 Siqi Cheng
Siqi Cheng Weihong Chen1
Weihong Chen1 Xing Xing
Xing Xing
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 20 October 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1012558
This article is part of the Research Topic Genetics of Inborn Errors of Metabolism View all 30 articles
Background: The cobalamin C (cblC) defect, a common inborn disorder of cobalamin metabolism due to a genetic mutation in MMACHC, can cause combined methylmalonic acid and homocysteine accumulation in blood, urine, or both. In this article, a late-onset case was reported, and the patient first presented with depression identified with the MMACHC gene. We summarized the clinical features of the cblC defect, the relationship between genotype and phenotype, and the clinical experience concerning the diagnosis and treatment of the cblC defect.
Case presentation: Initially presented with depression, the 16-year-old female patient showed progressive abnormal gait and bilateral lower limb weakness after 3 months. Blood routine examination suggested severe hyperhomocysteinemia, and screening for urine organic acids found elevated methylmalonic acid. Family gene sequencing showed mutations detected in MMACHC. She had a compound heterozygous mutation, while the c.271dupA (p.R91Kfs∗14) was only detected in her father and the c.482 G>A (p.R161Q) was only detected in her mother. Hence, she was diagnosed with a cblC defect and treated with B vitamin supplements. The muscle strength of both lower limbs improved notably.
Conclusion: This case indicated that depression could be a presenting sign of cblC-type methylmalonic aciduria and homocysteinemia, and enhanced the genotype–phenotype relationship of the cblC defect, which will contribute to further understanding of this emerging disease.
Cobalamin C defect (cblC, OMIM 277400), a kind of complementation group disease of defective cblC metabolism described by Rosenberg in 1975 (Gravel et al., 1975), can result in both methylmalonic acid (MMA) and homocysteine (Hcy) accumulation in vivo, which was first reported by professor Mudd in 1969 (Mudd et al., 1969). It is an inherited metabolic disease caused by a mutation in MMACHC (OMIM 609831) (Lerner-Ellis et al., 2006). A CblC defect is the most common genetically inherited reason for combined methylmalonic aciduria and homocysteinemia, constituting the main biochemical phenotype of China and accounting for 60%∼80% of the domestic methylmalonic aciduria (Liu et al., 2012; Zhang et al., 2007).
The clinical features of the cblC defect vary by age of disease onset. Differing from the early-onset type (<1 year old), the late-onset cases (>4 years old) usually have milder clinical manifestations and a better prognosis after early intervention (Martinelli et al., 2011).
Sometimes there is a failure to detect cblC defects early due to their heterogeneous phenotypic spectrum of presentations. Normally, the cblC defect produces a series of multisystem neurological symptoms, such as psychological and behavioral abnormalities, leukoencephalopathy, encephalopathy, subacute combined degeneration of the spinal cord, lower extremity weakness, gait disorder, and seizures (Martinelli et al., 2011; Carrillo-Carrasco et al., 2012). However, there are few case reports focusing on cblC patients initially manifesting as depression. Here, we reported one novel late-onset cblC case, where the patient first presented with depression, carrying heterozygous variants in MMACHC, and also explored the pathophysiological mechanism. To date, only a small number of studies have found a potential association between the cblC genotype and its phenotype, which can help predict the age of disease onset and severity (Matos et al., 2013; Yu et al., 2015). Therefore, more comprehensive studies on the clinical and genetic features of cblC patients are warranted.
The 16-year-old girl was born at full term with a spontaneous delivery from a non-consanguineous Chinese Han family. Her parents were healthy and denied a family history of neurologic illness. The girl first suffered from low mood, low self-esteem, self-blame, and visual hallucinations. Additionally, the neuropsychological scale at admission, particularly self-reporting inventory-90 and patient health questionnaire-9, pointed toward the occurrence of depression. A routine blood examination suggested moderate iron-deficiency anemia (hemoglobin of 86 g/L) and moderate homocysteinemia (Hcy of 62 μmol/L, reference range: 5–15 μmol/L). Brain MRI showed patchy and symmetric white matter hyperintensities in the posterior horn of the bilateral lateral ventricles. She was diagnosed with depression by her psychiatrist and was orally administered antidepressants, iron, and methylcobalamin (Mecbl). The depression and hallucination symptoms were alleviated after pharmacological therapy for only 2 weeks.
After 3 months, she showed gradually progressive abnormal gait and bilateral lower limb weakness without depression relapse, and she could hardly even stand without assistance. Physical examination also revealed hyperactive reflexes, bilateral ankle clonus, a positive Babinski sign, and suspicious deep sensory impairment. Iron-deficiency anemia and homocysteinemia were still detected with hyperuricemia. A review of the cerebral MRI found that the previous lesions displayed no change and were not enhanced on the spoiled gradient recalled (SPGR) sequence. The results of the special blood test were negative for pepsinogen I and II, pro-gastrin-releasing peptide, immune, and rheumatic factors. Moreover, neither serum nor cerebrospinal fluid discovered positive results of myelin oligodendrocyte glycoprotein antibody, aquaporin 4 antibody, glial fibrillary acidic protein antibody, and autoimmune encephalitis-related auto-antibodies.
After excluding other diseases leading to multisystem involvement, inborn metabolic diseases were considered. The detailed information of this patient is shown in Table 1. Screening for urine organic acids was performed after 3 months of cobalamin supplement and still, elevated methylmalonic acid of 21.2 mmol/mol creatinine (reference range: 0.0–4.0 mmol/mol creatinine) was found. Amino acids and acylcarnitines analysis in blood were normal. Notably, with family whole exome sequencing and Sanger sequencing performed, it was detected that a compound heterozygous mutation existed in the MMACHC gene. One frameshift variant was c.271dupA (p.R91Kfs∗14), inherited from her mother, and the other variant, belonging to the missense type, was c.482 G>A (p.R161Q) from her father. According to the American College of Medical Genetics and Genomics (ACMG), both variants are pathogenic (Lerner-Ellis et al., 2006; Lerner-Ellis et al., 2009; Richards et al., 2015) (Figure 1).
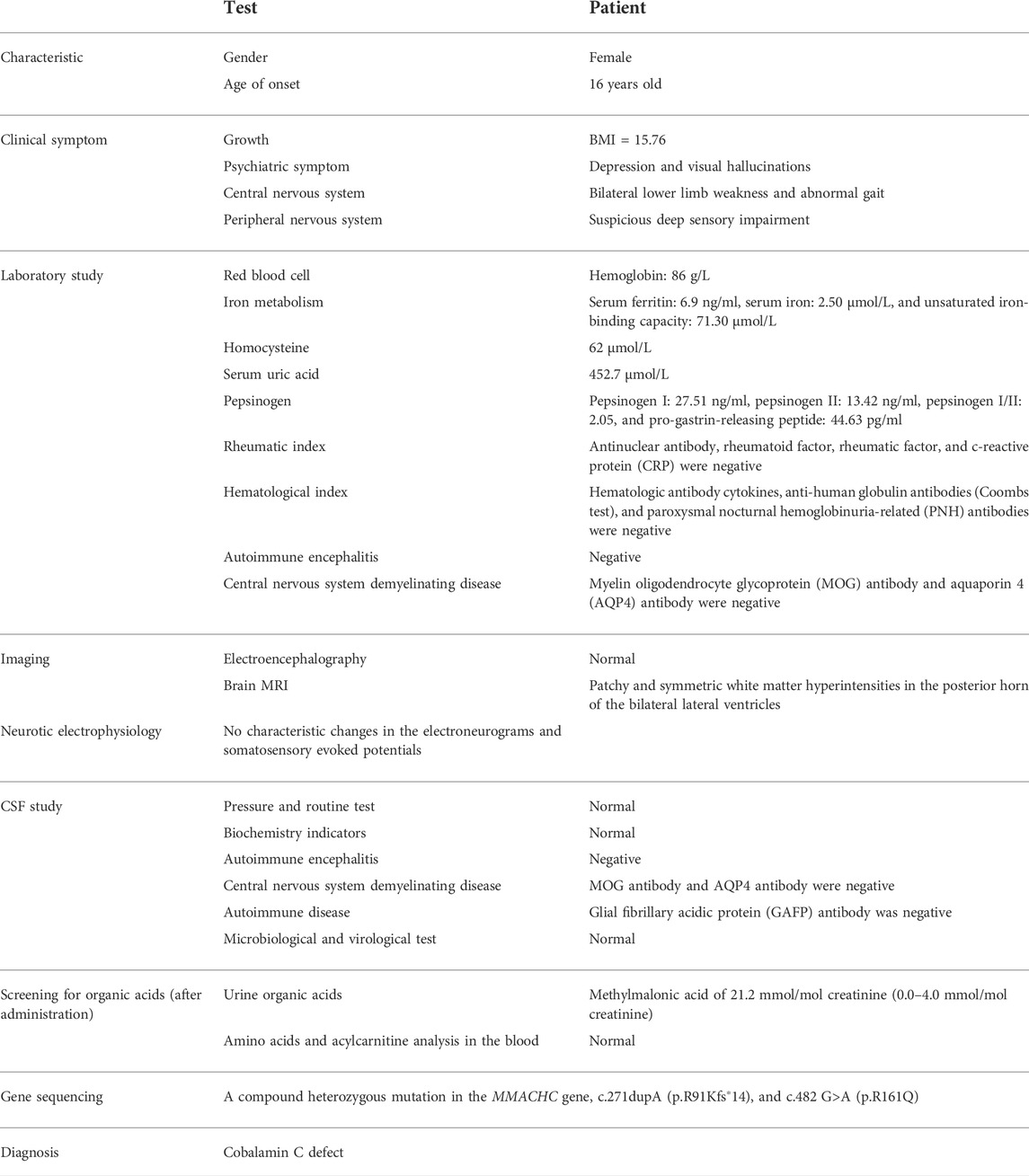
TABLE 1. Clinical profiles of this patient.
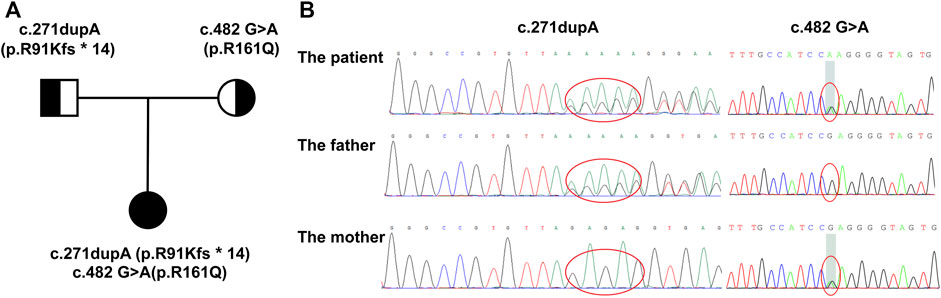
FIGURE 1. Family pedigree displaying the mutations detected in MMACHC. (A) The pedigree of the family with cblC defects. The proband is the daughter, and her parents have no signs of MMA. (B) Mutations detected in the family. The proband has a compound heterozygous mutation, while c.271dupA was inherited from her mother and c.482 G>A was from her father.
From the second onset, the patient got an intramuscular injection of vitamin B1 (50 mg/d) and cyanocobalamin (0.5 mg/d), intravenous injection of Mecbl (1 mg/d), and oral medication of vitamin B6 and folic acid tablets. The patient was not treated with betaine, for betaine is not available in our hospital. After 1 month of treatment, she could stand and walk for a short distance by herself; thus, she was allowed to be discharged from the hospital. The long-term oral administration of vitamin B and intermittent injections were executed after discharge. In the following 2 months, the muscles of both lower limbs gradually strengthened, so that she could walk indoors. The concentrations of MMA and Hcy were lowered.
As expected, the cblC defect is characterized by a heterogeneous clinical picture involving multipart nervous system symptoms, such as psychological and behavioral abnormalities, leukoencephalopathy, encephalopathy, and subacute combined degeneration of the spinal cord, extremity weakness, ataxia, and seizures. Notably, some studies have indicated that psychological and behavioral abnormalities are a presenting sign of late-onset cblC defect, among which schizophrenia and auditory hallucinations were common, rather than depression (Martinelli et al., 2011; Carrillo-Carrasco et al., 2012; Wei et al., 2019).
Our case report demonstrates that depression is also one of the initial manifestations of the late-onset cblC, which usually misleads psychologists or psychiatrists to diagnose it as a plain mood disorder. However, the mechanism of depression in cblC patients remains unclear.
Recently, some studies have pointed out that the underlying pathogenesis of depression might involve neurotransmitters and the related amino acid metabolism disorder (Wang et al., 2021), oxidative stress and inflammation (Behl et al., 2022), mitochondrial dysfunction, and energy metabolism disturbance (Weger et al., 2020). Some biochemical alterations in cblC patients may have harmful effects on the development of depression in multiple ways (Figure 2). First, Hcy elevation and vitamin B12 deficiency of cblC defect can result in depression via inhibiting the S-adenosyl-methionine-dependent synthesis of variable neurotransmitters and their biological activities, such as catecholamines, namely, dopamine, norepinephrine, epinephrine, and noncatecholamines, namely, serotonin, due to impairment in the methylation pathway (Bhatia and Singh, 2015). Additionally, rising Hcy and cysteine sulfinic acid can produce N-methyl-D-aspartate receptor agonists and reactive oxygen species, both of which have neurotoxic effects on dopaminergic neurons, thus inducing depression (Bhatia and Singh, 2015). Second, the dysfunction of mitochondrial energy metabolism and tricarboxylic acid cycle, caused by the elevated blood MMA level, can reduce neuroplasticity and impair hippocampal neurogenesis (Proctor et al., 2020; Forny et al., 2021), which is widely acknowledged to play a vital role in depression. Third, Hcy, together with MMA, also exacerbates oxidative stress and inflammation that are involved in key depressive pathogenic pathways. Thus, we deemed that the elevated Hcy and MMA levels in cblC patients, especially the former one (Moradi et al., 2021), are linked to depression. This connection is consistent with the fact that depression occurs in late-onset methylmalonic aciduria and homocysteinemia patients more frequently than in early-onset methylmalonic aciduria patients. Understanding the association between depression and cblC disease and its influence on the pathophysiologic mechanism of depression will be helpful in the early diagnosis of cblC disease and the etiology-based treatment of uncommon depression.
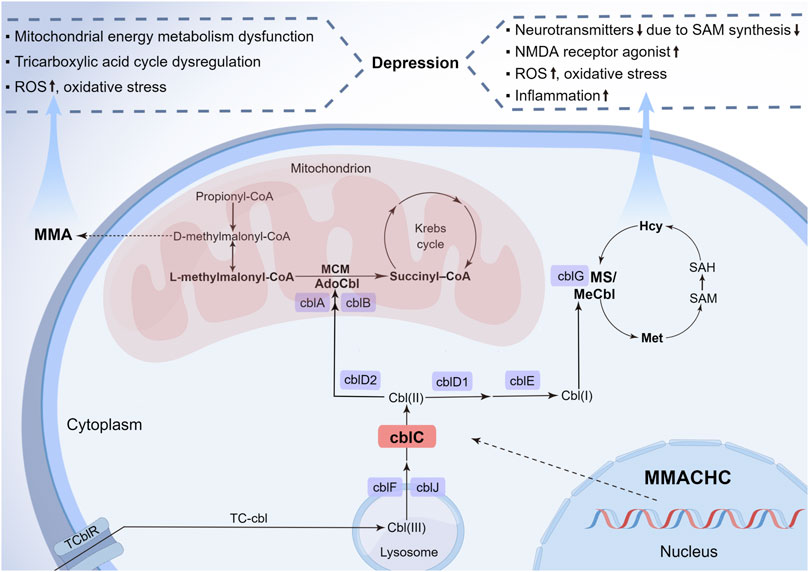
FIGURE 2. Metabolic pathways of cblC defects and its relationship with depression. Nine complementation group defects of the cobalamin pathways and the metabolic interrelationship of MMA and Hcy are summarized. The neurotoxicity of MMA and Hcy due to the cblC defect is described for their importance in the pathophysiology of depression. cbl, cobalamin; cblA-G, cblJ, cbl complementation group diseases; TC-cbl, transcobalamin–cobalamin complex; TC-cblR, transcobalamin receptor; AdoCbl, 5′-deoxyadenosylcobalamin; MCM, methylmalonyl-CoA mutase; MMA, methylmalonic acid; MeCbl, methylcobalamin; Hcy, homocysteine; Ms, methionine synthesis; Met, methionine; SAM, S-adenosyl-methionine; SAH, S-adenosyl-L-homocysteine; MMACHC, gene responsible for methylmalonic acidemia and homocysteinemia; ROS, reactive oxygen species; NMDA, N-methyl-D-aspartate (By Figdraw, ID: UPAOR62009).
Cobalamin, essential for human growth and development, is converted to two active forms: adenosylcobalamin is the coenzyme of mitochondrial methylmalonyl-CoA mutase, converting methylmalonyl-CoA to succinyl-coenzyme A, whose inborn error can cause MMA accumulation due to methylmalonyl-CoA increase; and Mecbl serves as the coenzyme of cytoplasmic methionine synthase, associated with the synthesis of methionine from Hcy, whose congenital disorder can lead to Hcy accumulation and methionine reduction. Details of the metabolic pathways of the cblC defect are shown in Figure 2.
To date, inherited cobalamin deficiencies mainly contain more than 10 types, such as methylmalonyl-CoA deficiency, haptocorrin deficiency, cblA-G, cblJ, cblX, and cblK. Except for cblX, which is X-linked recessive, all the other recognized types are autosomal recessive inheritance. Some types generate a block in the synthesis pathway of both adenosylcobalamin and Mecbl, such as cblC, cblD, cblF, and cblJ, leading to combined methylmalonic aciduria and homocysteinemia (Watkins and Rosenblatt, 2022). Among these types, cblC is the most common inborn disorder. The MMACHC mutation was first identified in 2006 (Lerner-Ellis et al., 2006) and consists of nearly 100 variants recorded in the Human Gene Mutation Database (HGMD) up to now, which are responsible for the cblC defect. Many variants differ in RNA stability or residual function of the protein, which may produce, at least, a certain discrepancy in the phenotype and severity of the disease (Maquat, 2004).
Previous published evidence indicated that the mutation spectrum of the MMACHC gene seemed to vary in different regions across the world. A study by Wang et al. suggested that c.609G>A (p.W203X), c.658_660delAAG (p.K220del), c.80A>G {p.Q27R [r.(spl?)]}, and c.482 G>A (p.R161Q) mutations were the most common mutations in the Chinese population, while the most frequent mutations in the Caucasian population were c.271dupA (p.R91Kfs∗14), c.394C>T (p.R132X), and c.331C>T (p.R111X) (Wang et al., 2019). However, little difference in the genotype–phenotype correlations of the different regions was observed. Present studies conformably believed that c.271dupA (p.R91Kfs∗14), c.609G>A (p.W203X), and c.331C>T (p.R111X) were mainly related to an early-onset and severe presentation, whilst c.482 G>A (p.R161Q) mainly caused the late onset and mild phenotype in any region (Carrillo-Carrasco et al., 2012; Morel et al., 2006; Wang et al., 2019; Yu et al., 2015).
The mutation in our case was a compound heterozygous mutation in the MMACHC gene, that is, c.271dupA (p.R91Kfs∗14) and c.482 G>A (p.R161Q), causing a late onset with mild symptoms, which does not prove the aforementioned correlation. The reason for that may be two points. On one hand, it seems that the compound heterozygotes could result in quite a moderate disease, between the severe early-onset form associated with homozygosity for c.271dupA (p.R91Kfs∗14) and the slight late-onset phenotype related to homozygosity for c.482 G>A (p.R161Q); on the other hand, the late-onset form of the disease usually occurs in patients who possess compound heterozygotes for the c.271dupA (p.R91Kfs∗14) mutation and a missense mutation (Morel et al., 2006). Moreover, the fact that the diverse genetic constitutions in the cblC patients make it difficult to establish the entire connection between genotype and phenotype. Despite this, with the rising awareness of newborn screening and prenatal diagnosis, this genotype–phenotype relationship is still expected to forecast the onset age and disease severity and provide a guide for treatment strategy proposal. Furthermore, case series need to focus on their connection, even on specific clinical manifestations, such as depression, encephalopathy, extremity weakness, gait abnormalities, and seizures, which help to predict the delayed presentation and prognosis.
CblC diagnosis typically depends on either biochemical examination or genetic analysis. Homocysteine in blood or urine, blood amino acids, free carnitine, acylcarnitines, and urine organic acids are vital biochemical assays. Importantly, mass spectrometry and gas chromatography-mass spectrometry are recommended to, respectively, detect blood levels of acylcarnitines, including propionylcarnitine (C3) and acetylcarnitine (C2), and urinary organic acids, including MMA and methylcitric acid. Particularly, using combined mass spectrometry and gas chromatography-mass spectrometry, newborn screening has effective identification ability, boosts early diagnosis, and allows better prognosis (Jin et al., 2021). At the same time, blood vitamin B12, folic acid, and antibody to intrinsic factor need to be detected to exclude the diagnosis of cobalamin disturbance due to secondary etiologies. Finally, genetic sequencing could facilitate the achievement of a definitive diagnosis and detailed genotyping, such as cblC disease based on the MMACHC gene mutation. With the improvement of detection means, cblC can be rapidly diagnosed if the physician is conscious of it. However, in the past reported cases, the time to unequivocal diagnosis needs more than 2 months, even 2 years (Wei et al., 2019). Unfortunately, it took us 3 months to make a clear diagnosis, although some points in this immature patient, for example, abnormal homocysteinemia and multiple neuropsychiatric symptoms, have provided a clue of the inherited metabolic disease for us. The major reason for the prolonged time to diagnosis is misdiagnosis or missed diagnosis during the initial visit. Thereby, physicians need to pay more attention to the understanding of cblC symptoms and heterogeneous clinical spectrum and enhance the awareness of disease screening.
Currently, depression is a very common disorder characterized by paroxysm and easy recurrence, and is closely related to living difficulties, abnormal personality development, and suicide in adolescents, which also, inevitably, increase family and social burdens. Although the possibility of a coincidence cannot be completely excluded in this case, the clinical features of this patient’s depression were not typical, for only one attack occurred and the condition improved quickly. Thence, we considered that depression could be a presenting sign of cblC-type methylmalonic aciduria and homocysteinemia, which also indicated that cblC disease might be a cause of depression. Psychiatrists should extend biochemical or genetic analysis looking for this rare disease to those patients with atypical depression, for example, patients with markedly elevated Hcy levels, anemia, intelligence, or fitness decline, which provide crucial clues for genetic metabolic disease. Moreover, extremely elevated serum Hcy levels, anemia, and intelligence or fitness decline provide crucial clues for genetic metabolic disease. In this case, the acylcarnitine profile was normal, which is, indeed, totally beyond our expectations, as the test was conducted after medication treatment for several weeks. It suggested that we conduct the blood and urinary biochemical examinations as early as possible, for an improper test time could lead to completely confusing results. White matter hyperintensity shown in brain MRI is also one of the common image presentations. Fortunately, in this case, when homocysteinemia was detected, the patient was immediately administered a cyanocobalamin and Mecbl supplement, so that she could have a better prognosis. Thence, once cblC is considered, treatment with parenteral cobalamin and betaine is suggested as soon as possible after the biochemical examination, which helps to correct the metabolic disorder. Most notably, hydroxocobalamin is the only form of cobalamin proven to benefit patients with cblC disease, and cyanocobalamin/Mecbl is not recommended as a regular course of treatment (Carrillo-Carrasco et al., 2012; Baumgartner et al., 2014). The treatment of cyanocobalamin/Mecbl can only be applied to the patient with the presence of a partially functional MMACHC protein in heterozygosity (Anna et al., 2022) Previous studies and our case study revealed that although the late-onset cblC patient could show improved clinical manifestation and long-life span through active treatments, urine MMA and blood Hcy levels of the patients could not be reduced to normal (Matos et al., 2013; Wei et al., 2019), which indicated that MMA metabolic disturbance would last for a relatively long period, and the delayed complication could follow as well.
These complications include encephalopathy, hydrocephalus (Zhang et al., 2019), optic neuropathy and retinopathy (Alowain et al., 2019), pulmonary hypertension, late-onset diffuse lung disease, cardiorespiratory disease (Liu et al., 2017; Zhang et al., 2020), proteinuria, renal thrombotic microangiopathy, and kidney function decline (Chen et al., 2020; Liu et al., 2017). Thereby, the late-onset cblC patients should be treated perpetually with hydroxocobalamin supplement and routine metabolic monitoring. During the follow-up, the physician needs to pay attention to the signs of these complications and conduct relevant imaging examinations if necessary. Except for the regular therapy, a lot of novel gene therapies, including adenovirus gene addition, genome editing, lentiviral gene therapy, and systemic mRNA therapy, would be effective for permanent long-term correction of metabolic disorders caused by methylmalonyl-CoA mutase gene mutation (Chandler and Venditti, 2019; Schneller et al., 2021). However, it should be noted that there have been no gene therapies available for cblC at present, and more attempts can be conducted to seize the appropriate genomic therapies.
Collectively, our case report has expanded the cblC clinical symptom spectrum, indicating that the consideration of depression as one kind of the initial sign of cblC would be helpful in the early diagnosis. Literature review about genotype–phenotype correlation and clinical experience could also remarkably improve physicians’ perspectives and patient prognosis for early diagnosis and treatment.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Hebei General Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Written informed consent was obtained from the individual(s) and minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
SC collected case data and wrote the manuscript. WC was the doctor-in-charge. MZ assisted in writing the manuscript. XX, LZ, and NL supervised and gave advise on the writing of the manuscript. BR was the adviser.
This work was supported by the Clinical Medicine Excellent Talents Government-funded Project of Hebei Province, China (2022-180-361003-21).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alowain, M., Khalifa, O. A., Al Sahlawi, Z., Hussein, M. H., Sulaiman, R. A., Al-Sayed, M., et al. (2019). Optic neuropathy in classical methylmalonic acidemia. Ophthalmic Genet. 40 (4), 313–322. doi:10.1080/13816810.2019.1634740
Anna, J. E., Srijan, M., Ilia, A. D., Sergei, V. M., Donald, W. J., Ute, S., et al. (2022). Versatile enzymology and heterogeneous phenotypes in cobalamin complementation type C disease. Iscience 25 (9), 104981. doi:10.1016/j.isci.2022.104981
Baumgartner, M. R., Horster, F., Dionisi-Vici, C., Haliloglu, G., Karall, D., Chapman, K. A., et al. (2014). Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 9, 130. doi:10.1186/s13023-014-0130-8
Behl, T., Rana, T., Alotaibi, G. H., Shamsuzzaman, M., Naqvi, M., Sehgal, A., et al. (2022). Polyphenols inhibiting MAPK signalling pathway mediated oxidative stress and inflammation in depression. Biomed. Pharmacother. 146, 112545. doi:10.1016/j.biopha.2021.112545
Bhatia, P., and Singh, N. (2015). Homocysteine excess: Delineating the possible mechanism of neurotoxicity and depression. Fundam. Clin. Pharmacol. 29 (6), 522–528. doi:10.1111/fcp.12145
Carrillo-Carrasco, N., Chandler, R. J., and Venditti, C. P. (2012). Combined methylmalonic acidemia and homocystinuria, cblC type I. Clinical presentations, diagnosis and management. J. Inherit. Metab. Dis. 35 (1), 91–102. doi:10.1007/s10545-011-9364-y
Chandler, R. J., and Venditti, C. P. (2019). Gene therapy for methylmalonic acidemia: Past, present, and future. Hum. Gene Ther. 30 (10), 1236–1244. doi:10.1089/hum.2019.113
Chen, R. Y., Li, X. Z., Lin, Q., Zhu, Y., Shen, Y. Y., Xu, Q. Y., et al. (2020). Proteinuria as a presenting sign of combined methylmalonic acidemia and homocysteinemia: Case report. BMC Med. Genet. 21 (1), 183. doi:10.1186/s12881-020-01122-x
Forny, P., Horster, F., Ballhausen, D., Chakrapani, A., Chapman, K. A., Dionisi-Vici, C., et al. (2021). Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: First revision. J. Inherit. Metab. Dis. 44 (3), 566–592. doi:10.1002/jimd.12370
Gravel, R. A., Mahoney, M. J., Ruddle, F. H., and Rosenberg, L. E. (1975). Genetic complementation in heterokaryons of human fibroblasts defective in cobalamin metabolism. Proc. Natl. Acad. Sci. U. S. A. 72 (8), 3181–3185. doi:10.1073/pnas.72.8.3181
Jin, L., Han, X., He, F., and Zhang, C. (2021). Prevalence of methylmalonic acidemia among newborns and the clinical-suspected population: A meta-analyse. J. matern-fetal neo M. 30, 1–16. doi:10.1080/14767058.2021.2008351
Lerner-Ellis, J. P., Anastasio, N., Liu, J., Coelho, D., Suormala, T., Stucki, M., et al. (2009). Spectrum of mutations in MMACHC, allelic expression, and evidence for genotype-phenotype correlations. Hum. Mutat. 30 (7), 1072–1081. doi:10.1002/humu.21001
Lerner-Ellis, J. P., Tirone, J. C., Pawelek, P. D., Dore, C., Atkinson, J. L., Watkins, D., et al. (2006). Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat. Genet. 38 (1), 93–100. doi:10.1038/ng1683
Liu, J., Peng, Y., Zhou, N., Liu, X., Meng, Q., Xu, H., et al. (2017). Combined methylmalonic acidemia and homocysteinemia presenting predominantly with late-onset diffuse lung disease: A case series of four patients. Orphanet J. Rare Dis. 12 (1), 58. doi:10.1186/s13023-017-0610-8
Liu, Y. P., Ma, Y. Y., Wu, T. F., Wang, Q., Li, X. Y., Ding, Y., et al. (2012). Abnormal findings during newborn period of 160 patients with early-onset methylmalonic aciduria. Zhonghua Er Ke Za Zhi 50 (6), 410–414.
Maquat, L. E. (2004). Nonsense-mediated mRNA decay: Splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 5 (2), 89–99. doi:10.1038/nrm1310
Martinelli, D., Deodato, F., and Dionisi-Vici, C. (2011). Cobalamin C defect: Natural history, pathophysiology, and treatment. J. Inherit. Metab. Dis. 34 (1), 127–135. doi:10.1007/s10545-010-9161-z
Matos, I. V., Castejon, E., Meavilla, S., O'Callaghan, M., Garcia-Villoria, J., Lopez-Sala, A., et al. (2013). Clinical and biochemical outcome after hydroxocobalamin dose escalation in a series of patients with cobalamin C deficiency. Mol. Genet. Metab. 109 (4), 360–365. doi:10.1016/j.ymgme.2013.05.007
Moradi, F., Lotfi, K., Armin, M., Clark, C. C. T., Askari, G., and Rouhani, M. H. (2021). The association between serum homocysteine and depression: A systematic review and meta-analysis of o bservational studies. Eur. J. Clin. Invest. 51 (5), e13486. doi:10.1111/eci.13486
Morel, C. F., Lerner-Ellis, J. P., and andRosenblatt, D. S. (2006). Combined methylmalonic aciduria and homocystinuria (cblC): Phenotype-genotype correlations and ethnic-specific observations. Mol. Genet. Metab. 88 (4), 315–321. doi:10.1016/j.ymgme.2006.04.001
Mudd, S. H., Levy, H. L., Abeles, R. H., and Jennedy, J. P. (1969). A derangement in B 12 metabolism leading to homocystinemia, cystathioninemia and methylmalonic aciduria. Biochem. Biophys. Res. Commun. 35 (1), 121–126. doi:10.1016/0006-291x(69)90491-4
Proctor, E. C., Turton, N., Boan, E. J., Bennett, E., Philips, S., Heaton, R. A., et al. (2020). The effect of methylmalonic acid treatment on human neuronal cell coenzyme Q(10) status and mitochondrial function. Int. J. Mol. Sci. 21 (23), E9137. doi:10.3390/ijms21239137
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Schneller, J. L., Lee, C. M., Venturoni, L. E., Chandler, R. J., Li, A., Myung, S., et al. (2021). In vivo genome editing at the albumin locus to treat methylmalonic acidemia. Mol. Ther. Methods Clin. Dev. 23, 619–632. doi:10.1016/j.omtm.2021.11.004
Wang, C., Li, D., Cai, F., Zhang, X., Xu, X., Liu, X., et al. (2019). Mutation spectrum of MMACHC in Chinese pediatric patients with cobalamin C disease: A case series and literature review. Eur. J. Med. Genet. 62 (10), 103713. doi:10.1016/j.ejmg.2019.103713
Wang, H. Q., Wang, Z. Z., and Chen, N. H. (2021). The receptor hypothesis and the pathogenesis of depression: Genetic bases and biological correlates. Pharmacol. Res. 167, 105542. doi:10.1016/j.phrs.2021.105542
Watkins, D., and Rosenblatt, D. S. (2022). Inherited defects of cobalamin metabolism. Vitam. Horm. 119, 355–376. doi:10.1016/bs.vh.2022.01.010
Weger, M., Alpern, D., Cherix, A., Ghosal, S., Grosse, J., Russeil, J., et al. (2020). Mitochondrial gene signature in the prefrontal cortex for differential susceptibility to chronic stress. Sci. Rep. 10 (1), 18308. doi:10.1038/s41598-020-75326-9
Wei, Y., Zhou, Y., Yuan, J., Ni, J., Qian, M., Cui, L., et al. (2019). Treatable cause of hereditary spastic paraplegia: Eight cases of combined homocysteinaemia with methylmalonic aciduria. J. Neurol. 266 (10), 2434–2439. doi:10.1007/s00415-019-09432-8
Yu, Y. F., Li, F., and Ma, H. W. (2015). Relationship of genotypes with clinical phenotypes and outcomes in children with cobalamin C type combined methylmalonic aciduria and homocystinuria. Zhongguo Dang Dai Er Ke Za Zhi 17 (8), 769–774.
Zhang, K., Gao, M., Wang, G., Shi, Y., Li, X., Lv, Y., et al. (2019). Hydrocephalus in cblC type methylmalonic acidemia. Metab. Brain Dis. 34 (2), 451–458. doi:10.1007/s11011-018-0351-y
Zhang, Y. N., Pi, Y. L., Yan, X., Li, Y. Q., Qi, Z. J., and Zhang, H. F. (2020). Methylmalonic acidemia complicated by homocystinuria diseases: A report of three cases. Adv. Ther. 37 (1), 630–636. doi:10.1007/s12325-019-01149-4
Keywords: CblC defect, MMACHC, depression, genotype–phenotype correlation, case report
Citation: Cheng S, Chen W, Zhao M, Xing X, Zhao L, Ren B and Li N (2022) Case report: A late-onset cobalamin C defect first presenting as a depression in a teenager. Front. Genet. 13:1012558. doi: 10.3389/fgene.2022.1012558
Received: 05 August 2022; Accepted: 06 October 2022;
Published: 20 October 2022.
Edited by:
Elsayed Abdelkreem, Sohag University, EgyptReviewed by:
Curtis R. Coughlin II, University of Colorado Anschutz Medical Campus, United StatesCopyright © 2022 Cheng, Chen, Zhao, Xing, Zhao, Ren and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Na Li, MzYxNjkzMTE0QHFxLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.