-y
31
Guo Y. Y. Yang J. X. Li H. K. Zhao H. S. (2021b). Chloroplast genomes of two species of Cypripedium: Expanded genome size and proliferation of AT-biased repeat sequences. Front. Plant Sci.12, 609729. 10.3389/fpls.2021.609729
32
Hahn C. Bachmann L. Chevreux B. (2013). Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads – a baiting and iterative mapping approach. Nucleic Acids Res.41, e129. 10.1093/nar/gkt371
33
Hansen D. R. Dastidar S. G. Cai Z. Penaflor C. Kuehl J. V. Boore J. L. et al (2007). Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae). Mol. Phylogenet. Evol.45, 547–563. 10.1016/j.ympev.2007.06.004
34
Henriquez C. L. Mehmood F. Carlsen M. M. Islam M. Waheed M. T. Poczai P. et al (2020). Complete chloroplast genomes of Anthurium huixtlense and Pothos scandens (pothoideae, araceae): unique inverted repeat expansion and contraction affect rate of evolution. J. Mol. Evol.88, 562–574. 10.1007/s00239-020-09958-w
35
Jansen R. K. Ruhlman T. A. (2012). “Plastid genomes of seed plants,” in Genomics of chloroplasts and mitochondria. Editor BockR. (Dordrecht: Springer Press), 103–126.
36
Jansen R. K. Raubeson L. A. Boore J. L. DePamphilis C. W. Chumley T. W. Haberle R. C. et al (2005). Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol.395, 348–384. 10.1016/S0076-6879(05)95020-9
37
Katoh K. Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol.30, 772–780. 10.1093/molbev/mst010
38
Kim K. J. Lee H. L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res.11, 247–261. 10.1093/dnares/11.4.247
39
Kumar M. Choi J. Y. Kumari N. Pareek A. Kim S. R. (2015). Molecular breeding in Brassica for salt tolerance: importance of microsatellite (SSR) markers for molecular breeding in Brassica. Front. Plant Sci.6, 688. 10.3389/fpls.2015.00688
40
Kurtz S. Choudhuri J. V. Ohlebusch E. Schleiermacher C. Stoye J. Giegerich R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res.29, 4633–4642. 10.1093/nar/29.22.4633
41
Leigh J. W. Bryant D. (2015). POPART: full-feature software for haplotype network construction. Methods Ecol. Evol.6, 1110–1116. 10.1111/2041-210X.12410
42
Li H. T. Yi T. S. Gao L. M. Ma P. F. Zhang T. Yang J. B. et al (2019). Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants5, 461–470. 10.1038/s41477-019-0421-0
43
Li G. Zhang L. Xue P. (2021). Codon usage pattern and genetic diversity in chloroplast genomes of Panicum species. Gene802, 145866. 10.1016/j.gene.2021.145866
44
Librado P. Rozas J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics25, 1451–1452. 10.1093/bioinformatics/btp187
45
Liou Y. X. (1998). “Zygophyllaceae,” in Flora reipublicae popularis sinicae. Editors XuL. R.HuangC. C. (Beijing: Science Press), 116–145.
46
Ma P. F. Zhang Y. X. Zeng C. X. Guo Z. H. Li D. Z. (2014). Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol.63, 933–950. 10.1093/sysbio/syu054
47
Martín M. Sabater B. (2010). Plastid ndh genes in plant evolution. Plant Physiol. biochem.48, 636–645. 10.1016/j.plaphy.2010.04.009
48
Mouillot D. Bellwood D. R. Baraloto C. Chave J. Galzin R. Harmelin-Vivien M. et al (2013). Rare species support vulnerable functions in high-diversity ecosystems. PLoS Biol.11, e1001569. 10.1371/journal.pbio.1001569
49
Peltier G. Cournac L. (2002). Chlororespiration. Annu. Rev. Plant Biol.53, 523–550. 10.1146/annurev.arplant.53.100301.135242
50
Peng L. Yamamoto H. Shikanai T. (2011). Structure and biogenesis of the chloroplast NAD(P)H dehydrogenase complex. Biochim. Biophys. Acta1807, 945–953. 10.1016/j.bbabio.2010.10.015
51
Posada D. Crandall K. A. (1998). Modeltest: testing the model of DNA substitution. Bioinformatics14, 817–818. 10.1093/bioinformatics/14.9.817
52
Ronquist F. Huelsenbeck J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics19, 1572–1574. 10.1093/bioinformatics/btg180
53
Ruhlman T. A. Jansen R. K. (2014). “The plastid genomes of flowering plants,” in Chloroplast biotechnology. Editor MaligaP. (Totowa, NJ: Humana Press), 3–38.
54
Sabater B. (2021). On the edge of dispensability, the chloroplast ndh genes. Int. J. Mol. Sci.22, 12505. 10.3390/ijms222212505
55
Sanderson M. J. Copetti D. Búrquez A. Bustamante E. Charboneau J. L. Eguiarte L. E. et al (2015). Exceptional reduction of the plastid genome of saguaro cactus (Carnegiea gigantea): Loss of the ndh gene suite and inverted repeat. Am. J. Bot.102, 1115–1127. 10.3732/ajb.1500184
56
Sharp P. M. Li W. H. (1986). An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol.24, 28–38. 10.1007/BF02099948
57
Sheahan M. C. Chase M. W. (2000). Phylogenetic relationships within Zygophyllaceae based on DNA sequences of three plastid regions, with special emphasis on Zygophylloideae. Syst. Bot.25, 371–384. 10.2307/2666648
58
Sinn B. T. Sedmak D. D. Kelly L. M. Freudenstein J. V. (2018). Total duplication of the small single copy region in the angiosperm plastome: rearrangement and inverted repeat instability in asarum. Am. J. Bot.105, 71–84. 10.1002/ajb2.1001
59
Song Y. Yu W. B. Tan Y. Liu B. Yao X. Jin J. et al (2017). Evolutionary comparisons of the chloroplast genome in Lauraceae and insights into loss events in the Magnoliids. Genome Biol. Evol.9, 2354–2364. 10.1093/gbe/evx180
60
Stamatakis A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics22, 2688–2690. 10.1093/bioinformatics/btl446
61
Sullivan A. R. Schiffthaler B. Thompson S. L. Street N. R. Wang X. R. (2017). Interspecific plastome recombination reflects ancient reticulate evolution in Picea (Pinaceae). Mol. Biol. Evol.34, 1689–1701. 10.1093/molbev/msx111
62
Sun Y. Moore M. J. Lin N. Adelalu K. F. Meng A. Jian S. et al (2017). Complete plastome sequencing of both living species of Circaeasteraceae (Ranunculales) reveals unusual rearrangements and the loss of the ndh gene family. BMC Genomics18, 592–610. 10.1186/s12864-017-3956-3
63
Sun J. Wang Y. Garran T. A. Qiao P. Wang M. Yuan Q. et al (2021). Heterogeneous genetic diversity estimation of a promising domestication medicinal motherwort Leonurus Cardiaca based on chloroplast genome resources. Front. Genet.12, 721022. 10.3389/fgene.2021.721022
64
Tamura K. Peterson D. Peterson N. Stecher G. Nei M. Kumar S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol.28, 2731–2739. 10.1093/molbev/msr121
65
Timme R. E. Kuehl J. V. Boore J. L. Jansen R. K. (2007). A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot.94, 302–312. 10.3732/ajb.94.3.302
66
Wang X. Dorjee T. Chen Y. Gao F. Zhou Y. (2022). The complete chloroplast genome sequencing analysis revealed an unusual IRs reduction in three species of subfamily Zygophylloideae. Plos One17, e0263253. 10.1371/journal.pone.0263253
67
Wang G. H. (2005). The Western Ordos plateau as a biodiversity center of relic shrubs in arid areas of China. Biodivers. Conserv.14, 3187–3200. 10.1007/s10531-004-0386-8
68
Wei N. Pérez-Escobar O. A. Musili P. M. Huang W. C. Yang J. B. Hu A. Q. et al (2021). Plastome evolution in the hyperdiverse genus Euphorbia (Euphorbiaceae) using phylogenomic and comparative analyses: Large-scale expansion and contraction of the inverted repeat region. Front. Plant Sci.1555, 712064. 10.3389/fpls.2021.712064
69
Weng M. L. Blazier J. C. Govindu M. Jansen R. K. (2014). Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol.31, 645–659. 10.1093/molbev/mst257
70
Wolfe A. D. Randle C. P. (2004). Recombination, heteroplasmy, haplotype polymorphism, and paralogy in plastid genes: implications for plant molecular systematics. Syst. Bot.29, 1011–1020. 10.1600/0363644042451008
71
Wright F. (1990). The ‘effective number of codons’ used in a gene. Gene87, 23–29. 10.1016/0378-1119(90)90491-9
72
Wu S. D. Lin L. Li H. L. Yu S. X. Zhang L. J. Wang W. (2015). Evolution of asian interior arid-zone biota: evidence from the diversification of asian Zygophyllum (Zygophyllaceae). PloS One10, e0138697. 10.1371/journal.pone.0138697
73
Wu S. D. Zhang L. J. Lin L. Yu S. X. Chen Z. D. Wang W. (2018). Insights into the historical assembly of global dryland floras: the diversification of Zygophyllaceae. BMC Evol. Biol.18, 166–210. 10.1186/s12862-018-1277-z
74
Xu C. Cai X. Chen Q. Zhou H. Cai Y. Ben A. (2011). Factors affecting synonymous codon usage bias in chloroplast genome of oncidium gower ramsey. Evol. Bioinform. Online7, 271–278. 10.4137/EBO.S8092
75
Yan J. Zhang N. Duan Y. (2019). The complete chloroplast genome sequence of Tribulus terrestris, an important traditional Chinese medicine. Mitochondrial DNA. B Resour.4, 3108–3109. 10.1080/23802359.2019.1667891
76
Yang Y. Zhou T. Duan D. Yang J. Feng L. Zhao G. (2016). Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci.7, 959. 10.3389/fpls.2016.00959
77
Yang Y. Zhu J. Feng L. Zhou T. Bai G. Yang J. et al (2018). Plastid genome comparative and phylogenetic analyses of the key genera in Fagaceae: Highlighting the effect of codon composition bias in phylogenetic inference. Front. Plant Sci.9, 82. 10.3389/fpls.2018.00082
78
Yang Y. Zhou T. Qian Z. Zhao G. (2021). Phylogenetic relationships in Chinese oaks (Fagaceae, Quercus): Evidence from plastid genome using low-coverage whole genome sequencing. Genomics113, 1438–1447. 10.1016/j.ygeno.2021.03.013
79
Younis A. Ramzan F. Ramzan Y. Zulfiqar F. Ahsan M. Lim K. B. (2020). Molecular markers improve abiotic stress tolerance in crops: a review. Plants9, 1374. 10.3390/plants9101374
80
Zhai W. Duan X. Zhang R. Guo C. Li L. Xu G. et al (2019). Chloroplast genomic data provide new and robust insights into the phylogeny and evolution of the Ranunculaceae. Mol. Phylogenet. Evol.135, 12–21. 10.1016/j.ympev.2019.02.024
81
Zhang S. D. Jin J. J. Chen S. Y. Chase M. W. Soltis D. E. Li H. T. et al (2017). Diversification of rosaceae since the late cretaceous based on plastid phylogenomics. New Phytol.214, 1355–1367. 10.1111/nph.14461
82
Zhang L. Wang S. Su C. Harris A. J. Zhao L. Su N. et al (2021). Comparative chloroplast genomics and phylogenetic analysis of Zygophyllum (Zygophyllaceae) of China. Front. Plant Sci.12, 723622. 10.3389/fpls.2021.723622
83
Zhao Y. Z. (1997). Endemic genera and their basic characteristics of the Mongolian Planteau plants. J. Inn. Mong. Univ. Nat. Sci. Ed.28, 547–552.
84
Zhi Y. Sun Z. Sun P. Zhao K. Guo Y. Zhang D. et al (2018). How much genetic variation is stored in the endangered and fragmented shrub Tetraena mongolica Maxim?PeerJ6, e5645. 10.7717/peerj.5645
85
Zhu A. Guo W. Gupta S. Fan W. Mower J. P. (2016). Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol.209, 1747–1756. 10.1111/nph.13743
86
Zhu S. Chen J. Zhao J. Comes H. P. Li P. Fu C. et al (2020). Genomic insights on the contribution of balancing selection and local adaptation to the long-term survival of a widespread living fossil tree, Cercidiphyllum japonicum. New Phytol.228, 1674–1689. 10.1111/nph.16798
Summary
Keywords
chloroplast evolution, phylogenetic inference, Tetraena mongolica , ndh gene family, Zygophyllaceae
Citation
Yang Y, Jia Y, Zhao Y, Wang Y and Zhou T (2022) Comparative chloroplast genomics provides insights into the genealogical relationships of endangered Tetraena mongolica and the chloroplast genome evolution of related Zygophyllaceae species. Front. Genet. 13:1026919. doi: 10.3389/fgene.2022.1026919
Received
24 August 2022
Accepted
18 November 2022
Published
08 December 2022
Volume
13 - 2022
Edited by
Wenqin Wang, Shanghai Normal University, China
Reviewed by
Yongji Huang, Fujian Agriculture and Forestry University, China
Yongrui Wu, Chinese Academy of Sciences (CAS), China
Updates
Copyright
© 2022 Yang, Jia, Zhao, Wang and Zhou.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yonglong Wang, wylongceltics@163.com; Tao Zhou, woody196@163.com
This article was submitted to Evolutionary and Population Genetics, a section of the journal Frontiers in Genetics
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.