 Eryk Duński
Eryk Duński Aleksandra Pękowska
Aleksandra Pękowska- Dioscuri Centre for Chromatin Biology and Epigenomics, Nencki Institute of Experimental Biology, Polish Academy of Sciences, Warsaw, Poland
The unique qualities of the human brain are a product of a complex evolutionary process. Evolution, famously described by François Jacob as a “tinkerer,” builds upon existing genetic elements by modifying and repurposing them for new functions. Genetic changes in DNA may lead to the emergence of new genes or cause altered gene expression patterns. Both gene and regulatory element mutations may lead to new functions. Yet, this process may lead to side-effects. An evolutionary trade-off occurs when an otherwise beneficial change, which is important for evolutionary success and is under strong positive selection, concurrently results in a detrimental change in another trait. Pleiotropy occurs when a gene affects multiple traits. Antagonistic pleiotropy is a phenomenon whereby a genetic variant leads to an increase in fitness at one life-stage or in a specific environment, but simultaneously decreases fitness in another respect. Therefore, it is conceivable that the molecular underpinnings of evolution of highly complex traits, including brain size or cognitive ability, under certain conditions could result in deleterious effects, which would increase the susceptibility to psychiatric or neurodevelopmental diseases. Here, we discuss possible trade-offs and antagonistic pleiotropies between evolutionary change in a gene sequence, dosage or activity and the susceptibility of individuals to autism spectrum disorders and schizophrenia. We present current knowledge about genes and alterations in gene regulatory landscapes, which have likely played a role in establishing human-specific traits and have been implicated in those diseases.
1 Introduction
Consciousness, self-awareness, and abstract thinking are the essential features of the human mind. One of the most fascinating questions in neurobiology is what changes in the genome have led to the emergence of unique properties of the human brain. However, the same shifts that have led to the development of the unprecedented capacities of the human brain may also underlie its susceptibility to disease. Neuropsychiatric disorders (NDs), in particular schizophrenia (SCZ) or autism spectrum disorder (ASD), specifically affect self-awareness, cognition, and the ability for social interaction (Geschwind and Rakic, 2013; Lai et al., 2014; Crespi and Ph, 2016; Owen et al., 2016; Benítez-Burraco et al., 2022), which were under positive pressure in human lineage (Kaczanowska et al., 2022). Remarkably, numerous genes that might underlie the unique features of the human brain are often at the same time risk genes of ASD or SCZ (see below). Loci associated with NDs are frequently expressed early in the brain development (O’Brien et al., 2018; Polioudakis et al., 2019; Ball et al., 2020; Satterstrom et al., 2020), the timepoint when the largest divergence of transcriptomic profiles between humans and macaques is also observed (Zhu et al., 2018). Together, these observations point to a possible link between evolutionary changes in brain biology and the emergence of SCZ and ASD.
ASD encompasses a broad group of conditions characterized by defective social communication, repetitive behaviour, and restricted interests or activities (Grzadzinski et al., 2013). Psychosis, which consists of altered perception of what is real and what is not, constitutes the primary feature of SCZ (Owen et al., 2016). It can be accompanied by social cognitive impairment and neurocognitive disfunctions (Stratton et al., 2017; Engelstad et al., 2019). Both SCZ and ASD affect the essential features of the human mind – intelligence and self-awareness (Keefe et al., 2005; Lord et al., 2018). Both diseases feature a strong genetic component. Heritability of SCZ is estimated between 47.3% and 81% (Owen et al., 2016; Chou et al., 2017) while for the ASD it is estimated between 64% and 91% (Tick et al., 2016). Despite a detrimental impact on reproductive fitness (Van Dongen and Boomsma, 2013) both SCZ and ASD are highly frequent in the human population and affect almost 1% (SCZ) (McGrath et al., 2008), and 1%–2% (ASD) of adults worldwide (Baio, 2012; Lai et al., 2014). These observations suggest that genes and gene variants linked to SCZ and ASD may play crucial roles in human brain biology; benefits from their functions may outweigh the negative selective pressure and the loss of fitness due to disease risk. Indeed, while ASD patients frequently display below-average intelligence quotient, they also feature enhanced perception, visuospatial performance, increased attention to detail and more focused concentration (Belmonte et al., 2004; Mottron et al., 2006; Kelleher and Bear, 2008; Muth et al., 2014; Sacco et al., 2015). Altogether, these observations led to a proposal that ASD may in fact, be considered a “disorder of high intelligence” (Crespi, 2016), which shares genetic basis with high cognitive abilities (Hagenaars et al., 2016). Furthermore, there is increasing evidence of overlapping genetic bases of ASD and SCZ (Fromer et al., 2014; Satterstrom et al., 2020), and that some aspects of ASD and SCZ may represent opposite ends of the same gene dosage spectrum. Therefore, it is interesting to consider the common genetic bases of evolution and susceptibility to SCZ and ASD (Crespi and Ph, 2016). Multiple microdeletion syndromes which are associated with both SCZ and ASD (Crespi et al., 2010) contain genes which have undergone an evolutionary change in humans (1q21.1 – NOTCH2NL, NBPF genes (Fiddes et al., 2018, 2019); 15q13.3–GOLGA8 (Hahn et al., 2007; Bekpen and Tautz, 2019), 16p11.2–BOLA2 (Zufferey et al., 2012; Nuttle et al., 2016); 16p13.1–NDE1 (Mosca et al., 2017; Monda and Cheeseman, 2018)) (Table 1). Taken together, the evidence points to the possibility that mechanisms which rendered human brain unique during evolution entail a trade-off of increased neuropsychiatric disease risk. In this mini-review article, we focus on the genetic link between evolution of the human brain and SCZ and ASD.
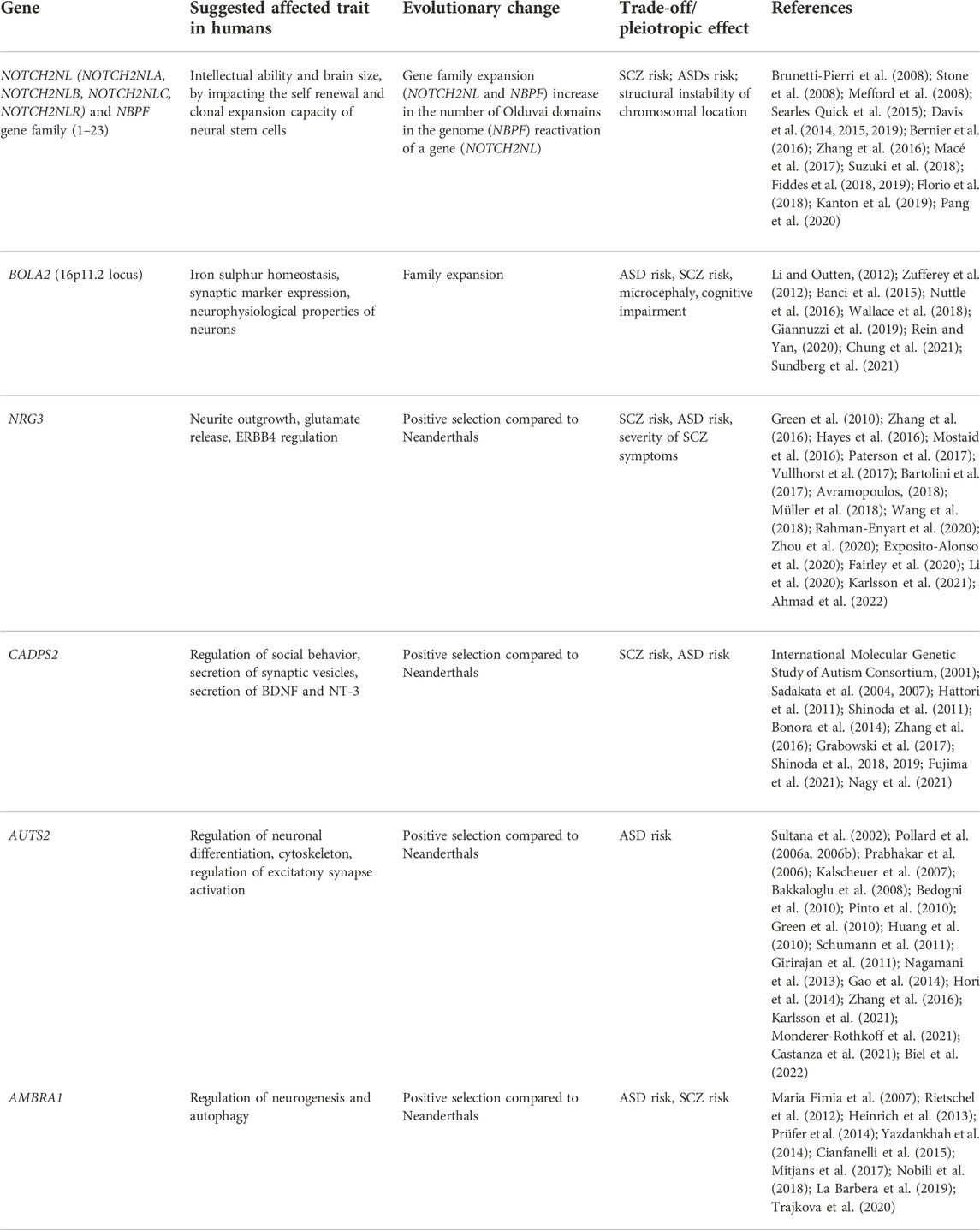
TABLE 1. Summary of genes which are implicated in ASD and SCZ, and bear signs of recent evolution in humans. Observed or proposed evolutionary trade-offs and antagonistic pleiotropies caused by evolution of those genes are listed.
2 Brain evolution: Size and connectivity
Cortical expansion, which denotes a disproportionate gain of the surface area of the cortex as compared to other regions of the brain, marks brain evolution in primates (Geschwind and Rakic, 2013; Fernández et al., 2016; Franchini, 2021). The increase in the absolute number of neurons contributes to the expansion of the cortex during evolution and is associated with restructuring of the network of connections between neuronal cells (“connectome” Figure 1A). Change in the architecture of the connectome is most likely the essential driver of the enhanced computational power of the human brain (van den Heuvel et al., 2016; Sousa et al., 2017; Ardesch et al., 2019).

FIGURE 1. Genetic alterations in loci related to human brain evolution can lead to neuropsychiatric disorders specifically affecting self-awareness and cognition including schizophrenia (SCZ) and autism spectrum disorder (ASD). (A). Human brain is larger and contains more neurons than the brain of our closest living relative the chimpanzee (Pan troglodytes). Evolution led to the enhancement of several features of human brain including cognition and self-awareness. There is also a considerable degree of convergence between genetic changes that underlie brain evolution and loci related to neuropsychiatric disorders. (B). Examples of genetic alterations in the human lineage related at the same time to evolution of SCZ or ASD.
Neurogenesis occurs primarily in the prenatal period of life (Villalba et al., 2021) and is ensured by activity of the neural precursors (NP), including the outer Radial Glial (oRG) cells (Rakic, 2009; Pinson and Huttner, 2021). As compared to their counterparts in the non-human primates (NHP), human oRG cells feature an enhanced capacity to self-renew. Hence, each human oRG cell can, in principle, produce more neurons than its counterpart in developing NHP brain (Florio and Huttner, 2014; Mora-Bermúdez et al., 2016; Pinson and Huttner, 2021; Pinson et al., 2022). Increase in the number of neurons is likely the cornerstone of brain evolution and is intimately linked to the reshaping of the connectome. Human connectome features larger number and diversity of areas in the cortex, and most likely evolved by enhancing the modularity, sparsity and segregation of the network (Changeux et al., 2021). The relationship between cortical expansion and changes in human connectome is, however not fully understood. According to the tethering hypothesis (Buckner and Krienen, 2013), the rapid expansion of the human cortex disrupted the organization of the connectome, and led to the establishment of new networks, which, in turn, enabled the emergence of the enhanced cognitive abilities of the human mind. Further changes in connectome were probably related to an increased intricacy of the cytoarchitecture of neurons as, for instance, in the case of pyramidal neurons (Galakhova et al., 2022). Likewise, the further specialisation and perhaps also the emergence of unique neuronal cell types (Allman et al., 2010; Banovac et al., 2021) and evolutionary changes in non-neuronal cells, including neuroglial cells, also likely contributed to changes in connectome during human brain evolution (Oberheim et al., 2009; Fields et al., 2015; Oberheim Bush and Nedergaard, 2017).
3 Genetic changes that link brain evolution, schizophrenia and autism spectrum disorder
Evolutionary changes in gene activity may emerge from alterations in the sequence of the gene (for review see Espinós et al. (2022)) or of the DNA regulatory elements including promoters, enhancers, silencers or insulators (for reviews see Bae et al. (2015); Silver, (2016); Liu et al. (2021)). As a result, genes which are similarly expressed across species may still differ in function due to divergence in coding sequences (Staes et al., 2017), while genes with conserved coding sequence may vary in expression level between species due to alterations in regulatory program (Prescott et al., 2015; Kanton et al., 2019; Wang et al., 2020).
According to the most recent estimates, 856 genes have undergone changes specific to the human lineage when compared to great apes (Bitar et al., 2019). Genes affected by evolution are primarily related to brain biology (71 of these loci are related to nervous system development and functions), immune system and metabolism. This most up-to-date account, is, however, most likely far from complete. Multiple gene duplications have been uncovered in the most recent human genome assembly (Nurk et al., 2022; Vollger et al., 2022). Remarkably, coding sequences of genes related to brain biology including loci implicated in nervous system development and synaptic transmission are generally conserved between humans and NHP (Dumas et al., 2021). This suggests that brain evolution might be driven primarily by differences in transcriptomes. Comparison of RNA-seq profiles of neuronal cells from the human and NHP stem cell-derived organoids and brain specimens (Kanton et al., 2019; Pollen et al., 2019; Ma et al., 2022), revealed hundreds of genes featuring altered expression in human, as compared to the NHP brain cells; these differences affected both neurons and glial cells including astrocytes, and oligodendrocytes (Kanton et al., 2019; Ma et al., 2022). While we anticipate the discovery of many more loci related to brain biology and evolution (Yamasaki et al., 2020), the already available analyses help identify several remarkable examples of human specific genes which may contribute to, and therefore bridge, human brain evolution, cognitive traits, and NDs (Figure 1).
3.1 Evolutionary hotspot at chromosome 1 region q21.1-2 contains genes linked with brain size, schizophrenia, and autism spectrum disorder
The cytoband q21.1-2 region at chromosome 1 is a site of a significant gene number expansion in the recent human evolution. Likewise, 1q21.1-2 appears polymorphic in the human population. Based on United Kingdom Biobank data, prevalence of copy number variations at this region is estimated at 0.027% for deletions and 0.044% for duplications (Owen et al., 2018). 1q21.1-2 encodes genes from the NOTCH2NL (Fiddes et al., 2018; Florio et al., 2018; Suzuki et al., 2018) and neuroblastoma breakpoint (NBPF) families (O’Bleness et al., 2014), both of which are genetically linked to SCZ and ASDs (Searles Quick et al., 2015; Fiddes et al., 2018; Davis et al., 2019). The 1q21.1-2 is most probably the best described example of a locus harboring genes amplified in the human lineage, that is also linked to changes in brain size and ND risk (see below).
3.1.1 NOTCH2NL gene family
The human specific NOTCH2NL gene family comprises four paralogs: three protein-coding genes (NOTCH2NL A, B, and C) and a pseudogene (NOTCH2NLR) (Suzuki et al., 2018). NOTCH2NL is absent in the macaques, while gorilla and chimpanzee genomes feature only NOTCH2NL pseudogenes (Fiddes et al., 2018; Suzuki et al., 2018), which is most likely a product of an incomplete duplication of an essential developmental regulator NOTCH2 (Fiddes et al., 2018). The current data suggests that, in the human lineage, the non-functional ancestral copy of NOTCH2 was first “re-activated” to form a functional gene and subsequently multiplied producing the NOTCH2NL A, B, and C genes (Fiddes et al., 2018).
Expression of NOTCH2NLB favors activation of the NOTCH pathway by inhibiting cis DELTA-NOTCH interactions (Fiddes et al., 2018). Experimental data in a mouse model and in organoid system show that NOTCH2NL genes may have contributed to cortical expansion by enhancing the proliferative capacity of the RG cells, and by delaying the differentiation of the RG cells to neurons (Fiddes et al., 2018; Florio et al., 2018; Suzuki et al., 2018). In human adult brain, both the ancestral gene NOTCH2 and members of NOTCH2NL family are expressed most strongly in astrocytes (Zhang et al., 2016; Kanton et al., 2019). Search for “NOTCH2NL” at https://brainrnaseq.org/ and https://bioinf.eva.mpg.de/shiny/sample-apps/scApeX/, Accessed September 29, 2022.
Alterations of the in the copy number of NOTCH2NL family genes are linked to congenital syndromes and NDs (Stone et al., 2008; Bernier et al., 2016; Macé et al., 2017). Microdeletions and microduplications of NOTCH2NL relate to micro- and macrocephaly respectively (Brunetti-Pierri et al., 2008; Mefford et al., 2008); likewise, loss or amplification of NOTCH2NL is detected in SCZ and ASD patients respectively (Pang et al., 2020).
Taken together, the emergence of the NOTCH2NL gene family increased the activity of NOTCH signaling pathway in the human neural stem cells which, likely by endowing them with an enhanced capacity to self-renew, contributed to the evolutionary expansion of the brain cortex in humans. Correct gene dosage of NOTCH2NL is essential not only to ensure appropriate brain size but also to allow proper neuronal activity (Chapman et al., 2022). Hence, the evolutionary expansion of NOTCH2NL gene family would result in a trade-off between an advantageous increase in one trait—a gain in brain size, and a detrimental change in another—an increased susceptibility to SCZ and ASD. It will be interesting to determine how NOTCH2NL gene dosage is distributed across a large human population and how the numbers (and isoforms) of NOTCH2NL genes correlate with brain structure and activity in ASD and SCZ.
3.1.2 NBPF gene family
The neuroblastoma breakpoint (NBPF) genes contain the Olduvai domain (OD), which underwent a remarkable expansion during recent human evolution (Popesco et al., 2006). Genome of the last common ancestor of humans and chimpanzees featured a total of 102 copies of ODs, chimpanzee genome contains 125 copies, while human genome contains 272 copies of ODs (O’Bleness et al., 2012). Expansion of such magnitude suggests an important function of genes containing ODs. Indeed, OD-containing proteins are known to contribute to neural progenitor proliferation (Keeney et al., 2015), and the number of OD repeats in the genome is related to severity of both ASD (Davis et al., 2014, 2015, 2019) and SCZ (Searles Quick et al., 2015). Similarly to NOTCH2NL genes, members of the NBFP gene family are expressed in astrocytes in humans, especially in fetal astrocytes (observation based on transcriptomic databases published in Zhang et al., (2016), Kanton et al., (2019)). Yet, the molecular function of ODs remains to be determined (Sikela and Searles Quick, 2018).
1q21.1-2 region harbors 13 genes from the NBPF family (O’Bleness et al., 2012). NBPF genes at the 1q21.1-2 region contain most (119) of the human-specific Olduvai domain repeats (Fiddes et al., 2019). NBPF genes lie proximally to each other at the 1q21.1-2 locus and are most likely transcriptionally co-regulated with NOTCH2NL genes (Fiddes et al., 2019). This genomic configuration seems to be inherently genetically unstable (Fiddes et al., 2018) and highly prone to non-allelic homologous recombination events (Chapman et al., 2022). In fact, the marked chromosomal instability of the 1q21.1-2 locus is likely caused by the presence of amplified NOTCH2NL and NBPF. This genetic configuration in turn, causes alterations in the copy number of other genes lying in q21.1-2 region including HYDIN2 PRKAB2, FMO5, CHD1L, BCL9, ACP6, GJA5, GJA8, GPR89B and PDZK1, which likely drives additional detrimental effects (Brunetti-Pierri et al., 2008; Yoon and Mao, 2021). Therefore, the local expansion of NOTCH2NL and NBPF genes may have exerted a direct effect through altering the dosage of the NOTCH2NL and NBPF genes but also an indirect impact by enhancing the probability of genetic alterations at the locus manifesting as copy number variants at the 1q21.1-2.
3.2 Copy number variants within the human-specific 16p11.2 locus are related to multiple NDs, including schizophrenia and autism spectrum disorder
The 600,000 bp region between breakpoints 4 and 5 (BP4-BP5) at chromosome 16 cytoband p11.2 (16p11.2) carries multiple genes, including BOLA2, SLX1, MAZ, and SULT1A3. A 95 kbp part of the region carrying BOLA2 has undergone a human-specific duplication in the most recent human evolution, from one copy in Neanderthals and Denisovans to three to eight diploid copies in modern humans (Nuttle et al., 2016). BOLA2 appears to be important from the evolutionary standpoint. BOLA2 encodes a cytosolic protein implicated in the maturation of iron-sulfur proteins; it is more expressed in the human embryonic stem cells and induced pluripotent stem (iPS) cells than in their chimpanzee counterparts (Li and Outten, 2012; Banci et al., 2015). Amplification of BOLA2 in the human lineage might protect from iron deficiency (Giannuzzi et al., 2019). A study using iPS cell models with altered copy number of the 16p11.2 region revealed that neurons obtained from iPS cells featuring 16p11.2 deletion are hyperactivated and overexpress synaptic markers compared to the isogenic control cells; neurons with 16p11.2 duplication display largely opposite characteristics (Sundberg et al., 2021).
Genes located in BP4-BP5 region of 16p11.2 are essential for proper brain activity, as evidenced by the fact that CNV in 16p11.2 are among the most frequent causes of neurodevelopmental disorders (Rein and Yan, 2020; Chung et al., 2021). Prevalence of reciprocal deletions of 16p11.2 is estimated at 1/2000, and of and reciprocal duplications at 1/1100 (Chung et al., 2021); triplications are very rare (Wallace et al., 2018).
Dosage of genes located in 16p11.2 has been linked to brain structure. Number of copies of the 16p11.2 region anticorrelates with the gray matter volume and is related to the microstructure of the regions implicated in reward, language and social cognition (Maillard et al., 2015). CNV in 16p11.2 are related to ASD, SCZ, intellectual disability, epilepsy, macrocephaly, depression, anxiety and attention deficit hyperactivity disorder (ADHD) (Rein and Yan, 2020). The nature of the mechanistic implication of the 16p11.2 locus in brain biology is, however, not clear. Despite this knowledge gap, a treatment for ASD symptoms induced by 16p11.2 deletion is currently under development. R-Baclofen has been successfully used to treat symptoms of ASD in mouse model of 16p11.2 deletion, and its clinical trials are in Phase II (Parellada et al., 2021, ClinicalTrials.gov Identifier: NCT03682978). It is likely that by determining how the evolutionary changes in the DNA sequence of 16p11.2 impacted brain biology, we will gain critical new insights into the mechanisms of NDs related to genes encoded at 16p11.2.
3.3 Recently evolved genes related to NDs
Comparison of the genome sequences of modern humans with Neandertals and Denisovans, the two best characterized archaic forms of humans (Green et al., 2010; Prüfer et al., 2014; Peyregne et al., 2017), allowed identification of a number of genes which underwent positive selection in humans. These include loci affected by the selective sweep, a process of genetic hitchhiking accompanied by directional selection (Green et al., 2010). Of those, NRG3, CADPS2, AUTS2, (Green et al., 2010) and AMBRA1 (Prüfer et al., 2014) seem particularly interesting in the context of this review, as they have also been linked to both SCZ and ASD (Figure 1B)
Neuregulin 3 (NRG3) is located in a region which has been under strong selective pressure after divergence between humans and Neanderthals (Green et al., 2010). NRG3 is an epidermal growth factor-like signaling protein acting on receptor tyrosine kinase ERBB4 (Avramopoulos, 2018). NRG3 is expressed in the developing and adult brain, in both glial and neuronal cells (Zhang et al., 2016; Karlsson et al., 2021). The NRG3-mediated signaling is important for brain development (Bartolini et al., 2017; Müller et al., 2018), and the formation of neural circuits (Exposito-Alonso et al., 2020). NRG3 was shown to promote neurite outgrowth (Rahman-Enyart et al., 2020) and control glutamate release (Wang et al., 2018). Interestingly, distinct NRG3 isoforms are expressed in the human brain at different developmental stages (Paterson et al., 2017). Importance of NRG3 for healthy brain functioning has been confirmed in vivo in mice with NRG3 knock-out, which display psychotic syndromes (Hayes et al., 2016). However, the mechanism by which NRG3 might affect behavior is still not fully understood.
There are 11 single nucleotide polymorphisms (SNPs) at the NRG3 locus, of which 5 have been directly linked to risk of SCZ; 3 SNPs seem to associate with the strength of delusion symptoms in SCZ patients, but not risk of developing SCZ (Avramopoulos, 2018). The rs10748842 lies in the region affected by a human-specific selective sweep. T/T variant of this SNP is positively related to the strength of delusions in SCZ; it has also been shown to correlate with elevated expression of particular NRG3 isoform classes (II and III) in patients with mood disorders (Paterson et al., 2017). Furthermore, the genotype at rs10748842 is related to cognitive functions in SCZ patients, individuals with SCZ who carry T/T variant of this SNP display decreased cognitive deficit than patients with the T/C or C/C genotypes (Li et al., 2020; Zhou et al., 2020). According to the 1000 genomes project (www.internationalgenome.org, retrieved on 29-09-22), the T allele is the most prevalent haplotype in the human population (frequency of T = 86%, frequency of C = 14%) (Fairley et al., 2020). Such high frequency suggests that the effect of T allele is otherwise neutral or beneficial, and only exerts its effect in brains of diseased individuals.
Interestingly, SCZ patients also feature disfunctions of ERBB4 which is regulated by both NRG3 and NRG1, which has also been implicated in SCZ (Mostaid et al., 2016). EGF domains of both Nrg1 and Nrg3 promote synapse formation in mouse pyramidal neurons further highlighting the functional convergence of NRG1 and 3 (Exposito-Alonso et al., 2020). Location, and hence the activity, of NRG3 depends in its processing (Vullhorst et al., 2017). In the mouse, the unprocessed isoform accumulates in the cell body, while the processed protein, including proteolytically cleaved portions of it, are transported to other subcellular locations, the N-terminal fragment of NRG3 can for instance be transported to the cell membrane of the neurites, where it can interact with the postsynaptic ErbB4 receptors (Ahmad et al., 2022). Interestingly, the N-terminus of NRG3 is also the site of the selective sweep in modern humans as compared to Neanderthals (Green et al., 2010).
The 7q31–q33 constitutes another example of a region that underwent a positive selection in the recent human genome history. There are several protein-coding loci within the 7q31–q33 region including RNF148, RNF133, and CADPS2; the latter being particularly interesting from the clinical and evolutionary standpoints. In human, deletions as well as amplifications of the genetic interval containing CADPS2 (Ca2+-dependent activator protein for secretion 2) have been associated to ASD (Bonora et al., 2014; Grabowski et al., 2017; Nagy et al., 2021). Interestingly, CADPS2 can be aberrantly spliced in ASD in autistic patients (Sadakata et al., 2007). On the other hand, CADPS2 is transcriptionally upregulated in SCZ patient brains (Hattori et al., 2011).
CADPS2, located in a region previously referred to as autism susceptibility locus 1 (AUTS1) (International Molecular Genetic Study of Autism Consortium, 2001). Based on the transcriptomic data from the human brain, CADPS2 is highly expressed in mature astrocytes and neurons (Zhang et al., 2016). CADPS2 is implicated in the regulation of secretion of synaptic and dense core vesicles (Sadakata et al., 2004). By regulating the release of vesicles, CADPS2 plays a role in the secretion of brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) thereby impacting the development and perhaps the balance between activatory and inhibitory synapses in the central nervous system (Sadakata et al., 2004, 2007; Shinoda et al., 2011). Interestingly, deletion of CADPS2 exerts brain region specific effects in the mouse (Shinoda et al., 2018, 2019) and affects social behavior of the animals leading to ASD-like behavior (Sadakata et al., 2007). Furthermore, the effects of CADPS2 loss might be caused by an altered release of oxytocin in the knockout animals (Fujima et al., 2021). Altogether, gene dosage of CADPS2 might have contributed to the evolutionary changes of brain morphology and activity. The alterations in the level of CADPS2 might have been linked to the establishment of more intricate social structure of the human population. The defects in the proper activity CADPS2 may alter the capacity for inter-individual interaction as observed in ASD patients.
The 5′ end of AUTS2 (autism-susceptibility-gene-2) has been under strong selection after divergence between humans and Neanderthals – this portion of the AUTS2 locus overlaps three evolutionarily accelerated non-coding regions featuring a large number (293) of SNPs differentiating modern from archaic humans (Green et al., 2010). Genetic alterations in the 5′ end of AUTS2 locus are related to numerous neurological disorders including ASD, ADHD, micro- and macrocephaly, intellectual disability, mental retardation (Sultana et al., 2002; Kalscheuer et al., 2007; Bakkaloglu et al., 2008; Huang et al., 2010; Pinto et al., 2010; Girirajan et al., 2011; Nagamani et al., 2013) or alcohol consumption (Schumann et al., 2011). The known SNPs at the 5′ end of AUTS2 are by and large non-coding (Oksenberg et al., 2013) and feature accelerated evolution in humans (Pollard et al., 2006b, 2006a; Prabhakar et al., 2006, see also below). It is important to stress that genetic alterations in the 3′ of AUTS2, with a yet unknown relevance from the evolutionary standpoint, are also related to neurological disorders and seem more penetrant than alterations in the 5’ end of the gene (Biel et al., 2022).
There are multiple animal models, including mouse and the zebrafish, amenable to study the functions of AUST2 and this area of research experiences a rapid development with inclusion of organoid systems (Biel et al., 2022). In the mouse, Auts2 is expressed during brain development and in multiple brain regions. AUTS2 is abundant in the cell nucleus and was shown to localize to gene promoter regions thereby suggesting its role in transcriptional regulation (Bedogni et al., 2010; Gao et al., 2014; Castanza et al., 2021). Furthermore, AUTS2 associates with RNA binding proteins in the developing mouse brain (Castanza et al., 2021). Other studies revealed the implication of AUTS2 in the regulation of cytoskeleton (Hori et al., 2014).
Experiments in mouse cell lines suggest that correct expression of AUTS2 is essential for the regulation of neuronal differentiation (Monderer-Rothkoff et al., 2021) and functions (Hori et al., 2014). Auts2-deficient mice feature hyperactivation of excitatory synapses and display behavioral and cognitive changes which partially mirror symptoms observed in human autistic patients (Hori et al., 2015), providing a strong link between AUTS2 and ASD. Transcriptomic data from acutely isolated human tissue showed strong expression of AUTS2 in neurons as well as in astrocytes (Zhang et al., 2016; Karlsson et al., 2021), in human cerebral organoids, AUTS2 is expressed in excitatory neurons, radial glia and intermediate progenitors (Fair et al., 2022). Heterozygous de novo missense mutation in AUTS2 detected in ASD patients alters the cell cycle dynamics of neural progenitors, which could hint at the mechanism by which this gene may contribute to the regulation of brain size (Fair et al., 2022). In humans, there are two major protein isoforms of AUTS2 (Biel et al., 2022). It will be interesting to determine the molecular mechanism by which the two AUTS2 isoforms regulate gene expression in the human lineage and how they might have contributed to the evolution of the human brain.
The activating molecule in Beclin-1-regulated autophagy (AMBRA1) lies in a region which has undergone a selective sweep between modern and archaic humans (Prüfer et al., 2014). AMBRA1 is an autophagy regulator (Cianfanelli et al., 2015) implicated in the development of the murine nervous system (La Barbera et al., 2019). AMBRA1 deficiency causes neural tube malformations (Maria Fimia et al., 2007). AMBRA1 also contributes to postnatal brain biology – neurospheres derived from Abra+/− mouse adult brain neural progenitors in the subventricular zone showed a decreased proliferation, and hence the number of neural precursor cells indicating a role of AMBRA1 in regulation of adult neurogenesis (Yazdankhah et al., 2014). AMBRA1+/− mice feature increased apoptosis of neuronal precursors in the medial ganglionic eminence and loss of parvalbumin interneurons in the hippocampus, phenomena which correlate with a decreased inhibition/excitation ratio in female animals, most likely underlying the altered social behavior of the heterozygous animals (Nobili et al., 2018; La Barbera et al., 2019).
Based on GWAS studies, AMBRA1 is associated with both ASD (Mitjans et al., 2017) and SCZ (Rietschel et al., 2012). The precise mechanism linking AMBRA1 to SCZ and ASD are not fully defined but most likely entail gene dosage effects as suggested by studies in the mouse model described above. An intronic SNP in AMBRA1 (rs11819869) is related to both SCZ and impulsivity (Heinrich et al., 2013) but the functional consequences of nucleotide changes at this site are not well understood. Multiple lines of evidence suggest that gene dosage of AMBRA1 impacts brain functions in a sexually dimorphic manner. Loss of one copy of AMBRA1 affects primarly females and leads to ASD (Mitjans et al., 2017). SNP rs3802890 related to loss of AMBRA1 expression in female individuals lies in an intron, which suggests it has a DNA regulatory function (Mitjans et al., 2017). Interestingly, AMBRA1 is lost in some of microdeletions classified as cases of rare Potocki-Shaffer Syndrome, which features intellectual disability, further linking this gene to cognitive abilities (Trajkova et al., 2020).
Taken together, there is a group of genes related to ASD and SCZ that feature evolutionary changes in the coding and non-coding portions of their DNA. The sequences that link these loci to evolution are also risk loci for ASD and SCZ. Below, we outline a class of particularly interesting non-coding regulatory element variants that are linked to ASD and SCZ and may contribute to brain evolution.
4 Human accelerated cis-regulatory elements as drivers of NDs
The changes in gene regulation have long been proposed to contribute to the human-specific features of the brain (King and Wilson, 1975; Geschwind and Rakic, 2013; Reilly et al., 2015). Since genes related to brain are more conserved than other loci (Dumas et al., 2021), brain evolution might be primarily driven by transcriptome differences. Human-specific changes in regulome have been linked to corticogenesis (Reilly et al., 2015). Furthermore, regions that have undergone a selective sweep in humans after divergence from Neanderthals are enriched in enhancers (Peyregne et al., 2017). Interestingly, active chromatin regions in humans, especially those elements which are active earlier in fetal development, have been linked to cognitive ability (Yao et al., 2022).
Human-accelerated regions (HAR) are genomic segments which have undergone accelerated nucleotide sequence change in humans relative to other primates. The most recent analyses suggest that there are 3,171 HARs in the human genome (Pollard et al., 2006a, 2006b; Prabhakar et al., 2006; Bird et al., 2007; Bush and Lahn, 2008; Lindblad-Toh et al., 2011; Hubisz and Pollard, 2014; Gittelman et al., 2015) HARs overlap regulatory elements important for brain development and are particularly related to elements active in the fetal brain (Won et al., 2019; Bhattacharyya et al., 2021, 2022; Cheung et al., 2022). Recently, massively parallel assay for regulatory element activity showed that 49% of HARs feature neurodevelopmental enhancer activity in human neural cells in vitro (Girskis et al., 2021).
Chromatin conformation capture experiments help to link putative enhancers with their cognate promoters and can help to predict the functions of enhancers. HARs are not only preferentially located near genes which are dosage-sensitive, sequence-constrained, and associated with ASD and SCZ (Xu et al., 2015; Doan et al., 2016), but also physically interact with promoters of genes implicated in brain development (Won et al., 2019). Prominent examples of loci likely regulated by HARs and implicated in ASD and SCZ include AUTS2, CDKL5, FOXP1, MEF2C, NRXN1, and SMARCA2 (Doan et al., 2016).
A more detailed analysis of the evolutionarily changed HARs can improve our understanding of the interplay between evolution and NDs. Three intronic regions between exons 1 and 4 at the AUTS2 locus: HAR31, HACNS174, and HACNS369 have been found to be significantly accelerated in humans when compared to primates (Pollard et al., 2006a, 2006b; Prabhakar et al., 2006). HACNS369 is of particular interest as it lies within a region that features signs of human-Neandertal sweep and at the same time alteration in ASD patients (Oksenberg et al., 2013). Transgene reporter assays indicate that HACNS369 can act as an enhancer in the developing midbrain of zebrafish (Oksenberg et al., 2013). Altogether, this data suggest that human specific regulatory element at the AUTS2 locus may contribute to brain evolution and susceptibility to ASD.
Some studies highlight that HARs seem to relate more prominently to genes previously implicated in SCZ than with other NDs such as ASD (Cheung et al., 2022). However, the meaning and relevance of this observation seems unclear. While genome-wide studies discussed above present interesting observations on a link between evolution of human regulome and SCZ and ADSs, it is also important to stress that the species-specific, recently evolved enhancers are variable between individuals and tend to have minor impact on gene expression (Castelijns et al., 2020). Altogether these observations reinforce the need for extensive functional validation of the human specific and disease associated regulatory variants in the human genome.
5 Summary—Perspectives and future avenues for research
One of the essential questions in neurobiology is how the genome changed to allow the increased intellectual abilities of humans compared to NHP. The discovery of the human-specific regulome of the nervous system is likely to provide important piece of information to solve this question (Yao et al., 2022). Addressing it will also likely help us understand the genetic bases of diseases that affect human specific features of mind. Above, we outlined several genes that are related to human brain evolution and which at the same time are liked to ASD and SCZ. These loci affect neurogenesis (NOTCHNLA2, AUTS2, AMBRA1), brain connectivity and synaptic transmission (NRG3, CADPS2), it will be fascinating to decipher the transcriptional regulatory networks that orchestrate the expression of these genes in various neural cells and understand the contribution of these loci to the development and the functions of the central nervous system in humans.
There are multiple newly discovered coding and non-coding regions implicated in SCZ, ASD and other ND that await further analysis from the evolutionary and functional standpoints (Demontis et al., 2019; Grove et al., 2019; Howard et al., 2019; Mullins et al., 2021; Wightman et al., 2021; Trubetskoy et al., 2022). However, to achieve an in-depth understanding of the contribution of non-coding sequences to NDs (and any other diseases for that matter) it will be essential to improve assemblies and annotations of the NHP genomes (Nurk et al., 2022; Vollger et al., 2022). Likewise, it will be beneficial to include a larger cohort of human and NHP genomes in the analysis (Castelijns et al., 2020), and improve protocols of in-vitro and in-vivo systems to assess the contribution of genetic variants in distinct cell types present in the human brain.
Author contributions
ED and AP—conceptualization, writing of the manuscript and manuscript revision.
Funding
This work was supported by Dioscuri Grant, (Dioscuri is a program initiated by the Max Planck Society, managed jointly with the National Science Centre (NCN, Poland) and mutually funded by the Polish Ministry of Education and Science and the German Federal Ministry of Education and Research), by the OPUS17 (UMO-2019/33/B/NZ2/02437) and the Sonata Bis (UMO-2021/42/E/NZ2/00392) grants from the NCN, and by the EMBO Installation Grant.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmad, T., Vullhorst, D., Chaudhuri, R., Guardia, C. M., Chaudhary, N., Karavanova, I., et al. (2022). Transcytosis and trans-synaptic retention by postsynaptic ErbB4 underlie axonal accumulation of NRG3. J. Cell. Biol. 221, e202110167. doi:10.1083/jcb.202110167
Allman, J. M., Tetreault, N. A., Hakeem, A. Y., Manaye, K. F., Semendeferi, K., Erwin, J. M., et al. (2010). The von Economo neurons in frontoinsular and anterior cingulate cortex in great apes and humans. Brain Struct. Funct. 214, 495–517. doi:10.1007/s00429-010-0254-0
Ardesch, D. J., Scholtens, L. H., Li, L., Preuss, T. M., Rilling, J. K., and van den Heuvel, M. P. (2019). Evolutionary expansion of connectivity between multimodal association areas in the human brain compared with chimpanzees. Proc. Natl. Acad. Sci. U. S. A. 116, 7101–7106. doi:10.1073/pnas.1818512116
Avramopoulos, D. (2018). Neuregulin 3 and its roles in schizophrenia risk and presentation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 177, 257–266. doi:10.1002/ajmg.b.32552
Bae, B., Jayaraman, D., and Walsh, C. A. (2015). Genetic changes shaping the human brain. Dev. Cell. 32, 423–434. doi:10.1016/j.devcel.2015.01.035
Baio, J. (2012). Prevalence of autism spectrum disorders-autism and developmental disabilities monitoring network. Centers Dis. Contreol Prev. MMWR Surveill. Summ. 61, 1–19.
Bakkaloglu, B., O’Roak, B. J., Louvi, A., Gupta, A. R., Abelson, J. F., Morgan, T. M., et al. (2008). Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 82, 165–173. doi:10.1016/j.ajhg.2007.09.017
Ball, G., Seidlitz, J., Beare, R., and Seal, M. L. (2020). Cortical remodelling in childhood is associated with genes enriched for neurodevelopmental disorders. Neuroimage 215, 116803. doi:10.1016/j.neuroimage.2020.116803
Banci, L., Camponeschi, F., Ciofi-Baffoni, S., and Muzzioli, R. (2015). Elucidating the molecular function of human BOLA2 in GRX3-dependent anamorsin maturation pathway. J. Am. Chem. Soc. 137, 16133–16143. doi:10.1021/jacs.5b10592
Banovac, I., Sedmak, D., Judaš, M., and Petanjek, Z. (2021). Von Economo neurons – primate-specific or commonplace in the mammalian brain? Front. Neural Circuits 15, 714611. doi:10.3389/fncir.2021.714611
Bartolini, G., Sánchez-Alcañiz, J. A., Osório, C., Valiente, M., García-Frigola, C., and Marín, O. (2017). Neuregulin 3 mediates cortical plate invasion and laminar allocation of GABAergic interneurons. Cell. Rep. 18, 1157–1170. doi:10.1016/j.celrep.2016.12.089
Bedogni, F., Hodge, R. D., Nelson, B. R., Frederick, E. A., Shiba, N., Daza, R. A., et al. (2010). Autism susceptibility candidate 2 (Auts2) encodes a nuclear protein expressed in developing brain regions implicated in autism neuropathology. Gene Expr. Patterns 10, 9–15. doi:10.1016/j.gep.2009.11.005
Bekpen, C., and Tautz, D. (2019). Human core duplicon gene families: Game changers or game players? Brief. Funct. Genomics 18, 402–411. doi:10.1093/bfgp/elz016
Belmonte, M. K., Allen, G., Beckel-Mitchener, A., Boulanger, L. M., Carper, R. A., and Webb, S. J. (2004). Autism and abnormal development of brain connectivity. J. Neurosci. 24, 9228–9231. doi:10.1523/JNEUROSCI.3340-04.2004
Benítez-Burraco, A., Ines, B., Francesco, F., and Progovac, L. (2022). An evolutionary account of impairment of self in cognitive disorders. Cogn. Process. [Epub ahead of print]. doi:10.1007/s10339-022-01110-4
Bernier, R., Steinman, K. J., Reilly, B., Wallace, A. S., Sherr, E. H., Pojman, N., et al. (2016). Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet. Med. 18, 341–349. doi:10.1038/gim.2015.78
Bhattacharyya, U., Bhatia, T., Deshpande, S. N., and Thelma, B. K. (2022). Genetic variations in evolutionary accelerated regions disrupt cognition in schizophrenia. Psychiatry Res. 314, 114586. doi:10.1016/j.psychres.2022.114586
Bhattacharyya, U., Deshpande, S. N., Bhatia, T., and Thelma, B. K. (2021). Revisiting schizophrenia from an evolutionary perspective: An association study of recent evolutionary markers and schizophrenia. Schizophr. Bull. 47, 827–836. doi:10.1093/schbul/sbaa179
Biel, A., Castanza, A. S., Rutherford, R., Fair, S. R., Chifamba, L., Wester, J. C., et al. (2022). AUTS2 syndrome: Molecular mechanisms and model systems. Front. Mol. Neurosci. 15, 858582. doi:10.3389/fnmol.2022.858582
Bird, C. P., Stranger, B. E., Liu, M., Thomas, D. J., Ingle, C. E., Beazley, C., et al. (2007). Fast-evolving noncoding sequences in the human genome. Genome Biol. 8, 1188–R212. doi:10.1186/gb-2007-8-6-r118
Bitar, M., Kuiper, S., O’Brien, E. A., and Barry, G. (2019). Genes with human-specific features are primarily involved with brain, immune and metabolic evolution. BMC Bioinforma. 20, 406–412. doi:10.1186/s12859-019-2886-2
Bonora, E., Graziano, C., Minopoli, F., Bacchelli, E., Magini, P., Diquigiovanni, C., et al. (2014). Maternally inherited genetic variants of CADPS 2 are present in Autism Spectrum Disorders and Intellectual Disability patients. EMBO Mol. Med. 6, 795–809. doi:10.1002/emmm.201303235
Brunetti-Pierri, N., Berg, J. S., Scaglia, F., Belmont, J., Bacino, C. A., Sahoo, T., et al. (2008). Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 40, 1466–1471. doi:10.1038/ng.279
Buckner, R. L., and Krienen, F. M. (2013). The evolution of distributed association networks in the human brain. Trends Cogn. Sci. 17, 648–665. doi:10.1016/j.tics.2013.09.017
Bush, E. C., and Lahn, B. T. (2008). A genome-wide screen for noncoding elements important in primate evolution. BMC Evol. Biol. 8, 17–10. doi:10.1186/1471-2148-8-17
Castanza, A. S., Ramirez, S., Tripathi, P. P., Daza, R. A. M., Kalume, F. K., Ramirez, J. M., et al. (2021). AUTS2 regulates RNA metabolism and dentate gyrus development in mice. Cereb. Cortex 31, 4808–4824. doi:10.1093/cercor/bhab124
Castelijns, B., Baak, M. L., Geeven, G., Vermunt, M. W., Wiggers, C. R. M., Timpanaro, I. S., et al. (2020). Recently evolved enhancers emerge with high interindividual variability and less frequently associate with disease. Cell. Rep. 31, 107799. doi:10.1016/j.celrep.2020.107799
Changeux, J. P., Goulas, A., and Hilgetag, C. C. (2021). A connectomic hypothesis for the hominization of the brain. Cereb. Cortex 31, 2425–2449. doi:10.1093/cercor/bhaa365
Chapman, G., Alsaqati, M., Lunn, S., Singh, T., Linden, S. C., Linden, D. E. J., et al. (2022). Using induced pluripotent stem cells to investigate human neuronal phenotypes in 1q21.1 deletion and duplication syndrome. Mol. Psychiatry 27, 819–830. doi:10.1038/s41380-021-01182-2
Cheung, J. P., Tubbs, J. D., and Sham, P. C. (2022). Extended gene set analysis of human neuro-psychiatric traits shows enrichment in brain-expressed human accelerated regions across development. Schizophr. Res. 246, 148–155. doi:10.1016/j.schres.2022.06.023
Chou, I. J., Kuo, C. F., Huang, Y. S., Grainge, M. J., Valdes, A. M., See, L. C., et al. (2017). Familial aggregation and heritability of schizophrenia and co-aggregation of psychiatric illnesses in affected families. Schizophr. Bull. 43, 1070–1078. doi:10.1093/schbul/sbw159
Chung, W. K., Roberts, T. P., Sherr, E. H., Snyder, L. A. G., and Spiro, J. E. (2021). 16P11.2 deletion syndrome. Curr. Opin. Genet. Dev. 68, 49–56. doi:10.1016/j.gde.2021.01.011
Cianfanelli, V., De Zio, D., Di Bartolomeo, S., Nazio, F., Strappazzon, F., and Cecconi, F. (2015). Ambra1 at a glance. J. Cell. Sci. 128, 2003–2008. doi:10.1242/jcs.168153
Crespi, B. J. (2016). Autism as a disorder of high intelligence. Front. Neurosci. 10, 300–317. doi:10.3389/fnins.2016.00300
Crespi, B., Stead, P., and Elliot, M. (2010). Evolution in health and medicine Sackler colloquium: Comparative genomics of autism and schizophrenia Proc. Natl. Acad. Sci. U. S. A. 107, 1736–1741. doi:10.1073/pnas.0906080106
Crespi, P. B. J., and Ph, D. (2016). Evolutionary thinking in medicine. Editors A. Alvergne, C. Jenkinson, and C. Faurie (Cham: Springer International Publishing). doi:10.1007/978-3-319-29716-3
Davis, J. M., Heft, I., Scherer, S. W., and Sikela, J. M. (2019). A third linear association between Olduvai (DUF1220) copy number and severity of the classic symptoms of inherited autism. Am. J. Psychiatry 176, 643–650. doi:10.1176/appi.ajp.2018.18080993
Davis, J. M., Searles Quick, V. B., and Sikela, J. M. (2015). Replicated linear association between DUF1220 copy number and severity of social impairment in autism. Hum. Genet. 134, 569–575. doi:10.1007/s00439-015-1537-6
Davis, J. M., Searles, V. B., Anderson, N., Keeney, J., Dumas, L., and Sikela, J. M. (2014). DUF1220 dosage is linearly associated with increasing severity of the three primary symptoms of autism. PLoS Genet. 10, e1004241–e1004245. doi:10.1371/journal.pgen.1004241
Demontis, D., Walters, R. K., Martin, J., Mattheisen, M., Als, T. D., Agerbo, E., et al. (2019). Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 51, 63–75. doi:10.1038/s41588-018-0269-7
Doan, R. N., Bae, B. Il, Cubelos, B., Chang, C., Hossain, A. A., Al-Saad, S., et al. (2016). Mutations in human accelerated regions disrupt cognition and social behavior. Cell. 167, 341–354. e12. doi:10.1016/j.cell.2016.08.071
Dumas, G., Malesys, S., and Bourgeron, T. (2021). Systematic detection of brain protein-coding genes under positive selection during primate evolution and their roles in cognition. Genome Res. 31, 484–496. doi:10.1101/GR.262113.120
Engelstad, K. N., Rund, B. R., Torgalsbøen, A. K., Lau, B., Ueland, T., and Vaskinn, A. (2019). Large social cognitive impairments characterize homicide offenders with schizophrenia. Psychiatry Res. 272, 209–215. doi:10.1016/j.psychres.2018.12.087
Espinós, A., Fernández-Ortuño, E., Negri, E., and Borrell, V. (2022). Evolution of genetic mechanisms regulating cortical neurogenesis. Dev. Neurobiol. 82, 428–453. doi:10.1002/dneu.22891
Exposito-Alonso, D., Osório, C., Bernard, C., Pascual-García, S., del Pino, I., Marín, O., et al. (2020). Subcellular sorting of neuregulins controls the assembly of excitatory-inhibitory cortical circuits. Elife 9, e57000–e57028. doi:10.7554/eLife.57000
Fair, S. R., Schwind, W., Julian, D., Biel, A., Guo, G., Rutherford, R., et al. (2022). Cerebral organoids containing an AUTS2 missense variant model microcephaly. Brain, awac244. doi:10.1093/brain/awac244
Fairley, S., Lowy-Gallego, E., Perry, E., and Flicek, P. (2020). The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 48, D941–D947. doi:10.1093/nar/gkz836
Fernández, V., Llinares-Benadero, C., and Borrell, V. (2016). Cerebral cortex expansion and folding: What have we learned? EMBO J. 35, 1021–1044. doi:10.15252/embj.201593701
Fiddes, I. T., Lodewijk, G. A., Mooring, M., Bosworth, C. M., Ewing, A. D., Mantalas, G. L., et al. (2018). Human-specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell. 173, 1356–1369. e22. doi:10.1016/j.cell.2018.03.051
Fiddes, I. T., Pollen, A. A., Davis, J. M., and Sikela, J. M. (2019). Paired involvement of human-specific Olduvai domains and NOTCH2NL genes in human brain evolution. Hum. Genet. 138, 715–721. doi:10.1007/s00439-019-02018-4
Fields, R. D., Woo, D. H., and Basser, P. J. (2015). Glial regulation of the neuronal connectome through local and long-distant communication. Neuron 86, 374–386. doi:10.1016/j.neuron.2015.01.014
Florio, M., Heide, M., Pinson, A., Brandl, H., Albert, M., Winkler, S., et al. (2018). Evolution and cell-type specificity of human-specific genes preferentially expressed in progenitors of fetal neocortex. Elife 7, e32332–e32337. doi:10.7554/eLife.32332
Florio, M., and Huttner, W. B. (2014). Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141, 2182–2194. doi:10.1242/dev.090571
Franchini, L. F. (2021). Genetic mechanisms underlying cortical evolution in mammals. Front. Cell. Dev. Biol. 9, 591017–591027. doi:10.3389/fcell.2021.591017
Fromer, M., Pocklington, A. J., Kavanagh, D. H., Williams, H. J., Dwyer, S., Gormley, P., et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. doi:10.1038/nature12929
Fujima, S., Yamaga, R., Minami, H., Mizuno, S., Shinoda, Y., Sadakata, T., et al. (2021). CAPS2 deficiency impairs the release of the social peptide oxytocin, as well as oxytocin-associated social behavior. J. Neurosci. 41, 4524–4535. doi:10.1523/JNEUROSCI.3240-20.2021
Galakhova, A. A., Hunt, S., Wilbers, R., Heyer, D. B., Kock, C. P. J. De, Mansvelder, H. D., et al. (2022). Evolution of cortical neurons supporting human cognition. Trends Cogn. Sci. xx, 909–922. doi:10.1016/j.tics.2022.08.012
Gao, Z., Lee, P., Stafford, J. M., Schimmelmann, M., Von, Schaefer, A., and Reinberg, D. (2014). An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 516, 349–354. doi:10.1038/nature13921
Geschwind, D. H., and Rakic, P. (2013). Cortical evolution: Judge the brain by its cover. Neuron 80, 633–647. doi:10.1016/j.neuron.2013.10.045
Giannuzzi, G., Schmidt, P. J., Porcu, E., Willemin, G., Munson, K. M., Nuttle, X., et al. (2019). The human-specific BOLA2 duplication modifies iron homeostasis and anemia predisposition in chromosome 16p11.2 autism individuals. Am. J. Hum. Genet. 105, 947–958. doi:10.1016/j.ajhg.2019.09.023
Girirajan, S., Brkanac, Z., Coe, B. P., Baker, C., Vives, L., Vu, T. H., et al. (2011). Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 7, e1002334. doi:10.1371/journal.pgen.1002334
Girskis, K. M., Stergachis, A. B., DeGennaro, E. M., Doan, R. N., Qian, X., Johnson, M. B., et al. (2021). Rewiring of human neurodevelopmental gene regulatory programs by human accelerated regions. Neuron 109, 3239–3251.e7. e7. doi:10.1016/j.neuron.2021.08.005
Gittelman, R. M., Hun, E., Ay, F., Madeoy, J., Pennacchio, L., Noble, W. S., et al. (2015). Comprehensive identification and analysis of human accelerated regulatory DNA. Genome Res. 25, 1245–1255. doi:10.1101/gr.192591.115
Grabowski, P. A. P., Bello, A. F., Rodrigues, D. L., Forbeci, M. J., Motter, V., and Raskin, S. (2017). Deletion involving the 7q31-32 band at the CADPS2 gene locus in a patient with autism spectrum disorder and recurrent psychotic syndrome triggered by stress. Case Rep. Psychiatry 2017, 4254152–4254154. doi:10.1155/2017/4254152
Green, R. E., Krause, J., Briggs, A. W., Maricic, T., Stenzel, U., Kircher, M., et al. (2010). A draft sequence of the neandertal genome. Sci. (80-. ) 328, 710–722. doi:10.1126/science.1188021
Grove, J., Ripke, S., Als, T. D., Mattheisen, M., Walters, R. K., Won, H., et al. (2019). Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444. doi:10.1038/s41588-019-0344-8
Grzadzinski, R., Huerta, M., Lord, C., Wagner, A. P., Baron-Cohen, S., and Ring, H. (2013). Task-related functional connectivity in autism spectrum conditions: An EEG study using wavelet transform coherence. Mol. Autism 4, 1. doi:10.1186/2040-2392-4-1
Hagenaars, S. P., Harris, S. E., Davies, G., Hill, W. D., Liewald, D. C. M., Ritchie, S. J., et al. (2016). Shared genetic aetiology between cognitive functions and physical and mental health in UK Biobank (N=112 151) and 24 GWAS consortia. Mol. Psychiatry 21, 1624–1632. doi:10.1038/mp.2015.225
Hahn, M. W., Demuth, J. P., and Han, S. G. (2007). Accelerated rate of gene gain and loss in primates. Genetics 177, 1941–1949. doi:10.1534/genetics.107.080077
Hattori, K., Tanaka, H., Wakabayashi, C., Yamamoto, N., Uchiyama, H., Teraishi, T., et al. (2011). Expression of Ca²⁺-dependent activator protein for secretion 2 is increased in the brains of schizophrenic patients Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 1738–1743. doi:10.1016/j.pnpbp.2011.05.004
Hayes, L. N., Shevelkin, A., Zeledon, M., Steel, G., Chen, P.-L., Obie, C., et al. (2016). Neuregulin 3 knockout mice exhibit behaviors consistent with psychotic disorders. Mol. Neuropsychiatry 2, 79–87. doi:10.1159/000445836
Heinrich, A., Nees, F., Lourdusamy, A., Tzschoppe, J., Meier, S., Vollstädt-Klein, S., et al. (2013). From gene to brain to behavior: Schizophrenia-associated variation in AMBRA1 alters impulsivity-related traits. Eur. J. Neurosci. 38, 2941–2945. doi:10.1111/ejn.12201
Hori, K., Nagai, T., Shan, W., Sakamoto, A., Abe, M., Yamazaki, M., et al. (2015). Heterozygous disruption of autism susceptibility candidate 2 causes impaired emotional control and cognitive memory. PLoS One 10, 01459799–e146013. doi:10.1371/journal.pone.0145979
Hori, K., Nagai, T., Shan, W., Sakamoto, A., Taya, S., Hashimoto, R., et al. (2014). Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell. Rep. 9, 2166–2179. doi:10.1016/j.celrep.2014.11.045
Howard, D. M., Adams, M. J., Clarke, T. K., Hafferty, J. D., Gibson, J., Shirali, M., et al. (2019). Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352. doi:10.1038/s41593-018-0326-7
Huang, X. L., Zou, Y. S., Maher, T. A., Newton, S., and Milunsky, J. M. (2010). A de novo balanced translocation breakpoint truncating the autism susceptibility candidate 2 (AUTS2) gene in a patient with autism. Am. J. Med. Genet. A 152, 2112–2114. doi:10.1002/ajmg.a.33497
Hubisz, M. J., and Pollard, K. S. (2014). Exploring the Genesis and functions of Human Accelerated Regions sheds light on their role in human evolution. Curr. Opin. Genet. Dev. 29, 15–21. doi:10.1016/j.gde.2014.07.005
International Molecular Genetic Study of Autism Consortium (2001). Further characterization of the autism susceptibility locus AUTS1 on chromosome 7q. Hum. Mol. Genet. 10, 973–982. doi:10.1093/hmg/10.9.973
Kaczanowska, J., Ganglberger, F., Chernomor, O., Kargl, D., Galik, B., Hess, A., et al. (2022). Molecular archaeology of human cognitive traits. Cell. Rep. 40, 111287. doi:10.1016/j.celrep.2022.111287
Kalscheuer, V. M., FitzPatrick, D., Tommerup, N., Bugge, M., Niebuhr, E., Neumann, L. M., et al. (2007). Mutations in autism susceptibility candidate 2 (AUTS2) in patients with mental retardation. Hum. Genet. 121, 501–509. doi:10.1007/s00439-006-0284-0
Kanton, S., Boyle, M. J., He, Z., Santel, M., Weigert, A., Sanchís-Calleja, F., et al. (2019). Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422. doi:10.1038/s41586-019-1654-9
Karlsson, M., Zhang, C., Méar, L., Zhong, W., Digre, A., Katona, B., et al. (2021). A single–cell type transcriptomics map of human tissues. Sci. Adv. 7, eabh2169–9. doi:10.1126/sciadv.abh2169
Keefe, R. S. E., Eesley, C. E., and Poe, M. P. (2005). Defining a cognitive function decrement in schizophrenia. Biol. Psychiatry 57, 688–691. doi:10.1016/j.biopsych.2005.01.003
Keeney, J. G., Davis, J. M., Siegenthaler, J., Post, M. D., Nielsen, B. S., Hopkins, W. D., et al. (2015). DUF1220 protein domains drive proliferation in human neural stem cells and are associated with increased cortical volume in anthropoid primates. Brain Struct. Funct. 220, 3053–3060. doi:10.1007/s00429-014-0814-9
Kelleher, R. J., and Bear, M. F. (2008). The autistic neuron: Troubled translation? Cell. 135, 401–406. doi:10.1016/j.cell.2008.10.017
King, M.-C., and Wilson, A. C. (1975). Evolution at two levels in humans and chimpanzees. Science 188, 107–116. doi:10.1126/science.1090005
La Barbera, L., Vedele, F., Nobili, A., D’Amelio, M., and Krashia, P. (2019). Neurodevelopmental disorders: Functional role of Ambra1 in autism and schizophrenia. Mol. Neurobiol. 56, 6716–6724. doi:10.1007/s12035-019-1557-7
Lai, M. C., Lombardo, M. V., and Baron-Cohen, S. (2014). Autism. Lancet 383, 896–910. doi:10.1016/S0140-6736(13)61539-1
Li, H., and Outten, C. E. (2012). Monothiol CGFS glutaredoxins and BolA-like proteins: [2Fe-2S] binding partners in iron homeostasis. Biochemistry 51, 4377–4389. doi:10.1021/bi300393z
Li, Z., Liu, L., Lin, W., Zhou, Y., Zhang, G., Du, X., et al. (2020). NRG3 contributes to cognitive deficits in chronic patients with schizophrenia. Schizophr. Res. 215, 134–139. doi:10.1016/j.schres.2019.10.060
Lindblad-Toh, K., Garber, M., Zuk, O., Lin, M. F., Parker, B. J., Washietl, S., et al. (2011). A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482. doi:10.1038/nature10530
Liu, J., Mosti, F., and Silver, D. L. (2021). Human brain evolution: Emerging roles for regulatory DNA and RNA. Curr. Opin. Neurobiol. 71, 170–177. doi:10.1016/j.conb.2021.11.005
Lord, C., Elsabbagh, M., Baird, G., and Veenstra-Vanderweele, J. (2018). Autism spectrum disorder. Lancet 392, 508–520. doi:10.1016/S0140-6736(18)31129-2
Ma, S., Skarica, M., Li, Q., Xu, C., Risgaard, R. D., Tebbenkamp, A. T. N., et al. (2022). Molecular and cellular evolution of the primate dorsolateral prefrontal cortex. Science 80, 7257. doi:10.1126/science.abo7257
Macé, A., Tuke, M. A., Deelen, P., Kristiansson, K., Mattsson, H., Nõukas, M., et al. (2017). CNV-association meta-analysis in 191, 161 European adults reveals new loci associated with anthropometric traits. Nat. Commun. 8, 744–811. doi:10.1038/s41467-017-00556-x
Maillard, A. M., Ruef, A., Pizzagalli, F., Migliavacca, E., Hippolyte, L., Adaszewski, S., et al. (2015). The 16p11.2 locus modulates brain structures common to autism, schizophrenia and obesity. Mol. Psychiatry 20, 140–147. doi:10.1038/mp.2014.145
Maria Fimia, G., Stoykova, A., Romagnoli, A., Giunta, L., Di Bartolomeo, S., Nardacci, R., et al. (2007). Ambra1 regulates autophagy and development of the nervous system. Nature 447, 1121–1125. doi:10.1038/nature05925
McGrath, J., Saha, S., Chant, D., and Welham, J. (2008). Schizophrenia: A concise overview of incidence, prevalence, and mortality. Epidemiol. Rev. 30, 67–76. doi:10.1093/epirev/mxn001
Mefford, H. C., Sharp, A. J., Baker, C., Itsara, A., Jiang, Z., Buysse, K., et al. (2008). Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 359, 1685–1699. doi:10.1056/nejmoa0805384
Mitjans, M., Begemann, M., Ju, A., Dere, E., Wüstefeld, L., Hofer, S., et al. (2017). Sexual dimorphism of AMBRA1-related autistic features in human and mouse. Transl. Psychiatry 7, e1247–e1249. doi:10.1038/TP.2017.213
Monda, J. K., and Cheeseman, I. M. (2018). Nde1 promotes diverse dynein functions through differential interactions and exhibits an isoform-specific proteasome association. Mol. Biol. Cell. 29, 2336–2345. doi:10.1091/mbc.E18-07-0418
Monderer-Rothkoff, G., Tal, N., Risman, M., Shani, O., Nissim-Rafinia, M., Malki-Feldman, L., et al. (2021). AUTS2 isoforms control neuronal differentiation. Mol. Psychiatry 26, 666–681. doi:10.1038/s41380-019-0409-1
Mora-Bermúdez, F., Badsha, F., Kanton, S., Camp, J. G., Vernot, B., Köhler, K., et al. (2016). Differences and similarities between human and chimpanzee neural progenitors during cerebral cortex development. Elife 5, 186833–e18724. doi:10.7554/eLife.18683
Mosca, S., Raponi, M., Meneghello, A., Buratti, E., Woods, C. G., and Baralle, D. (2017). Human NDE1 splicing and mammalian brain development. Sci. Rep. 7, 43504–43509. doi:10.1038/srep43504
Mostaid, M. S., Lloyd, D., Liberg, B., Sundram, S., Pereira, A., Pantelis, C., et al. (2016). Neuregulin-1 and schizophrenia in the genome-wide association study era. Neurosci. Biobehav. Rev. 68, 387–409. doi:10.1016/j.neubiorev.2016.06.001
Mottron, L., Dawson, M., Soulières, I., Hubert, B., and Burack, J. (2006). Enhanced perceptual functioning in autism: An update, and eight principles of autistic perception. J. Autism Dev. Disord. 36, 27–43. doi:10.1007/s10803-005-0040-7
Müller, T., Braud, S., Jüttner, R., Voigt, B. C., Paulick, K., Sheean, M. E., et al. (2018). Neuregulin 3 promotes excitatory synapse formation on hippocampal interneurons. EMBO J. 37, 988588–e98919. doi:10.15252/embj.201798858
Mullins, N., Forstner, A. J., O’Connell, K. S., Coombes, B., Coleman, J. R. I., Qiao, Z., et al. (2021). Genome-wide association study of more than 40, 000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet. 53, 817–829. doi:10.1038/s41588-021-00857-4
Muth, A., Hönekopp, J., and Falter, C. M. (2014). Visuo-spatial performance in autism: A meta-analysis. J. Autism Dev. Disord. 44, 3245–3263. doi:10.1007/s10803-014-2188-5
Nagamani, S. C. S., Erez, A., Ben-Zeev, B., Frydman, M., Winter, S., Zeller, R., et al. (2013). Detection of copy-number variation in AUTS2 gene by targeted exonic array CGH in patients with developmental delay and autistic spectrum disorders. Eur. J. Hum. Genet. 21, 343–346. doi:10.1038/ejhg.2012.157
Nagy, O., Kárteszi, J., Elmont, B., and Ujfalusi, A. (2021). Case report: Expressive speech disorder in a family as a hallmark of 7q31 deletion involving the FOXP2 gene. Front. Pediatr. 9, 664548–8. doi:10.3389/fped.2021.664548
Nobili, A., Krashia, P., Cordella, A., La Barbera, L., Dell’Acqua, M. C., Caruso, A., et al. (2018). Ambra1 shapes hippocampal inhibition/excitation balance: Role in neurodevelopmental disorders. Mol. Neurobiol. 55, 7921–7940. doi:10.1007/s12035-018-0911-5
Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A. V., Mikheenko, A., et al. (2022). The complete sequence of a human genome. Sci. (80-. ) 376, 44–53. doi:10.1126/science.abj6987
Nuttle, X., Giannuzzi, G., Duyzend, M. H., Schraiber, J. G., Narvaiza, I., Sudmant, P. H., et al. (2016). Emergence of a Homo sapiens-specific gene family and chromosome 16p11.2 CNV susceptibility. Nature 536, 205–209. doi:10.1038/nature19075
Oberheim Bush, N. A., and Nedergaard, M. (2017). Do evolutionary changes in astrocytes contribute to the computational power of the hominid brain? Neurochem. Res. 42, 2577–2587. doi:10.1007/s11064-017-2363-0
Oberheim, N. A., Takano, T., Han, X., He, W., Lin, J. H. C., Wang, F., et al. (2009). Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287. doi:10.1523/JNEUROSCI.4707-08.2009
O’Bleness, M., Searles, V. B., Dickens, C. M., Astling, D., Albracht, D., Mak, A. C. Y., et al. (2014). Finished sequence and assembly of the DUF1220-rich 1q21 region using a haploid human genome. BMC Genomics 15, 387. doi:10.1186/1471-2164-15-387
O’Bleness, M., Searles, V. B., Varki, A., Gagneux, P., and Sikela, J. M. (2012). Evolution of genetic and genomic features unique to the human lineage. Nat. Rev. Genet. 13, 853–866. doi:10.1038/nrg3336
O’Brien, H. E., Hannon, E., Hill, M. J., Toste, C. C., Robertson, M. J., Morgan, J. E., et al. (2018). Expression quantitative trait loci in the developing human brain and their enrichment in neuropsychiatric disorders Genome Biol. 19, 194–213. doi:10.1186/s13059-018-1567-1
Oksenberg, N., Stevison, L., Wall, J. D., and Ahituv, N. (2013). Function and regulation of AUTS2, a gene implicated in autism and human evolution. PLoS Genet. 9, 1003221. doi:10.1371/journal.pgen.1003221
Owen, D., Bracher-Smith, M., Kendall, K. M., Rees, E., Einon, M., Escott-Price, V., et al. (2018). Effects of pathogenic CNVs on physical traits in participants of the UK Biobank. BMC Genomics 19, 867–869. doi:10.1186/s12864-018-5292-7
Owen, M. J., Sawa, A., and Mortensen, P. B. (2016). Schizophrenia. Lancet 388, 86–97. doi:10.1016/S0140-6736(15)01121-6
Pang, H., Yu, X., Kim, Y. M., Wang, X., Jinkins, J. K., Yin, J., et al. (2020). Disorders associated with diverse, recurrent deletions and duplications at 1q21.1. Front. Genet. 11, 577. doi:10.3389/fgene.2020.00577
Parellada, M., San José Cáceres, A., Palmer, M., Delorme, R., Jones, E. J. H., Parr, J. R., et al. (2021). A Phase II randomized, double-blind, placebo-controlled study of the efficacy, safety, and tolerability of arbaclofen administered for the treatment of social function in children and adolescents with autism spectrum disorders: Study protocol for AIMS-2-TRIALS-CT1 Front. Psychiatry 12, 701729. doi:10.3389/fpsyt.2021.701729
Paterson, C., Wang, Y., Hyde, T. M., Weinberger, D. R., Kleinman, J. E., and Law, A. J. (2017). Temporal, diagnostic, and tissue-specific regulation of NRG3 Isoform expression in human brain development and affective disorders. Am. J. Psychiatry 174, 256–265. doi:10.1176/appi.ajp.2016.16060721
Peyregne, S., Boyle, M. J., Dannemann, M., and Prufer, K. (2017). Detecting ancient positive selection in humans using extended lineage sorting. Genome Res. 27, 1563–1572. doi:10.1101/gr.219493.116
Pinson, A., and Huttner, W. B. (2021). Neocortex expansion in development and evolution—From genes to progenitor cell biology. Curr. Opin. Cell. Biol. 73, 9–18. doi:10.1016/j.ceb.2021.04.008
Pinson, A., Xing, L., Namba, T., Kalebic, N., Peters, J., Oegema, C. E., et al. (2022). Human TKTL1 implies greater neurogenesis in frontal neocortex of modern humans than Neanderthals. Science 80, eabl6422. doi:10.1126/science.abl6422
Pinto, D., Pagnamenta, A. T., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. doi:10.1038/nature09146
Polioudakis, D., de la Torre-Ubieta, L., Langerman, J., Elkins, A. G., Shi, X., Stein, J. L., et al. (2019). A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron 103, 785–801. e8. doi:10.1016/j.neuron.2019.06.011
Pollard, K. S., Salama, S. R., King, B., Kern, A. D., Dreszer, T., Katzman, S., et al. (2006a). Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2, e168–e1611. doi:10.1371/journal.pgen.0020168
Pollard, K. S., Salama, S. R., Lambert, N., Lambot, M. A., Coppens, S., Pedersen, J. S., et al. (2006b). An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443, 167–172. doi:10.1038/nature05113
Pollen, A. A., Bhaduri, A., Andrews, M. G., Nowakowski, T. J., Meyerson, O. S., Mostajo-Radji, M. A., et al. (2019). Establishing cerebral organoids as models of human-specific brain evolution. Cell. 176, 743–756. e17. doi:10.1016/j.cell.2019.01.017
Popesco, M. C., MacLaren, E. J., Hopkins, J., Dumas, L., Cox, M., Meltesen, L., et al. (2006). Human lineage-specific amplification, selection, and neuronal expression of DUF1220 domains. Science 313, 1304–1307. doi:10.1126/science.1127980
Prabhakar, S., Noonan, J. P., Pääbo, S., and Rubin, E. M. (2006). Accelerated evolution of conserved noncoding sequences in humans. Science 314, 786. doi:10.1126/science.1130738
Prescott, S. L., Srinivasan, R., Marchetto, M. C., Grishina, I., Narvaiza, I., Selleri, L., et al. (2015). Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell. 163, 68–83. doi:10.1016/j.cell.2015.08.036
Prüfer, K., Racimo, F., Patterson, N., Jay, F., Sankararaman, S., Sawyer, S., et al. (2014). The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49. doi:10.1038/nature12886
Rahman-Enyart, A., Lai, C., and Prieto, A. L. (2020). Neuregulins 1, 2, and 3 promote early neurite outgrowth in ErbB4-expressing cortical GABAergic interneurons. Mol. Neurobiol. 57, 3568–3588. doi:10.1007/s12035-020-01966-7
Rakic, P. (2009). Evolution of the neocortex: A perspective from developmental biology. Nat. Rev. Neurosci. 10, 724–735. doi:10.1038/nrn2719
Reilly, S. K., Yin, J., Ayoub, A. E., Emera, D., Leng, J., Cotney, J., et al. (2015). Evolutionary genomics. Evolutionary changes in promoter and enhancer activity during human corticogenesis Sci. (80-. ) 347, 1155–1159. doi:10.1126/science.1260943
Rein, B., and Yan, Z. (2020). 16p11.2 copy number variations and neurodevelopmental disorders. Trends Neurosci. 43, 886–901. doi:10.1016/j.tins.2020.09.001
Rietschel, M., Mattheisen, M., Degenhardt, F., Kahn, R. S., Linszen, D. H., Os, J. van, et al. (2012). Association between genetic variation in a region on chromosome 11 and schizophrenia in large samples from Europe. Mol. Psychiatry 17, 906–917. doi:10.1038/mp.2011.80
Sacco, R., Gabriele, S., and Persico, A. M. (2015). Head circumference and brain size in autism spectrum disorder: A systematic review and meta-analysis. Psychiatry Res. 234, 239–251. doi:10.1016/j.pscychresns.2015.08.016
Sadakata, T., Mizoguchi, A., Sato, Y., Katoh-Semba, R., Fukuda, M., Mikoshiba, K., et al. (2004). The secretory granule-associated protein CAPS2 regulates neurotrophin release and cell survival. J. Neurosci. 24, 43–52. doi:10.1523/JNEUROSCI.2528-03.2004
Sadakata, T., Washida, M., Iwayama, Y., Shoji, S., Sato, Y., Ohkura, T., et al. (2007). Autistic-like phenotypes in Cadps2-knockout mice and aberrant CADPS2 splicing in autistic patients. J. Clin. Invest. 117, 931–943. doi:10.1172/JCI29031
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., De Rubeis, S., An, J. Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 180, 568–584. e23. doi:10.1016/j.cell.2019.12.036
Schumann, G., Coin, L. J., Lourdusamy, A., Charoen, P., Berger, K. H., Stacey, D., et al. (2011). Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proc. Natl. Acad. Sci. U. S. A. 108, 7119–7124. doi:10.1073/pnas.1017288108
Searles Quick, V. B., Davis, J. M., Olincy, A., and Sikela, J. M. (2015). DUF1220 copy number is associated with schizophrenia risk and severity: Implications for understanding autism and schizophrenia as related diseases. Transl. Psychiatry 5, e697–7. doi:10.1038/tp.2015.192
Shinoda, Y., Sadakata, T., Akagi, T., Sakamaki, Y., Hashikawa, T., Sano, Y., et al. (2018). Calcium-dependent activator protein for secretion 2 (CADPS2) deficiency causes abnormal synapse development in hippocampal mossy fiber terminals. Neurosci. Lett. 677, 65–71. doi:10.1016/j.neulet.2018.04.036
Shinoda, Y., Sadakata, T., Nakao, K., Katoh-Semba, R., Kinameri, E., Furuya, A., et al. (2011). Calcium-dependent activator protein for secretion 2 (CAPS2) promotes BDNF secretion and is critical for the development of GABAergic interneuron network. Proc. Natl. Acad. Sci. U. S. A. 108, 373–378. doi:10.1073/pnas.1012220108
Shinoda, Y., Sadakata, T., Yagishita, K., Kinameri, E., Katoh-Semba, R., Sano, Y., et al. (2019). Aspects of excitatory/inhibitory synapses in multiple brain regions are correlated with levels of brain-derived neurotrophic factor/neurotrophin-3. Biochem. Biophys. Res. Commun. 509, 429–434. doi:10.1016/j.bbrc.2018.12.100
Sikela, J. M., and Searles Quick, V. B. (2018). Genomic trade-offs: Are autism and schizophrenia the steep price of the human brain? Hum. Genet. 137, 1–13. doi:10.1007/s00439-017-1865-9
Silver, D. L. (2016). Genomic divergence and brain evolution: How regulatory DNA influences development of the cerebral cortex. BioEssays 38, 162–171. doi:10.1002/bies.201500108
Sousa, A. M. M., Meyer, K. A., Santpere, G., Gulden, F. O., and Sestan, N. (2017). Evolution of the human nervous system function, structure, and development. Cell. 170, 226–247. doi:10.1016/j.cell.2017.06.036
Staes, N., Sherwood, C. C., Wright, K., De Manuel, M., Guevara, E. E., Marques-Bonet, T., et al. (2017). FOXP2 variation in great ape populations offers insight into the evolution of communication skills. Sci. Rep. 7, 16866–16910. doi:10.1038/s41598-017-16844-x
Stone, J. L., O’Donovan, M. C., Gurling, H., Kirov, G. K., Blackwood, D. H. R., Corvin, A., et al. (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241. doi:10.1038/nature07239
Stratton, J., Brook, M., and Hanlon, R. E. (2017). Murder and psychosis: Neuropsychological profiles of homicide offenders with schizophrenia. Crim. Behav. Ment. Health 27, 146–161. doi:10.1002/cbm.1990
Sultana, R., Yu, C. E., Yu, J., Munson, J., Chen, D., Hua, W., et al. (2002). Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics 80, 129–134. doi:10.1006/geno.2002.6810
Sundberg, M., Pinson, H., Smith, R. S., Winden, K. D., Venugopal, P., Tai, D. J. C., et al. (2021). 16p11.2 deletion is associated with hyperactivation of human iPSC-derived dopaminergic neuron networks and is rescued by RHOA inhibition in vitro. Nat. Commun. 12, 2897–2920. doi:10.1038/s41467-021-23113-z
Suzuki, I. K., Gacquer, D., Van Heurck, R., Kumar, D., Wojno, M., Bilheu, A., et al. (2018). Human-specific NOTCH2NL genes expand cortical neurogenesis through delta/notch regulation. Cell. 173, 1370–1384. e16. doi:10.1016/j.cell.2018.03.067
Tick, B., Bolton, P., Happé, F., Rutter, M., and Rijsdijk, F. (2016). Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child. Psychol. Psychiatry 57, 585–595. doi:10.1111/jcpp.12499
Trajkova, S., Di Gregorio, E., Ferrero, G. B., Carli, D., Pavinato, L., Delplancq, G., et al. (2020). New insights into potocki-shaffer syndrome: Report of two novel cases and literature review. Brain Sci. 10, 7888–E815. doi:10.3390/brainsci10110788
Trubetskoy, V., Pardiñas, A. F., Qi, T., Panagiotaropoulou, G., Awasthi, S., Bigdeli, T. B., et al. (2022). Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508. doi:10.1038/s41586-022-04434-5
van den Heuvel, M. P., Bullmore, E. T., and Sporns, O. (2016). Comparative connectomics. Trends Cogn. Sci. 20, 345–361. doi:10.1016/j.tics.2016.03.001
Van Dongen, J., and Boomsma, D. I. (2013). The evolutionary paradox and the missing heritability of schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 162, 122–136. doi:10.1002/ajmg.b.32135
Villalba, A., Götz, M., and Borrell, V. (2021). The regulation of cortical neurogenesis. Curr. Top. Dev. Biol. 142, 1–66. doi:10.1016/bs.ctdb.2020.10.003
Vollger, M. R., Guitart, X., Dishuck, P. C., Mercuri, L., Harvey, W. T., Gershman, A., et al. (2022). Segmental duplications and their variation in a complete human genome. Science 376, 445678. doi:10.1126/science.abj6965
Vullhorst, D., Ahmad, T., Karavanova, I., Keating, C., and Buonanno, A. (2017). Structural similarities between neuregulin 1–3 isoforms determine their subcellular distribution and signaling mode in central neurons. J. Neurosci. 37, 5232–5249. doi:10.1523/JNEUROSCI.2630-16.2017
Wallace, A. S., Hudac, C. M., Steinman, K. J., Peterson, J. L., DesChamps, T. D., Duyzend, M. H., et al. (2018). Longitudinal report of child with de novo 16p11.2 triplication. Clin. Case Rep. 6, 147–154. doi:10.1002/ccr3.1236
Wang, Y. N., Figueiredo, D., Sun, X. D., Dong, Z. Q., Chen, W. B., Cui, W. P., et al. (2018). Controlling of glutamate release by neuregulin3 via inhibiting the assembly of the SNARE complex. Proc. Natl. Acad. Sci. U. S. A. 115, 2508–2513. doi:10.1073/pnas.1716322115
Wang, Z. Y., Leushkin, E., Liechti, A., Ovchinnikova, S., Mößinger, K., Brüning, T., et al. (2020). Transcriptome and translatome co-evolution in mammals. Nature 588, 642–647. doi:10.1038/s41586-020-2899-z
Wightman, D. P., Jansen, I. E., Savage, J. E., Shadrin, A. A., Bahrami, S., Holland, D., et al. (2021). A genome-wide association study with 1, 126, 563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282. doi:10.1038/s41588-021-00921-z
Won, H., Huang, J., Opland, C. K., Hartl, C. L., and Geschwind, D. H. (2019). Human evolved regulatory elements modulate genes involved in cortical expansion and neurodevelopmental disease susceptibility. Nat. Commun. 10, 2396–2411. doi:10.1038/s41467-019-10248-3
Xu, K., Schadt, E. E., Pollard, K. S., Roussos, P., and Dudley, J. T. (2015). Genomic and network patterns of schizophrenia genetic variation in human evolutionary accelerated regions. Mol. Biol. Evol. 32, 1148–1160. doi:10.1093/molbev/msv031
Yamasaki, M., Makino, T., Khor, S. S., Toyoda, H., Miyagawa, T., Liu, X., et al. (2020). Sensitivity to gene dosage and gene expression affects genes with copy number variants observed among neuropsychiatric diseases. BMC Med. Genomics 13, 55–13. doi:10.1186/s12920-020-0699-9
Yao, X., Lu, Z., Feng, Z., Gao, L., Zhou, X., Li, M., et al. (2022). Comparison of chromatin accessibility landscapes during early development of prefrontal cortex between rhesus macaque and human. Nat. Commun. 13, 3883. doi:10.1038/s41467-022-31403-3
Yazdankhah, M., Farioli-Vecchioli, S., Tonchev, A. B., Stoykova, A., and Cecconi, F. (2014). The autophagy regulators Ambra1 and Beclin 1 are required for adult neurogenesis in the brain subventricular zone. Cell. Death Dis. 5, e1403–e1408. doi:10.1038/cddis.2014.358
Yoon, J., and Mao, Y. (2021). Dissecting molecular genetic mechanisms of 1q21.1 cnv in neuropsychiatric disorders. Int. J. Mol. Sci. 22, 5811. doi:10.3390/ijms22115811
Zhang, Y., Sloan, S. A., Clarke, L. E., Caneda, C., Plaza, C. A., Blumenthal, P. D., et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53. doi:10.1016/j.neuron.2015.11.013
Zhou, Y., Li, Y., Meng, Y., Wang, J., Wu, F., Ning, Y., et al. (2020). Neuregulin 3 rs10748842 polymorphism contributes to the effect of body mass index on cognitive impairment in patients with schizophrenia. Transl. Psychiatry 10, 62. doi:10.1038/s41398-020-0746-5
Zhu, Y., Sousa, A. M. M., Gao, T., Skarica, M., Li, M., Santpere, G., et al. (2018). Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 80, eaat8077. doi:10.1126/science.aat8077
Keywords: schizophrenia, ASD, cognition, evolution, psychiatric discorders
Citation: Duński E and Pękowska A (2022) Keeping the balance: Trade-offs between human brain evolution, autism, and schizophrenia. Front. Genet. 13:1009390. doi: 10.3389/fgene.2022.1009390
Received: 01 August 2022; Accepted: 12 October 2022;
Published: 21 November 2022.
Edited by:
Anthony John Hannan, University of Melbourne, AustraliaReviewed by:
Vijay Tiwari, Queen’s University Belfast, United KingdomLambert Zixin Li, Stanford University, United States
Copyright © 2022 Duński and Pękowska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aleksandra Pękowska, YS5wZWtvd3NrYUBuZW5ja2kuZWR1LnBs; Eryk Duński, ZS5kdW5za2lAbmVuY2tpLmVkdS5wbA==