 Alexander M. Kleschevnikov
Alexander M. Kleschevnikov- Department of Neurosciences, University of California San Diego, San Diego, CA, United States
The most distinctive feature of Down syndrome (DS) is moderate to severe cognitive impairment. Genetic, molecular, and neuronal mechanisms of this complex DS phenotype are currently under intensive investigation. It is becoming increasingly clear that the abnormalities arise from a combination of initial changes caused by triplication of genes on human chromosome 21 (HSA21) and later compensatory adaptations affecting multiple brain systems. Consequently, relatively mild initial cognitive deficits become pronounced with age. This pattern of changes suggests that one approach to improving cognitive function in DS is to target the earliest critical changes, the prevention of which can change the ‘trajectory’ of the brain development and reduce the destructive effects of the secondary alterations. Here, we review the experimental data on the role of KCNJ6 in DS-specific brain abnormalities, focusing on a putative role of this gene in the development of abnormal neural circuits in the hippocampus of genetic mouse models of DS. It is suggested that the prevention of these early abnormalities with pharmacological or genetic means can ameliorate cognitive impairment in DS.
Introduction
Down syndrome (DS), a genetic disorder caused by the triplication of genes on the human chromosome 21 (HSA21) (Lejeune et al., 1959), is characterized by moderate to severe cognitive impairment. The mechanisms underlying to cognitive impairment in DS are being methodically investigated (Lott, 2012; Contestabile et al., 2017; Rafii et al., 2019; Antonarakis et al., 2020; Botte and Potier, 2020). The abnormalities can be conditionally divided into two groups: primary changes, caused directly by overexpression of the HSA21 genes, and secondary changes, arising mainly as compensatory reactions to abnormal development (Sturgeon et al., 2012; Antonarakis, 2017; Hasina et al., 2022). Due to the accumulation of these secondary abnormalities, initially mild developmental deficits become severe with age. This suggests that one approach to improving cognitive function in DS is to target the early primary changes, which can reduce or prevent the development and, thus, the destructive effects of the secondary alterations. In this review, we focus on the role of the KCNJ6 gene in DS-specific brain abnormalities. In particular, we will discuss the putative role of KCNJ6 in the development of abnormal neural circuits in genetic mouse models of DS. It is suggested that prevention of these early abnormalities through pharmacological or genetic means may improve cognitive impairment in DS.
Role of KCNJ6 in Down syndrome and other developmental disorders
KCNJ6 is located in the middle of the “Down syndrome critical region” (DSCR) of HSA21, the triplication of which is necessary for the manifestation of cognitive impairment (McCormick et al., 1989; Korenberg et al., 1990; Peterson et al., 1994), and is sufficient in mouse genetic models to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of DS (Belichenko N. P. et al., 2009; Jiang et al., 2015). Recent reanalysis of all ‘partial trisomy 21’ cases described from 1973 to 2021 revealed an extremely narrow (34 kbp) subregion of DSCR, most closely related to the DS diagnosis (Pelleri et al., 2016; Pelleri et al., 2019; Antonaros et al., 2021). Only two genes, DSCR4 and KCNJ6, span across this subregion. DSCR4 is a de novo-originated protein-coding gene that is likely absent in mice (Toyoda et al., 2002). In people, it is predominantly expressed in the placenta (Asai et al., 2008) and is involved in the regulation of biological pathways related to cell migration, coagulation, and the immune system (Saber et al., 2021). The role of DSCR4 in cognition is unknown. In contrast, KCNJ6 has been implicated in several genetic abnormalities characterized by cognitive impairment, as well as in the modulation of a variety of higher brain functions. Thus, heterozygous mutations of KCNJ6, affecting the pore-forming domain of the GIRK2 channel, result in the Keppen-Lubinsky syndrome, a genetic disorder characterized by severe developmental delay, microcephaly, and intellectual disability (Basel-Vanagaite et al., 2009; Masotti et al., 2015). Genetic variations of KCNJ6 were implicated in the modulation of reward-related brain processes (Kamarajan et al., 2017; Ziegler et al., 2020) and pain sensitivity (Nishizawa et al., 2014; Langford et al., 2015; Elens et al., 2016). KCNJ6 may also play an important role in the generation of infantile spasms associated with cognitive impairment (Cortez et al., 2009; Blichowski et al., 2015; Joshi et al., 2016), as well as in other disorders (Mayfield et al., 2015; Jeremic et al., 2021). In rodents, a missense mutation of KCNJ6 is responsible for the ‘weaver’ phenotype, characterized by abnormal development of the cerebellum, ectopic cells in the CA3 region, and other developmental abnormalities (Patil et al., 1995; Sekiguchi et al., 1995; Ozaki et al., 2002; Schein et al., 2005). Overexpression of KCNJ6 impairs synaptic plasticity and provokes cognitive impairment resembling DS in wild type mice (Cooper et al., 2012). Thus, KCNJ6 abnormalities may influence brain development and lead to cognitive impairment, supporting the notion that triplication of this gene plays an important role in cognitive impairment in DS.
Properties of GIRK2 channels
The KCNJ6 gene encodes GIRK2 subunits of G protein-activated inwardly rectifying potassium channels. Alternative splicing of KNCJ6 results in the generation of four GIRK2 isoforms (GIRK2a, GIRK2b, GIRK2c, and GIRK2d), which differ in trafficking and therefore in the regional and subcellular distribution (Wei et al., 1998; Harashima et al., 2006b; Marron Fernandez de Velasco et al., 2017). Functional potassium channels are formed by GIRK2 as heterotetramers in combination with other subunits (GIRK1, GIRK3, and GIRK4) or as homotetramers (Yamada et al., 1998; Inanobe et al., 1999; Hibino et al., 2010). GIRK2 channels serve as effectors for a number of postsynaptic metabotropic receptors such as GABAB, A1, 5-HT1A, m2, and others (Luscher et al., 1997; Yamada et al., 1998; Dascal and Kahanovitch, 2015). In addition, GIRK2 channels are activated constitutively in an agonist-independent manner (Chen and Johnston, 2005; Kim and Johnston, 2015). High-resolution immunohistochemical studies showed that, in the hippocampus, GIRK2 channels are most abundant at the perisynaptic locations on dendritic shafts and spines of principle neurons, where they co-localized with GABAB receptors (Kulik et al., 2006). Activation of GIKR2 channels requires simultaneous action of beta-gamma subunits of G-proteins, which are produced following activation of the corresponding G protein-coupled metabotropic receptors, and PIP2 (Whorton and MacKinnon, 2011; Wang et al., 2014). Since GIRK2 channels are selective for K+ ions, opening of these channels shifts the membrane potential in the direction of equilibrium potential for potassium (∼ -94 mV), thus hyperpolarizing neurons. Both the hyperpolarization and the reduction of membrane resistance upon activation of these channels reduces neuronal excitability. Thus, GIRK2 channels play an important role in the maintenance of excitatory/inhibitory balance by effectively controlling the resting membrane potential and neuronal excitability.
GIRK2 channels in Down syndrome models
Mouse genetic models have been successfully used to examine structural and functional abnormalities of DS in invasive experiments (Dierssen et al., 2001; Belichenko et al., 2004; Salehi et al., 2006; Yu et al., 2010; Gotti et al., 2011; Liu et al., 2011; Popov et al., 2011; Kleschevnikov et al., 2012c; Cramer and Galdzicki, 2012; Haydar and Reeves, 2012; Ruparelia et al., 2012; Zhang et al., 2012; Zhang et al., 2014; Belichenko et al., 2015). In addition, genetic models of DS have been instrumental for the discovery and evaluation of potential treatments to improve the impaired cognition (Dierssen et al., 2001; Belichenko et al., 2007; Dierssen et al., 2009; Salehi et al., 2009; Faizi et al., 2011; Kleschevnikov et al., 2012a; Kleschevnikov et al., 2012c; Haydar and Reeves, 2012; Lott, 2012; Mohler, 2012; Rueda et al., 2012; Ruparelia et al., 2012; Lysenko et al., 2014; Martinez-Cue et al., 2014; Belichenko et al., 2016; Hamlett et al., 2016; Reeves et al., 2019; Tayebati et al., 2019; Chen et al., 2020). The most extensively used genetic model of DS, Ts65Dn mice, are segmentally trisomic for the mouse chromosome 16 region that is syntenic to a subregion of HSA21 containing DSCR. These mice exhibit multiple structural, physiological, and behavioral phenotypes characteristic of DS (Freeburn and Munn, 2021).
In Ts65Dn mice, levels of Girk2 are increased by about 50% in the hippocampus, neocortex, and other brain regions, consistent with the increased dose of KCNJ6 (Kleschevnikov et al., 2005; Harashima et al., 2006b; Kleschevnikov et al., 2012b). The expression of all splicing GIRK2 isoforms is increased (Harashima et al., 2006a). Regional distribution of GIRK2 is also altered with a disproportional increase of GIRK2 levels in the outer molecular layer of the dentate gyrus (DG) and the lacunosum moleculare of the CA3 (Harashima et al., 2006a). The targeting of GIRK2 channels to the cellular membrane is also enhanced in DS (Wang et al., 2013). This effect is mediated by an increased expression of miR-155, which reduces expression levels of sorting nexin 27, thereby reducing trafficking of GIRK2 from the cellular membrane to endosomes (Wang et al., 2013).
Increased membrane targeting and expression of GIRK2 channels implies that their efficiency is enhanced in DS models. Thus, the selective GABAB receptor agonist baclofen evoked larger whole-cell currents and caused a greater reduction of the input resistance thus signifying that GABAB/GIRK2 signaling is enhanced in cultured hippocampal neurons of Ts65Dn mice (Best et al., 2007). Baclofen-evoked currents were also increased, and the resting membrane potential of the DG granule neurons was excessively hyperpolarized, in acute hippocampal slices from Ts65Dn mice (Kleschevnikov et al., 2012b). These observations show that GIRK2 signaling is significantly enhanced thus contributing to the increased overall inhibitory efficiency in genetic models of DS.
Downregulation of GIRK2 channels improves synaptic plasticity and cognition in adult Ts65Dn mice
The increased GABAB/GIRK2 signaling can reduce synaptic plasticity and thus lead to deficient cognition in DS models. If so, downregulating GABAB/GIRK2 currents can improve both synaptic plasticity and cognition. This can be achieved either pharmacologically with GABAB receptor antagonists or GIRK2 channel blockers, or genetically by reducing KCNJ6 gene dose. Thus, long-term potentiation in the dentate gyrus (Kleschevnikov et al., 2012a) and perirhinal cortex (Roncace et al., 2017) of Ts65Dn mice was improved by selective GABAB receptor antagonists. Direct blocking of GIRK2 channels with fluoxetine, a serotonin reuptake inhibitor that effectively suppresses GIRK2 currents (Kobayashi et al., 2003), was also effective in the restoration of long-term potentiation (LTP) in Ts656Dn mice (Kleschevnikov et al., 2017). This action of fluoxetine cannot be attributed to an increase in serotonin level since this would suppress LTP in the dentate gyrus (Sakai and Tanaka, 1993) or to a promotion of adult neurogenesis (Clark et al., 2006) since this would require a longer amount of time. Ethosuximide, another partial blocker of GIRK2 channels (Kobayashi et al., 2009), was not effective in terms of cognitive recovery or other improvements in adult Ts65Dn mice (Vidal et al., 2012). However, this drug also inhibits low-threshold T-type Ca+2 channels, which may negatively affect synaptic plasticity and memory (Ponnusamy and Pradhan, 2006; Leresche and Lambert, 2017), thus mitigating the possible positive effects of GIRK2 channel blockade. Finally, LTP was restored in Ts65Dn mice that only had two functional copies of KCNJ6 (Kleschevnikov et al., 2017). All these treatments also improved the cognitive function in Ts65Dn mice. Chronic i. p. injections of the GABAB antagonist CGP55845 restored long-term memory in the Novel Object Recognition and Contextual Fear Conditioning tests (Kleschevnikov et al., 2012a). Interestingly, not all abnormal behavioral parameters were restored by this treatment. For example, abnormal working memory and increased locomotor activity were not affected (Kleschevnikov et al., 2012a). Similar to GABAB antagonists, chronic administration of fluoxetine also improved spatial memory in adult Ts65Dn mice (Begenisic et al., 2014). However, no improvement of cognition was observed in another study in which an excessively high dose of fluoxetine had been used (Heinen et al., 2012). These results indicate that the suppression of GABAB/GIRK2 signaling must be within certain limits to be effective. In addition, since fluoxetine increases the rate of adult neurogenesis mice (Clark et al., 2006), it is not clear to what degree the suppression of GIRK2 channels was responsible for the cognitive improvement in this study. Finally, restoration of long-term memory was observed in Ts65Dn mice that had only two copies of KCNJ6 (Kleschevnikov et al., 2017). Thus, moderate suppression of GIRK2 channels restores hippocampal synaptic plasticity and long-term memory in Ts65Dn model of DS. On the other hand, many aspects of abnormal behavior do not improve with such treatments. It is therefore important to understand how and when those abnormalities arise during the brain development in DS models.
Formation of nascent neuronal circuits
In rodents, the formation of neural circuits starts in the prenatal period and continues postnatally for several weeks. In the CA1 and CA3 regions, neurogenesis is mostly completed before birth (Figure 1A, black lines). However, astrogenesis and synaptogenesis last for several postnatal weeks (Figure 1A, green line). The formation of correct synaptic connections during this period critically depends on spontaneous neuronal activity (Kirkby et al., 2013; Kerschensteiner, 2014; Luhmann et al., 2016; Griguoli and Cherubini, 2017). Factors that affect neuronal excitability and neuronal firing can thus interfere with the formation of neural circuits.
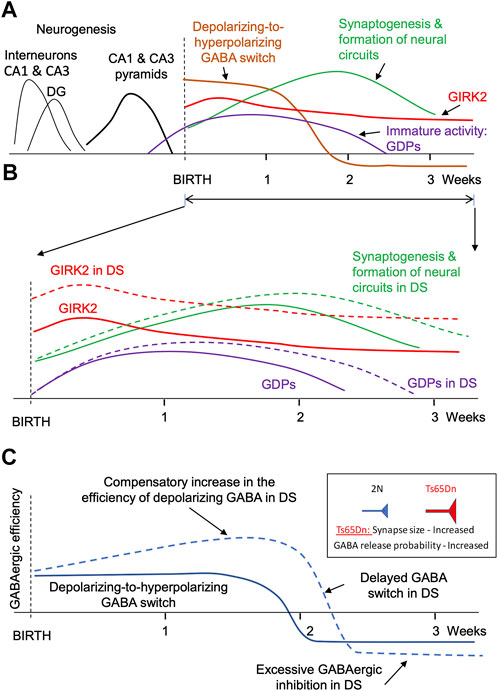
FIGURE 1. Major events during the formation of faulty neuronal circuits in the hippocampus of mouse genetic model of DS. (A). Main events in the hippocampus of normosomic mice. The time course of neurogenesis (black lines), synaptogenesis (green), GIRK2 expression (red), depolarizing-to-hyperpolarizing GABA switch (brown), and immature giant depolarizing potentials (GDP, purple). (B). Changes in GIRK2 expression levels (red), immature activity (violet), and neural circuit formation (green). Dashed lines show the changes in DS mice. Note the increase in GIRK2 levels and the prolongation of the period of immature activity in DS models. (C). Putative changes in the efficiency of GABAergic neurotransmission in the hippocampus of mouse genetic models during early postnatal development. Note a compensatory elevation of the depolarizing GABAergic efficiency before the GABA switch. The increased GABAergic excitation turns into an increased GABAergic inhibition after the GABA switch.
One factor affecting excitability and spontaneous activity of neonatal neurons is the depolarizing action of GABA. Thus, in early development GABA depolarizes neurons due to high intracellular chloride level (Rivera et al., 1999; Ben-Ari et al., 2007) (Figure 1A, brown line). Because glutamatergic synapses are not yet fully developed at this time (Tyzio et al., 1999), neuronal depolarization caused by the activation of GABAA receptors provides an important source of neuronal excitation (Rivera et al., 1999; Ben-Ari, 2002; Blankenship and Feller, 2010). Later in development, after the formation of glutamatergic synapses, the polarity of GABA action switches from depolarizing to hyperpolarizing (Ben-Ari et al., 2007) (Figure 1A, brown line). Therefore, changes in the duration or strength of the depolarizing GABA action in early development lead to the formation of abnormal dendrites, synaptic connections, faulty neural circuits (Cancedda et al., 2007; Levav et al., 2008; Levav-Rabkin et al., 2010; Chen and Kriegstein, 2015), and abnormal behavior in adulthood (Levav et al., 2004; Melamed et al., 2014).
Activation of GIRK2 channels represents another important factor affecting neuronal excitability and hence spontaneous activity. GIRK2 is expressed at high levels during the perinatal and early postnatal periods (Karschin and Karschin, 1997; Fernandez-Alacid et al., 2011; Aguado et al., 2013). In the cerebellum, GIRK2 levels peak at P5 and gradually decrease over several weeks to adult levels that are 3 times lower than in the early postnatal period (Aguado et al., 2013). GIRK2 levels are also high in the neocortex and hippocampus early in the postnatal period (Fernandez-Alacid et al., 2011) (Figure 1A, red line). High expression levels of GIRK2 during the perinatal and early postnatal periods suggest that altered signaling through these channels can interfere with the formation of nascent neural circuits in DS.
Early neuronal abnormalities in Down syndrome: Possible role of GIRK2 channels
The earliest brain abnormalities in DS are observed in prenatal development and include changes in the expression of proteins (Engidawork and Lubec, 2003; Lott, 2012), reduced neurogenesis (Sylvester, 1983), and disorganized cortical lamination (Golden and Hyman, 1994; Utagawa et al., 2022). However, the most noticeable physiological and morphological alterations occur after birth, when dendritic and synaptic abnormalities quickly accumulate in the cerebral cortex and hippocampus (Marin-Padilla, 1976; Takashima et al., 1989; Schmidt-Sidor et al., 1990; Engidawork and Lubec, 2003; Aldridge et al., 2007). At this period, GIRK2 is already expressed at high levels in the brain of DS individuals (Thiery et al., 2003). In genetic mouse models, the first abnormalities are also observed in the prenatal period (Haydar et al., 1996; Haydar et al., 2000; Cheng et al., 2004; Chakrabarti et al., 2007; Haydar and Reeves, 2012). In Ts65Dn mice, delays in growth of the neocortex and hippocampus have been noted with the first changes seen on E14.5 (Chakrabarti et al., 2007). However, like in people with DS, the most significant alterations have been observed in postnatal (P0-P16) development (Lorenzi and Reeves, 2006; Contestabile et al., 2007; Lysenko et al., 2018; Jain et al., 2020; Uguagliati et al., 2022). These postnatal changes were accompanied by significant delays in achieving developmental motor and sensory milestones (Holtzman et al., 1996; Toso et al., 2008; Olmos-Serrano et al., 2016).
Rapid accumulation of the DS-specific brain abnormalities in the early postnatal period suggests that some of these changes could be compensatory, caused by the changes in the expression of the genes present on HSA21. It is therefore feasible that these secondary changes can be mitigated by time-limited pharmacological or genetic interventions at the time or soon after their origin. One possible scenario could be the that an increased expression of KCNJ6 and miR-155, both on HSA21, increases the efficiency of the GIRK2 channel signaling (the primary change) (Figure 1B, red dashed line). This provokes excessive hyperpolarization and reduced excitability of neonatal neurons, which reduces their spontaneous activity (Madamba et al., 2021). Spontaneous activity plays an important role in synaptogenesis and in establishing correct synaptic connections (Kirkby et al., 2013; Kerschensteiner, 2014). Thus, a decrease in neonatal spontaneous activity will delay and disrupt the formation of neural circuits (Figure 1B, green dashed). In confirmation of this, a prolongation of the period of immature neuronal activity such as generation of the giant depolarizing potentials (GDPs) was observed in neonatal Ts65Dn mice (Lysenko et al., 2018) (Figure 1B, violet dashed line).
An additional outcome of the reduced neonatal spontaneous activity could be an increase in the efficiency of GABAergic neurotransmission in Ts65Dn mice (Figure 1C). Indeed, the depolarizing GABA action is one of the main sources of neonatal neuronal activation. A decrease in spontaneous activity usually triggers homeostatic compensatory changes to restore this activity (Turrigiano and Nelson, 2004; Wefelmeyer et al., 2016; Tien and Kerschensteiner, 2018). One such change may be an increase in the efficiency of depolarizing GABA (Figure 1C, upper part of the blue dashed line). For example, a decrease in spontaneous network activity causes an increase in the amplitude of the excitatory GABAergic postsynaptic currents in neonatal spinal motoneuron inputs (Gonzalez-Islas and Wenner, 2006). After switching the GABA action from depolarizing to hyperpolarizing, this change results in an increased GABAergic inhibition (Figure 1C, bottom part of the blue dashed line). The efficiency of GABAergic neurotransmission is increased in Ts65Dn and other genetic models of DS (Kleschevnikov et al., 2004; Belichenko P. V. et al., 2009; Kleschevnikov et al., 2012b; Kleschevnikov et al., 2012c). Interestingly, the increase in GABAergic efficiency is associated with an increase in the size of GABAergic synapses (Belichenko P. V. et al., 2009) and the presynaptic release probability of GABA (Kleschevnikov et al., 2004; Kleschevnikov et al., 2012b), which are characteristic of compensatory changes caused by a decrease in neuronal activity (Murthy et al., 2001; Han and Stevens, 2009).
Provided that this scenario is correct, one approach to improve cognitive impairment in DS is to reduce GIRK2 channel signaling during the formation of the nascent neural circuits. An indirect confirmation of the validity of such approach is the improvement of synaptic plasticity and cognition in Ts65Dn mice following prenatal (Guidi et al., 2014) and neonatal (Bianchi et al., 2010; Stagni et al., 2015) treatment with fluoxetine, which, among other effects, suppresses GIRK2 channels. However, since fluoxetine has many other effects, it is not clear if and whether some of the improvements were due to the suppression of GIRK2 channels in these studies. Additional investigations are needed to either prove or disprove the critical role of GIRK2 channel signaling in the formation of abnormal neural circuits in DS.
Conclusion: Methodology of cognitive correction in DS
Theoretically, two major strategies can be used to improve cognitive function in DS. The first strategy is to correct genetic abnormalities, on the assumption that this will eventually lead to the correction of various brain functions, including cognition. Several attempts have been made to implement this strategy. For example, silencing of genes on one chromosome 21 using the integrated XIST (X-Inactivation Specific Transcript) transgene broadly repressed this chromosome in DS pluripotent cells, partially correcting cell pathogenesis in an in vitro model of human fetal hematopoiesis (Li et al., 2012; Jiang et al., 2013; Plona et al., 2016; Chiang et al., 2018). In other studies, the corrections were restricted to one or several genes of interest. e.g., correction of the gene dose of Dyrk1a (Altafaj et al., 2013; Garcia-Cerro et al., 2017), Kcnj6 (Kleschevnikov et al., 2017), or App (Salehi et al., 2006; Mojabi et al., 2016) improved neuronal properties and behavior in mouse models of DS. The second strategy is to identify critical synaptic and cellular abnormalities leading to impaired brain development and then directly correct these abnormalities with pharmacological, genetic, or other means. This can be achieved not only in mouse genetic models but also in humans by comparing the development of cell lines or organoids derived from isogenic pairs of trisomy 21 and euploid induced pluripotent stem cells (iPSCs) (Weick et al., 2016; Araujo et al., 2018; Real et al., 2018; Kawatani et al., 2021; Tang et al., 2021; Nehra et al., 2022; Sharma et al., 2022; Xu et al., 2022). It is important to note that this second strategy does not require correction of all genetic abnormalities, and it does not target all DS-specific phenotypes. While implementing such strategy, it was previously shown that the efficiency of GABAergic inhibition is increased in the hippocampus of DS mice, resulting in decreased synaptic plasticity (Kleschevnikov et al., 2004). Correction of the increased inhibition with antagonists of GABAA (Kleschevnikov et al., 2004; Fernandez et al., 2007), GABAB (Kleschevnikov et al., 2012a), or inverse agonists of α5 subunit-containing GABAA receptors (Braudeau et al., 2011; Martinez-Cue et al., 2013) improved both synaptic plasticity and cognition in mouse genetic models of DS.
Although the first strategy looks preferable, it has several limitations restricting its practical use. Thus, not all genes on chromosome 21 could be suppressed by the XIST transgene or other methods, leaving many genes expressed at higher levels. This problem is especially noticeable in the case of correction of only one or several triplicated genes. Next, expression changes in DS are genome-wide and cannot be corrected by silencing of genes only on chromosome 21. Most importantly, such genetic therapy requires germline genetic alterations, which is highly controversial and currently ethically precluded. These limitations are absent in the second strategy.
In our view, the most promising approach is a combination of these two strategies. As described in this review, KCNJ6 gene was identified a critical target for genetic correction in DS, because: 1) Increased expression of KCNJ6 alone results in phenotypes reminiscent of DS (Cooper et al., 2012), and 2) Correction of KCNJ6 dose restores synaptic plasticity and cognition in the Ts65Dn model (Kleschevnikov et al., 2017). This change likely affects neonatal neuronal activity, thus resulting in the development of abnormal synaptic circuits. We can thus hypothesize that correcting these critical abnormalities in the neonatal brain by time-limited pharmacological or genetic interventions may normalize the development of nascent neural circuits and thereby improve cognitive function in DS. These expectations should be tested in the future studies.
Author contributions
AK wrote the review.
Funding
This publication was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health under award number R01HD100607.
Acknowledgments
The author thanks Mr. Joshua Jin and Mr. Nicholas Moy for discussions and technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aguado, C., Fernandez-Alacid, L., Cabanero, M. J., Yanagawa, Y., Schilling, K., Watanabe, M., et al. (2013). Differential maturation of GIRK2-expressing neurons in the mouse cerebellum. J. Chem. Neuroanat. 47, 79–89. doi:10.1016/j.jchemneu.2012.11.001
Aldridge, K., Reeves, R. H., Olson, L. E., and Richtsmeier, J. T. (2007). Differential effects of trisomy on brain shape and volume in related aneuploid mouse models. Am. J. Med. Genet. A 143A (10), 1060–1070. doi:10.1002/ajmg.a.31721
Altafaj, X., Martin, E. D., Ortiz-Abalia, J., Valderrama, A., Lao-Peregrin, C., Dierssen, M., et al. (2013). Normalization of Dyrk1A expression by AAV2/1-shDyrk1A attenuates hippocampal-dependent defects in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 52, 117–127. doi:10.1016/j.nbd.2012.11.017
Antonarakis, S. E. (2017). Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet. 18 (3), 147–163. doi:10.1038/nrg.2016.154
Antonarakis, S. E., Skotko, B. G., Rafii, M. S., Strydom, A., Pape, S. E., Bianchi, D. W., et al. (2020). Down syndrome. Nat. Rev. Dis. Prim. 6 (1), 9. doi:10.1038/s41572-019-0143-7
Antonaros, F., Pitocco, M., Abete, D., Vione, B., Piovesan, A., Vitale, L., et al. (2021). Structural characterization of the highly restricted down syndrome critical region on 21q22.13: New KCNJ6 and DSCR4 transcript isoforms. Front. Genet. 12, 770359. doi:10.3389/fgene.2021.770359
Araujo, B. H. S., Kaid, C., De Souza, J. S., Gomes da Silva, S., Goulart, E., Caires, L. C. J., et al. (2018). Down syndrome iPSC-derived astrocytes impair neuronal synaptogenesis and the mTOR pathway in vitro. Mol. Neurobiol. 55 (7), 5962–5975. doi:10.1007/s12035-017-0818-6
Asai, S., Yamaki, A., Kudoh, J., Shimizu, N., and Shimizu, Y. (2008). Analysis of the promoter region of human placenta-specific DSCR4 gene. Biochim. Biophys. Acta 1779 (1), 40–50. doi:10.1016/j.bbagrm.2007.09.005
Basel-Vanagaite, L., Shaffer, L., and Chitayat, D. (2009). Keppen-Lubinsky syndrome: Expanding the phenotype. Am. J. Med. Genet. A 149A (8), 1827–1829. doi:10.1002/ajmg.a.32975
Begenisic, T., Baroncelli, L., Sansevero, G., Milanese, M., Bonifacino, T., Bonanno, G., et al. (2014). Fluoxetine in adulthood normalizes GABA release and rescues hippocampal synaptic plasticity and spatial memory in a mouse model of Down syndrome. Neurobiol. Dis. 63, 12–19. doi:10.1016/j.nbd.2013.11.010
Belichenko, N. P., Belichenko, P. V., Kleschevnikov, A. M., Salehi, A., Reeves, R. H., and Mobley, W. C. (2009a). The "Down syndrome critical region" is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. J. Neurosci. 29 (18), 5938–5948. [pii]. doi:10.1523/JNEUROSCI.1547-09.2009
Belichenko, P. V., Kleschevnikov, A. M., Becker, A., Wagner, G. E., Lysenko, L. V., Yu, Y. E., et al. (2015). Down syndrome cognitive phenotypes modeled in mice trisomic for all HSA 21 homologues. PLoS One 10 (7), e0134861. doi:10.1371/journal.pone.0134861
Belichenko, P. V., Kleschevnikov, A. M., Masliah, E., Wu, C., Takimoto-Kimura, R., Salehi, A., et al. (2009b). Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of Down syndrome. J. Comp. Neurol. 512 (4), 453–466. doi:10.1002/cne.21895
Belichenko, P. V., Kleschevnikov, A. M., Salehi, A., Epstein, C. J., and Mobley, W. C. (2007). Synaptic and cognitive abnormalities in mouse models of down syndrome: Exploring genotype-phenotype relationships. J. Comp. Neurol. 504 (4), 329–345. doi:10.1002/cne.21433
Belichenko, P. V., Madani, R., Rey-Bellet, L., Pihlgren, M., Becker, A., Plassard, A., et al. (2016). An anti-beta-amyloid vaccine for treating cognitive deficits in a mouse model of down syndrome. PLoS One 11 (3), e0152471. doi:10.1371/journal.pone.0152471
Belichenko, P. V., Masliah, E., Kleschevnikov, A. M., Villar, A. J., Epstein, C. J., Salehi, A., et al. (2004). Synaptic structural abnormalities in the Ts65Dn mouse model of Down Syndrome. J. Comp. Neurol. 480 (3), 281–298. doi:10.1002/cne.20337
Ben-Ari, Y. (2002). Excitatory actions of gaba during development: The nature of the nurture. Nat. Rev. Neurosci. 3 (9), 728–739. doi:10.1038/nrn920
Ben-Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. (2007). Gaba: A pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87 (4), 1215–1284. doi:10.1152/physrev.00017.2006
Best, T. K., Siarey, R. J., and Galdzicki, Z. (2007). Ts65Dn, a mouse model of Down syndrome, exhibits increased GABAB-induced potassium current. J. Neurophysiol. 97 (1), 892–900. doi:10.1152/jn.00626.2006
Bianchi, P., Ciani, E., Guidi, S., Trazzi, S., Felice, D., Grossi, G., et al. (2010). Early pharmacotherapy restores neurogenesis and cognitive performance in the Ts65Dn mouse model for Down syndrome. J. Neurosci. 30 (26), 8769–8779. doi:10.1523/JNEUROSCI.0534-10.2010
Blankenship, A. G., and Feller, M. B. (2010). Mechanisms underlying spontaneous patterned activity in developing neural circuits. Nat. Rev. Neurosci. 11 (1), 18–29. doi:10.1038/nrn2759
Blichowski, M., Shephard, A., Armstrong, J., Shen, L., Cortez, M. A., Eubanks, J. H., et al. (2015). The GIRK2 subunit is involved in IS-like seizures induced by GABA(B) receptor agonists. Epilepsia 56 (7), 1081–1087. doi:10.1111/epi.13034
Botte, A., and Potier, M. C. (2020). Focusing on cellular biomarkers: The endo-lysosomal pathway in Down syndrome. Prog. Brain Res. 251, 209–243. doi:10.1016/bs.pbr.2019.10.002
Braudeau, J., Delatour, B., Duchon, A., Pereira, P. L., Dauphinot, L., de Chaumont, F., et al. (2011). Specific targeting of the GABA-A receptor α5 subtype by a selective inverse agonist restores cognitive deficits in Down syndrome mice. J. Psychopharmacol. 25 (8), 1030–1042. doi:10.1177/0269881111405366
Cancedda, L., Fiumelli, H., Chen, K., and Poo, M. M. (2007). Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J. Neurosci. 27 (19), 5224–5235. doi:10.1523/JNEUROSCI.5169-06.2007
Chakrabarti, L., Galdzicki, Z., and Haydar, T. F. (2007). Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J. Neurosci. 27 (43), 11483–11495. doi:10.1523/JNEUROSCI.3406-07.2007
Chen, J., and Kriegstein, A. R. (2015). A GABAergic projection from the zona incerta to cortex promotes cortical neuron development. Science 350 (6260), 554–558. doi:10.1126/science.aac6472
Chen, X., and Johnston, D. (2005). Constitutively active G-protein-gated inwardly rectifying K+ channels in dendrites of hippocampal CA1 pyramidal neurons. J. Neurosci. 25 (15), 3787–3792. doi:10.1523/JNEUROSCI.5312-04.2005
Chen, X. Q., Salehi, A., Pearn, M. L., Overk, C., Nguyen, P. D., Kleschevnikov, A. M., et al. (2020). Targeting increased levels of APP in Down syndrome: Posiphen-mediated reductions in APP and its products reverse endosomal phenotypes in the Ts65Dn mouse model. Alzheimers Dement. 17, 271–292. doi:10.1002/alz.12185
Cheng, A., Haydar, T. F., Yarowsky, P. J., and Krueger, B. K. (2004). Concurrent generation of subplate and cortical plate neurons in developing trisomy 16 mouse cortex. Dev. Neurosci. 26 (2-4), 255–265. doi:10.1159/000082142
Chiang, J. C., Jiang, J., Newburger, P. E., and Lawrence, J. B. (2018). Trisomy silencing by XIST normalizes Down syndrome cell pathogenesis demonstrated for hematopoietic defects in vitro. Nat. Commun. 9 (1), 5180. doi:10.1038/s41467-018-07630-y
Clark, S., Schwalbe, J., Stasko, M. R., Yarowsky, P. J., and Costa, A. C. (2006). Fluoxetine rescues deficient neurogenesis in hippocampus of the Ts65Dn mouse model for Down syndrome. Exp. Neurol. 200 (1), 256–261. doi:10.1016/j.expneurol.2006.02.005
Contestabile, A., Fila, T., Ceccarelli, C., Bonasoni, P., Bonapace, L., Santini, D., et al. (2007). Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with Down syndrome and in Ts65Dn mice. Hippocampus 17 (8), 665–678. doi:10.1002/hipo.20308
Contestabile, A., Magara, S., and Cancedda, L. (2017). The GABAergic hypothesis for cognitive disabilities in down syndrome. Front. Cell. Neurosci. 11, 54. doi:10.3389/fncel.2017.00054
Cooper, A., Grigoryan, G., Guy-David, L., Tsoory, M. M., Chen, A., and Reuveny, E. (2012). Trisomy of the G protein-coupled K+ channel gene, Kcnj6, affects reward mechanisms, cognitive functions, and synaptic plasticity in mice. Proc. Natl. Acad. Sci. U. S. A. 109 (7), 2642–2647. doi:10.1073/pnas.1109099109
Cortez, M. A., Shen, L., Wu, Y., Aleem, I. S., Trepanier, C. H., Sadeghnia, H. R., et al. (2009). Infantile spasms and down syndrome: A new animal model. Pediatr. Res. 65 (5), 499–503. doi:10.1203/PDR.0b013e31819d9076
Cramer, N., and Galdzicki, Z. (2012). From abnormal hippocampal synaptic plasticity in down syndrome mouse models to cognitive disability in down syndrome. Neural Plast., 101542. doi:10.1155/2012/101542
Dascal, N., and Kahanovitch, U. (2015). The roles of gβγ and gα in gating and regulation of GIRK channels. Int. Rev. Neurobiol. 123, 27–85. doi:10.1016/bs.irn.2015.06.001
Dierssen, M., Fillat, C., Crnic, L., Arbones, M., Florez, J., and Estivill, X. (2001). Murine models for Down syndrome. Physiol. Behav. 73 (5), 859–871. -6 [pii]. doi:10.1016/s0031-9384(01)00523-6
Dierssen, M., Herault, Y., and Estivill, X. (2009). Aneuploidy: From a physiological mechanism of variance to down syndrome. Physiol. Rev. 89 (3), 887–920. doi:10.1152/physrev.00032.2007
Elens, L., Norman, E., Matic, M., Rane, A., Fellman, V., and van Schaik, R. H. (2016). Genetic predisposition to poor opioid response in preterm infants: Impact of KCNJ6 and COMT polymorphisms on pain relief after endotracheal intubation. Ther. Drug Monit. 38 (4), 525–533. doi:10.1097/FTD.0000000000000301
Engidawork, E., and Lubec, G. (2003). Molecular changes in fetal Down syndrome brain. J. Neurochem. 84 (5), 8951614–8951904. [pii]. doi:10.1046/j.1471-4159.2003.01614.x
Faizi, M., Bader, P. L., Tun, C., Encarnacion, A., Kleschevnikov, A., Belichenko, P., et al. (2011). Comprehensive behavioral phenotyping of Ts65Dn mouse model of down syndrome: Activation of β1-adrenergic receptor by xamoterol as a potential cognitive enhancer. Neurobiol. Dis. 43 (211), 397–413. 00128-8 [pii]. doi:10.1016/j.nbd.2011.04.011
Fernandez, F., Morishita, W., Zuniga, E., Nguyen, J., Blank, M., Malenka, R. C., et al. (2007). Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat. Neurosci. 10 (4), 411–413. doi:10.1038/nn1860
Fernandez-Alacid, L., Watanabe, M., Molnar, E., Wickman, K., and Lujan, R. (2011). Developmental regulation of G protein-gated inwardly-rectifying K+ (GIRK/Kir3) channel subunits in the brain. Eur. J. Neurosci. 34 (11), 1724–1736. doi:10.1111/j.1460-9568.2011.07886.x
Freeburn, A., and Munn, R. G. K. (2021). Signalling pathways contributing to learning and memory deficits in the Ts65Dn mouse model of Down syndrome. Neuronal Signal. 5 (1), NS20200011. doi:10.1042/NS20200011
Garcia-Cerro, S., Rueda, N., Vidal, V., Lantigua, S., and Martinez-Cue, C. (2017). Normalizing the gene dosage of Dyrk1A in a mouse model of Down syndrome rescues several Alzheimer's disease phenotypes. Neurobiol. Dis. 106, 76–88. doi:10.1016/j.nbd.2017.06.010
Golden, J. A., and Hyman, B. T. (1994). Development of the superior temporal neocortex is anomalous in trisomy 21. J. Neuropathol. Exp. Neurol. 53 (5), 513–520. doi:10.1097/00005072-199409000-00011
Gonzalez-Islas, C., and Wenner, P. (2006). Spontaneous network activity in the embryonic spinal cord regulates AMPAergic and GABAergic synaptic strength. Neuron 49 (4), 563–575. doi:10.1016/j.neuron.2006.01.017
Gotti, S., Caricati, E., and Panzica, G. (2011). Alterations of brain circuits in Down syndrome murine models. J. Chem. Neuroanat. 42 (4), 317–326. doi:10.1016/j.jchemneu.2011.09.002
Griguoli, M., and Cherubini, E. (2017). Early correlated network activity in the Hippocampus: Its putative role in shaping neuronal circuits. Front. Cell. Neurosci. 11, 255. doi:10.3389/fncel.2017.00255
Guidi, S., Stagni, F., Bianchi, P., Ciani, E., Giacomini, A., De Franceschi, M., et al. (2014). Prenatal pharmacotherapy rescues brain development in a Down's syndrome mouse model. Brain 137, 380–401. doi:10.1093/brain/awt340
Hamlett, E. D., Boger, H. A., Ledreux, A., Kelley, C. M., Mufson, E. J., Falangola, M. F., et al. (2016). Cognitive impairment, neuroimaging, and alzheimer neuropathology in mouse models of down syndrome. Curr. Alzheimer Res. 13 (1), 35–52. doi:10.2174/1567205012666150921095505
Han, E. B., and Stevens, C. F. (2009). Development regulates a switch between post- and presynaptic strengthening in response to activity deprivation. Proc. Natl. Acad. Sci. U. S. A. 106 (26), 10817–10822. doi:10.1073/pnas.0903603106
Harashima, C., Jacobowitz, D. M., Stoffel, M., Chakrabarti, L., Haydar, T. F., Siarey, R. J., et al. (2006a). Elevated expression of the G-protein-activated inwardly rectifying potassium channel 2 (GIRK2) in cerebellar unipolar brush cells of a Down syndrome mouse model. Cell. Mol. Neurobiol. 26 (4-6), 719–734. doi:10.1007/s10571-006-9066-4
Harashima, C., Jacobowitz, D. M., Witta, J., Borke, R. C., Best, T. K., Siarey, R. J., et al. (2006b). Abnormal expression of the G-protein-activated inwardly rectifying potassium channel 2 (GIRK2) in hippocampus, frontal cortex, and substantia nigra of Ts65Dn mouse: A model of down syndrome. J. Comp. Neurol. 494 (5), 815–833. doi:10.1002/cne.20844
Hasina, Z., Wang, N., and Wang, C. C. (2022). Developmental neuropathology and neurodegeneration of down syndrome: Current knowledge in humans. Front. Cell Dev. Biol. 10, 877711. doi:10.3389/fcell.2022.877711
Haydar, T. F., Blue, M. E., Molliver, M. E., Krueger, B. K., and Yarowsky, P. J. (1996). Consequences of trisomy 16 for mouse brain development: Corticogenesis in a model of down syndrome. J. Neurosci. 16 (19), 6175–6182. doi:10.1523/jneurosci.16-19-06175.1996
Haydar, T. F., Nowakowski, R. S., Yarowsky, P. J., and Krueger, B. K. (2000). Role of founder cell deficit and delayed neuronogenesis in microencephaly of the trisomy 16 mouse. J. Neurosci. 20 (11), 4156–4164. [pii]. doi:10.1523/jneurosci.20-11-04156.2000
Haydar, T. F., and Reeves, R. H. (2012). Trisomy 21 and early brain development. Trends Neurosci. 35 (2), 81–91. doi:10.1016/j.tins.2011.11.001
Heinen, M., Hettich, M. M., Ryan, D. P., Schnell, S., Paesler, K., and Ehninger, D. (2012). Adult-onset fluoxetine treatment does not improve behavioral impairments and may have adverse effects on the Ts65Dn mouse model of Down syndrome. Neural Plast., 467251. doi:10.1155/2012/467251
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., and Kurachi, Y. (2010). Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 90 (1), 291–366. doi:10.1152/physrev.00021.2009
Holtzman, D. M., Santucci, D., Kilbridge, J., Chua-Couzens, J., Fontana, D. J., Daniels, S. E., et al. (1996). Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proc. Natl. Acad. Sci. U. S. A. 93 (23), 13333–13338. doi:10.1073/pnas.93.23.13333
Inanobe, A., Yoshimoto, Y., Horio, Y., Morishige, K. I., Hibino, H., Matsumoto, S., et al. (1999). Characterization of G-protein-gated K+ channels composed of Kir3.2 subunits in dopaminergic neurons of the substantia nigra. J. Neurosci. 19 (3), 1006–1017. doi:10.1523/jneurosci.19-03-01006.1999
Jain, S., Watts, C. A., Chung, W. C. J., and Welshhans, K. (2020). Neurodevelopmental wiring deficits in the Ts65Dn mouse model of Down syndrome. Neurosci. Lett. 714, 134569. doi:10.1016/j.neulet.2019.134569
Jeremic, D., Sanchez-Rodriguez, I., Jimenez-Diaz, L., and Navarro-Lopez, J. D. (2021). Therapeutic potential of targeting G protein-gated inwardly rectifying potassium (GIRK) channels in the central nervous system. Pharmacol. Ther. 223, 107808. doi:10.1016/j.pharmthera.2021.107808
Jiang, J., Jing, Y., Cost, G. J., Chiang, J. C., Kolpa, H. J., Cotton, A. M., et al. (2013). Translating dosage compensation to trisomy 21. Nature 500 (7462), 296–300. doi:10.1038/nature12394
Jiang, X., Liu, C., Yu, T., Zhang, L., Meng, K., Xing, Z., et al. (2015). Genetic dissection of the Down syndrome critical region. Hum. Mol. Genet. 24 (22), 6540–6551. doi:10.1093/hmg/ddv364
Joshi, K., Shen, L., Michaeli, A., Salter, M., Thibault-Messier, G., Hashmi, S., et al. (2016). Infantile spasms in down syndrome: Rescue by knockdown of the GIRK2 channel. Ann. Neurol. 80 (4), 511–521. doi:10.1002/ana.24749
Kamarajan, C., Pandey, A. K., Chorlian, D. B., Manz, N., Stimus, A. T., Edenberg, H. J., et al. (2017). A KCNJ6 gene polymorphism modulates theta oscillations during reward processing. Int. J. Psychophysiol. 115, 13–23. doi:10.1016/j.ijpsycho.2016.12.007
Karschin, C., and Karschin, A. (1997). Ontogeny of gene expression of Kir channel subunits in the rat. Mol. Cell. Neurosci. 10 (3-4), 131–148. doi:10.1006/mcne.1997.0655
Kawatani, K., Nambara, T., Nawa, N., Yoshimatsu, H., Kusakabe, H., Hirata, K., et al. (2021). A human isogenic iPSC-derived cell line panel identifies major regulators of aberrant astrocyte proliferation in Down syndrome. Commun. Biol. 4 (1), 730. doi:10.1038/s42003-021-02242-7
Kerschensteiner, D. (2014). Spontaneous network activity and synaptic development. Neuroscientist 20 (3), 272–290. doi:10.1177/1073858413510044
Kim, C. S., and Johnston, D. (2015). A1 adenosine receptor-mediated GIRK channels contribute to the resting conductance of CA1 neurons in the dorsal hippocampus. J. Neurophysiol. 113 (7), 2511–2523. doi:10.1152/jn.00951.2014
Kirkby, L. A., Sack, G. S., Firl, A., and Feller, M. B. (2013). A role for correlated spontaneous activity in the assembly of neural circuits. Neuron 80 (5), 1129–1144. doi:10.1016/j.neuron.2013.10.030
Kleschevnikov, A. M., Belichenko, P. V., Faizi, M., Jacobs, L. F., Htun, K., Shamloo, M., et al. (2012a). Deficits in cognition and synaptic plasticity in a mouse model of Down syndrome ameliorated by GABAB receptor antagonists. J. Neurosci. 32 (27), 9217–9227. doi:10.1523/JNEUROSCI.1673-12.2012
Kleschevnikov, A. M., Belichenko, P. V., Gall, J., George, L., Nosheny, R., Maloney, M. T., et al. (2012b). Increased efficiency of the GABAA and GABAB receptor-mediated neurotransmission in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 45 (2), 683–691. doi:10.1016/j.nbd.2011.10.009
Kleschevnikov, A. M., Belichenko, P. V., Salehi, A., and Wu, C. (2012c). Discoveries in down syndrome: Moving basic science to clinical care. Prog. Brain Res. 197, 199–221. doi:10.1016/B978-0-444-54299-1.00010-8
Kleschevnikov, A. M., Belichenko, P. V., Villar, A. J., Epstein, C. J., Malenka, R. C., and Mobley, W. C. (2004). Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J. Neurosci. 24 (37), 8153–8160. doi:10.1523/JNEUROSCI.1766-04.2004
Kleschevnikov, A. M., Van Volkinburg, J., Zhan, K., and Mobley, W. C. (2005). Expression of GIRK2 and related proteins after chronic administrations of Prozac in Ts65Dn mice, a genetic model of Down syndrome. Program No. 115.7. Washington, DC: Society for Neuroscience. Abstract Viewer/Itinerary Planner. doi:10.1523/JNEUROSCI.1766-04.2004
Kleschevnikov, A. M., Yu, J., Kim, J., Lysenko, L. V., Zhen, Z., Eugene Yu, Y., et al. (2017). Evidence that increased Kcnj6 gene dose is necessary for deficits in behavior and dentate gyrus synaptic plasticity in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 103, 1–10. doi:10.1016/j.nbd.2017.03.009
Kobayashi, T., Hirai, H., Iino, M., Fuse, I., Mitsumura, K., Washiyama, K., et al. (2009). Inhibitory effects of the antiepileptic drug ethosuximide on G protein-activated inwardly rectifying K+ channels. Neuropharmacology 56 (2), 499–506. doi:10.1016/j.neuropharm.2008.10.003
Kobayashi, T., Washiyama, K., and Ikeda, K. (2003). Inhibition of G protein-activated inwardly rectifying K+ channels by fluoxetine (Prozac). Br. J. Pharmacol. 138 (6), 1119–1128. doi:10.1038/sj.bjp.0705172
Korenberg, J. R., Kawashima, H., Pulst, S. M., Ikeuchi, T., Ogasawara, N., Yamamoto, K., et al. (1990). Molecular definition of a region of chromosome 21 that causes features of the Down syndrome phenotype. Am. J. Hum. Genet. 47 (2), 236–246.
Kulik, A., Vida, I., Fukazawa, Y., Guetg, N., Kasugai, Y., Marker, C. L., et al. (2006). Compartment-dependent colocalization of Kir3.2-containing K+ channels and GABAB receptors in hippocampal pyramidal cells. J. Neurosci. 26 (16), 4289–4297. doi:10.1523/JNEUROSCI.4178-05.2006
Langford, D. J., Paul, S. M., West, C. M., Dunn, L. B., Levine, J. D., Kober, K. M., et al. (2015). Variations in potassium channel genes are associated with distinct trajectories of persistent breast pain after breast cancer surgery. Pain 156 (3), 371–380. doi:10.1097/01.j.pain.0000460319.87643.11
Lejeune, J., Turpin, R., and Gautier, M. (1959). Mongolism; a chromosomal disease (trisomy). Bull. Acad. Natl. Med. 143 (11-12), 256–265.
Leresche, N., and Lambert, R. C. (2017). T-type calcium channels in synaptic plasticity. Channels (Austin) 11 (2), 121–139. doi:10.1080/19336950.2016.1238992
Levav, T., Saar, T., Berkovich, L., and Golan, H. (2004). Perinatal exposure to GABA-transaminase inhibitor impaired psychomotor function in the developing and adult mouse. Int. J. Dev. Neurosci. 22 (3), 137–147. doi:10.1016/j.ijdevneu.2004.03.004
Levav, T., Wirthaim, O., Weiss, R., Grossman, Y., and Golan, H. (2008). Impaired synaptogenesis and long-term modulation of behavior following postnatal elevation of GABA levels in mice. Neuropharmacology 54 (2), 387–398. doi:10.1016/j.neuropharm.2007.10.012
Levav-Rabkin, T., Melamed, O., Clarke, G., Farber, M., Cryan, J. F., Dinan, T. G., et al. (2010). A sensitive period of mice inhibitory system to neonatal GABA enhancement by vigabatrin is brain region dependent. Neuropsychopharmacology 35 (5), 1138–1154. doi:10.1038/npp.2009.219
Li, L. B., Chang, K. H., Wang, P. R., Hirata, R. K., Papayannopoulou, T., and Russell, D. W. (2012). Trisomy correction in Down syndrome induced pluripotent stem cells. Cell Stem Cell 11 (5), 615–619. doi:10.1016/j.stem.2012.08.004
Liu, C., Belichenko, P. V., Zhang, L., Fu, D., Kleschevnikov, A. M., Baldini, A., et al. (2011). Mouse models for down syndrome-associated developmental cognitive disabilities. Dev. Neurosci. 33, 404–413. [pii]. doi:10.1159/000329422
Lorenzi, H. A., and Reeves, R. H. (2006). Hippocampal hypocellularity in the Ts65Dn mouse originates early in development. Brain Res. 1104 (1), 153–159. doi:10.1016/j.brainres.2006.05.022
Lott, I. T. (2012). Neurological phenotypes for Down syndrome across the life span. Prog. Brain Res. 197, 101–121. doi:10.1016/B978-0-444-54299-1.00006-6
Luhmann, H. J., Sinning, A., Yang, J. W., Reyes-Puerta, V., Stuttgen, M. C., Kirischuk, S., et al. (2016). Spontaneous neuronal activity in developing neocortical networks: From single cells to large-scale interactions. Front. Neural Circuits 10, 40. doi:10.3389/fncir.2016.00040
Luscher, C., Jan, L. Y., Stoffel, M., Malenka, R. C., and Nicoll, R. A. (1997). G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19 (3), 687–695. 5 [pii]. doi:10.1016/s0896-6273(00)80381-5
Lysenko, L. V., Kim, J., Henry, C., Tyrtyshnaia, A., Kohnz, R. A., Madamba, F., et al. (2014). Monoacylglycerol lipase inhibitor JZL184 improves behavior and neural properties in Ts65Dn mice, a model of down syndrome. PLoS One 9 (12), e114521. doi:10.1371/journal.pone.0114521
Lysenko, L. V., Kim, J., Madamba, F., Tyrtyshnaia, A. A., Ruparelia, A., and Kleschevnikov, A. M. (2018). Developmental excitatory-to-inhibitory GABA polarity switch is delayed in Ts65Dn mice, a genetic model of Down syndrome. Neurobiol. Dis. 115, 1–8. doi:10.1016/j.nbd.2018.03.005
Madamba, F. , B. K., L., and Kleschevnikov, A. M. (2021). Spontaneous neuronal activity is reduced during the ‘vulnerable’ period of nascent neural circuit formation in the hippocampus of Ts65Dn mice, a genetic model of Down syndrome. Chicago: Society for Neuroscience Annual Meeting. Abstract P029.06.
Marin-Padilla, M. (1976). Pyramidal cell abnormalities in the motor cortex of a child with Down's syndrome. A Golgi study. J. Comp. Neurol. 167 (1), 63–81. doi:10.1002/cne.901670105
Marron Fernandez de Velasco, E., Zhang, L., , B, N. V., Tipps, M., Farris, S., Xia, Z., et al. (2017). GIRK2 splice variants and neuronal G protein-gated K+ channels: Implications for channel function and behavior. Sci. Rep. 7 (1), 1639. doi:10.1038/s41598-017-01820-2
Martinez-Cue, C., Delatour, B., and Potier, M. C. (2014). Treating enhanced GABAergic inhibition in down syndrome: Use of GABA α5-selective inverse agonists. Neurosci. Biobehav. Rev. 46 Pt 2, 218–227. doi:10.1016/j.neubiorev.2013.12.008
Martinez-Cue, C., Martinez, P., Rueda, N., Vidal, R., Garcia, S., Vidal, V., et al. (2013). Reducing GABAA α5 receptor-mediated inhibition rescues functional and neuromorphological deficits in a mouse model of down syndrome. J. Neurosci. 33 (9), 3953–3966. doi:10.1523/JNEUROSCI.1203-12.2013
Masotti, A., Uva, P., Davis-Keppen, L., Basel-Vanagaite, L., Cohen, L., Pisaneschi, E., et al. (2015). Keppen-Lubinsky syndrome is caused by mutations in the inwardly rectifying K+ channel encoded by KCNJ6. Am. J. Hum. Genet. 96 (2), 295–300. doi:10.1016/j.ajhg.2014.12.011
Mayfield, J., Blednov, Y. A., and Harris, R. A. (2015). Behavioral and genetic evidence for GIRK channels in the CNS: Role in physiology, pathophysiology, and drug addiction. Int. Rev. Neurobiol. 123, 279–313. doi:10.1016/bs.irn.2015.05.016
McCormick, M. K., Schinzel, A., Petersen, M. B., Stetten, G., Driscoll, D. J., Cantu, E. S., et al. (1989). Molecular genetic approach to the characterization of the "Down syndrome region" of chromosome 21. Genomics 5 (2), 325–331. doi:10.1016/0888-7543(89)90065-7
Melamed, O., Levav-Rabkin, T., Zukerman, C., Clarke, G., Cryan, J. F., Dinan, T. G., et al. (2014). Long-lasting glutamatergic modulation induced by neonatal GABA enhancement in mice. Neuropharmacology 79, 616–625. doi:10.1016/j.neuropharm.2013.12.015
Mohler, H. (2012). Cognitive enhancement by pharmacological and behavioral interventions: The murine down syndrome model. Biochem. Pharmacol. 84 (8), 994–999. doi:10.1016/j.bcp.2012.06.028
Mojabi, F. S., Fahimi, A., Moghadam, S., Moghadam, S., Windy McNerneny, M., Ponnusamy, R., et al. (2016). GABAergic hyperinnervation of dentate granule cells in the Ts65Dn mouse model of down syndrome: Exploring the role of App. Hippocampus 26 (12), 1641–1654. doi:10.1002/hipo.22664
Murthy, V. N., Schikorski, T., Stevens, C. F., and Zhu, Y. (2001). Inactivity produces increases in neurotransmitter release and synapse size. Neuron 32 (4), 673–682. doi:10.1016/s0896-6273(01)00500-1
Nehra, S., Sharma, V., Singh, M., Singhal, P., and Singhal, N. (2022). Generation of integration free hiPSCs clones, NSi001-A, NSi001-B, and NSi001-C from peripheral blood mononuclear cells of an individual with down syndrome having Robertsonian translocation. Stem Cell Res. 61, 102771. doi:10.1016/j.scr.2022.102771
Nishizawa, D., Fukuda, K., Kasai, S., Ogai, Y., Hasegawa, J., Sato, N., et al. (2014). Association between KCNJ6 (GIRK2) gene polymorphism rs2835859 and post-operative analgesia, pain sensitivity, and nicotine dependence. J. Pharmacol. Sci. 126 (3), 253–263. doi:10.1254/jphs.14189fp
Olmos-Serrano, J. L., Tyler, W. A., Cabral, H. J., and Haydar, T. F. (2016). Longitudinal measures of cognition in the Ts65Dn mouse: Refining windows and defining modalities for therapeutic intervention in Down syndrome. Exp. Neurol. 279, 40–56. doi:10.1016/j.expneurol.2016.02.005
Ozaki, M., Hashikawa, T., Ikeda, K., Miyakawa, Y., Ichikawa, T., Ishihara, Y., et al. (2002). Degeneration of pontine mossy fibres during cerebellar development in weaver mutant mice. Eur. J. Neurosci. 16 (4), 565–574. doi:10.1046/j.1460-9568.2002.02111.x
Patil, N., Cox, D. R., Bhat, D., Faham, M., Myers, R. M., and Peterson, A. S. (1995). A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat. Genet. 11 (2), 126–129. doi:10.1038/ng1095-126
Pelleri, M. C., Cicchini, E., Locatelli, C., Vitale, L., Caracausi, M., Piovesan, A., et al. (2016). Systematic reanalysis of partial trisomy 21 cases with or without Down syndrome suggests a small region on 21q22.13 as critical to the phenotype. Hum. Mol. Genet. 25 (12), 2525–2538. doi:10.1093/hmg/ddw116
Pelleri, M. C., Cicchini, E., Petersen, M. B., Tranebjaerg, L., Mattina, T., Magini, P., et al. (2019). Partial trisomy 21 map: Ten cases further supporting the highly restricted Down syndrome critical region (HR-DSCR) on human chromosome 21. Mol. Genet. Genomic Med. 7 (8), e797. doi:10.1002/mgg3.797
Peterson, A., Patil, N., Robbins, C., Wang, L., Cox, D. R., and Myers, R. M. (1994). A transcript map of the Down syndrome critical region on chromosome 21. Hum. Mol. Genet. 3 (10), 1735–1742. doi:10.1093/hmg/3.10.1735
Plona, K., Kim, T., Halloran, K., and Wynshaw-Boris, A. (2016). Chromosome therapy: Potential strategies for the correction of severe chromosome aberrations. Am. J. Med. Genet. C Semin. Med. Genet. 172 (4), 422–430. doi:10.1002/ajmg.c.31530
Ponnusamy, R., and Pradhan, N. (2006). The effects of chronic administration of ethosuximide on learning and memory: A behavioral and biochemical study on nonepileptic rats. Behav. Pharmacol. 17 (7), 573–580. doi:10.1097/01.fbp.0000236268.79923.18
Popov, V. I., Kleschevnikov, A. M., Klimenko, O. A., Stewart, M. G., and Belichenko, P. V. (2011). Three-dimensional synaptic ultrastructure in the dentate gyrus and hippocampal area CA3 in the Ts65Dn mouse model of Down syndrome. J. Comp. Neurol. 519 (7), 1338–1354. doi:10.1002/cne.22573
Rafii, M. S., Kleschevnikov, A. M., Sawa, M., and Mobley, W. C. (2019). Down syndrome. Handb. Clin. Neurol. 167, 321–336. doi:10.1016/B978-0-12-804766-8.00017-0
Real, R., Peter, M., Trabalza, A., Khan, S., Smith, M. A., Dopp, J., et al. (2018). In vivo modeling of human neuron dynamics and Down syndrome. Science 362 (6416), eaau1810. doi:10.1126/science.aau1810
Reeves, R. H., Delabar, J., Potier, M. C., Bhattacharyya, A., Head, E., Lemere, C., et al. (2019). Paving the way for therapy: The second international conference of the trisomy 21 research society. Mol. Syndromol. 9 (6), 279–286. doi:10.1159/000494231
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397 (6716), 251–255. doi:10.1038/16697
Roncace, V., Burattini, C., Stagni, F., Guidi, S., Giacomini, A., Emili, M., et al. (2017). Neuroanatomical alterations and synaptic plasticity impairment in the perirhinal cortex of the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 106, 89–100. doi:10.1016/j.nbd.2017.06.017
Rueda, N., Florez, J., and Martinez-Cue, C. (2012). Mouse models of Down syndrome as a tool to unravel the causes of mental disabilities. Neural Plast. 2012, 584071. doi:10.1155/2012/584071
Ruparelia, A., Pearn, M. L., and Mobley, W. C. (2012). Cognitive and pharmacological insights from the Ts65Dn mouse model of Down syndrome. Curr. Opin. Neurobiol. 22 (5), 880–886. doi:10.1016/j.conb.2012.05.002
Saber, M. M., Karimiavargani, M., Uzawa, T., Hettiarachchi, N., Hamada, M., Ito, Y., et al. (2021). Possible roles for the hominoid-specific DSCR4 gene in human cells. Genes Genet. Syst. 96 (1), 1–11. doi:10.1266/ggs.20-00012
Sakai, N., and Tanaka, C. (1993). Inhibitory modulation of long-term potentiation via the 5-HT1A receptor in slices of the rat hippocampal dentate gyrus. Brain Res. 613 (293), 326–330. 90921-9 [pii]. doi:10.1016/0006-8993(93)90921-9
Salehi, A., Delcroix, J. D., Belichenko, P. V., Zhan, K., Wu, C., Valletta, J. S., et al. (2006). Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51 (1), 29–42. doi:10.1016/j.neuron.2006.05.022
Salehi, A., Faizi, M., Colas, D., Valletta, J., Laguna, J., Takimoto-Kimura, R., et al. (2009). Restoration of norepinephrine-modulated contextual memory in a mouse model of Down syndrome. Sci. Transl. Med. 1 (7), 7ra17. doi:10.1126/scitranslmed.3000258
Schein, J. C., Wang, J. K., and Roffler-Tarlov, S. K. (2005). The effect of GIRK2(wv) on neurite growth, protein expression, and viability in the CNS-derived neuronal cell line, CATH.A-differentiated. Neuroscience 134 (1), 21–32. doi:10.1016/j.neuroscience.2005.03.043
Schmidt-Sidor, B., Wisniewski, K. E., Shepard, T. H., and Sersen, E. A. (1990). Brain growth in Down syndrome subjects 15 to 22 weeks of gestational age and birth to 60 months. Clin. Neuropathol. 9 (4), 181–190.
Sekiguchi, M., Nowakowski, R. S., Nagato, Y., Tanaka, O., Guo, H., Madoka, M., et al. (1995). Morphological abnormalities in the hippocampus of the weaver mutant mouse. Brain Res. 696 (1-2), 262–267. doi:10.1016/0006-8993(95)00974-u
Sharma, V., Nehra, S., and Singhal, N. (2022). Generation of AAVS1-EGFP reporter cell lines from an isogenic pair of trisomy 21 and euploid human iPSCs. Stem Cell Res. 64, 102890. doi:10.1016/j.scr.2022.102890
Stagni, F., Giacomini, A., Guidi, S., Ciani, E., Ragazzi, E., Filonzi, M., et al. (2015). Long-term effects of neonatal treatment with fluoxetine on cognitive performance in Ts65Dn mice. Neurobiol. Dis. 74, 204–218. doi:10.1016/j.nbd.2014.12.005
Sturgeon, X., Le, T., Ahmed, M. M., and Gardiner, K. J. (2012). Pathways to cognitive deficits in Down syndrome. Prog. Brain Res. 197, 73–100. doi:10.1016/B978-0-444-54299-1.00005-4
Sylvester, P. E. (1983). The hippocampus in Down's syndrome. J. Ment. Defic. Res. 27, 227–236. doi:10.1111/j.1365-2788.1983.tb00294.x
Takashima, S., Ieshima, A., Nakamura, H., and Becker, L. E. (1989). Dendrites, dementia and the Down syndrome. Brain Dev. 11 (2), 131–133. doi:10.1016/s0387-7604(89)80082-8
Tang, X. Y., Xu, L., Wang, J., Hong, Y., Wang, Y., Zhu, Q., et al. (2021). DSCAM/PAK1 pathway suppression reverses neurogenesis deficits in iPSC-derived cerebral organoids from patients with Down syndrome. J. Clin. Invest. 131 (12), 135763. doi:10.1172/JCI135763
Tayebati, S. K., Cecchi, A., Martinelli, I., Carboni, E., and Amenta, F. (2019). Pharmacotherapy of down's syndrome: When and which? CNS Neurol. Disord. Drug Targets 18 (10), 750–757. doi:10.2174/1871527318666191114092924
Thiery, E., Thomas, S., Vacher, S., Delezoide, A. L., Delabar, J. M., and Creau, N. (2003). Chromosome 21 KIR channels in brain development. J. Neural Transm. Suppl. Suppl (67), 105–115. doi:10.1007/978-3-7091-6721-2_9
Tien, N. W., and Kerschensteiner, D. (2018). Homeostatic plasticity in neural development. Neural Dev. 13 (1), 9. doi:10.1186/s13064-018-0105-x
Toso, L., Cameroni, I., Roberson, R., Abebe, D., Bissell, S., and Spong, C. Y. (2008). Prevention of developmental delays in a Down syndrome mouse model. Obstet. Gynecol. 112 (6), 1242–1251. doi:10.1097/AOG.0b013e31818c91dc
Toyoda, A., Noguchi, H., Taylor, T. D., Ito, T., Pletcher, M. T., Sakaki, Y., et al. (2002). Comparative genomic sequence analysis of the human chromosome 21 Down syndrome critical region. Genome Res. 12 (9), 1323–1332. doi:10.1101/gr.153702
Turrigiano, G. G., and Nelson, S. B. (2004). Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 5 (2), 97–107. doi:10.1038/nrn1327
Tyzio, R., Represa, A., Jorquera, I., Ben-Ari, Y., Gozlan, H., and Aniksztejn, L. (1999). The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of the apical dendrite. J. Neurosci. 19 (23), 10372–10382. doi:10.1523/jneurosci.19-23-10372.1999
Uguagliati, B., Stagni, F., Emili, M., Giacomini, A., Russo, C., Guidi, S., et al. (2022). Early appearance of dendritic alterations in neocortical pyramidal neurons of the Ts65Dn model of down syndrome. Dev. Neurosci. 44 (1), 23–38. doi:10.1159/000520925
Utagawa, E. C., Moreno, D. G., Schafernak, K. T., Arva, N. C., Malek-Ahmadi, M. H., Mufson, E. J., et al. (2022). Neurogenesis and neuronal differentiation in the postnatal frontal cortex in Down syndrome. Acta Neuropathol. Commun. 10 (1), 86. doi:10.1186/s40478-022-01385-w
Vidal, V., Garcia, S., Martinez, P., Corrales, A., Florez, J., Rueda, N., et al. (2012). Lack of behavioral and cognitive effects of chronic ethosuximide and gabapentin treatment in the Ts65Dn mouse model of Down syndrome. Neuroscience 220, 158–168. doi:10.1016/j.neuroscience.2012.06.031
Wang, W., Whorton, M. R., and MacKinnon, R. (2014). Quantitative analysis of mammalian GIRK2 channel regulation by G proteins, the signaling lipid PIP2 and Na+ in a reconstituted system. Elife 3, e03671. doi:10.7554/eLife.03671
Wang, X., Zhao, Y., Zhang, X., Badie, H., Zhou, Y., Mu, Y., et al. (2013). Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down's syndrome. Nat. Med. 19 (4), 473–480. doi:10.1038/nm.3117
Wefelmeyer, W., Puhl, C. J., and Burrone, J. (2016). Homeostatic plasticity of subcellular neuronal structures: From inputs to outputs. Trends Neurosci. 39 (10), 656–667. doi:10.1016/j.tins.2016.08.004
Wei, J., Hodes, M. E., Piva, R., Feng, Y., Wang, Y., Ghetti, B., et al. (1998). Characterization of murine Girk2 transcript isoforms: Structure and differential expression. Genomics 51 (3), 379–390. doi:10.1006/geno.1998.5369
Weick, J. P., Kang, H., Bonadurer, G. F., and Bhattacharyya, A. (2016). Gene expression studies on human trisomy 21 iPSCs and neurons: Towards mechanisms underlying down's syndrome and early alzheimer's disease-like pathologies. Methods Mol. Biol. 1303, 247–265. doi:10.1007/978-1-4939-2627-5_15
Whorton, M. R., and MacKinnon, R. (2011). Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147 (1), 199–208. doi:10.1016/j.cell.2011.07.046
Xu, L., Huo, H. Q., Lu, K. Q., Tang, X. Y., Hong, Y., Han, X., et al. (2022). Abnormal mitochondria in Down syndrome iPSC-derived GABAergic interneurons and organoids. Biochim. Biophys. Acta. Mol. Basis Dis. 1868 (6), 166388. doi:10.1016/j.bbadis.2022.166388
Yamada, M., Inanobe, A., and Kurachi, Y. (1998). G protein regulation of potassium ion channels. Pharmacol. Rev. 50 (4), 723–760.
Yu, T., Liu, C., Belichenko, P., Clapcote, S. J., Li, S., Pao, A., et al. (2010). Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Res. 1366, 162–171. doi:10.1016/j.brainres.2010.09.107
Zhang, L., Fu, D., Belichenko, P. V., Liu, C., Kleschevnikov, A. M., Pao, A., et al. (2012). Genetic analysis of Down syndrome facilitated by mouse chromosome engineering. Bioeng. Bugs 3 (1), 8–12. [pii]. doi:10.4161/bbug.3.1.17696
Zhang, L., Meng, K., Jiang, X., Liu, C., Pao, A., Belichenko, P. V., et al. (2014). Human chromosome 21 orthologous region on mouse chromosome 17 is a major determinant of Down syndrome-related developmental cognitive deficits. Hum. Mol. Genet. 23 (3), 578–589. doi:10.1093/hmg/ddt446
Ziegler, G. C., Roser, C., Renner, T., Hahn, T., Ehlis, A. C., Weber, H., et al. (2020). KCNJ6 variants modulate reward-related brain processes and impact executive functions in attention-deficit/hyperactivity disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 183 (5), 247–257. doi:10.1002/ajmg.b.32734
Keywords: KCNJ6, early postnatal development, miR-155, depolarizing GABA, developmental GABA switch, neuronal excitability, gabaergic neurotransmission, synaptogenesis
Citation: Kleschevnikov AM (2022) Enhanced GIRK2 channel signaling in Down syndrome: A feasible role in the development of abnormal nascent neural circuits. Front. Genet. 13:1006068. doi: 10.3389/fgene.2022.1006068
Received: 28 July 2022; Accepted: 24 August 2022;
Published: 12 September 2022.
Edited by:
Nishant Singhal, National Centre for Cell Science, IndiaReviewed by:
Tarik Haydar, Children’s National Hospital, United StatesMiguel Angel Cortez, University of Toronto, Canada
Copyright © 2022 Kleschevnikov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander M. Kleschevnikov, YWtsZXNjaGV2bmlrb3ZAdWNzZC5lZHU=