 Hao Wan
Hao Wan Jina Zhang2
Jina Zhang2- 1Institute of Advanced Cross-field Science, College of Life Science, Qingdao University, Qingdao, China
- 2Geneis (Beijing) Co., Ltd., Beijing, China
- 3Yangtze Delta Region Institute (Quzhou), University of Electronic Science and Technology of China, Quzhou, China
- 4Beidahuang Industry Group General Hospital, Harbin, China
Immunoglobulins have a pivotal role in disease regulation. Therefore, it is vital to accurately identify immunoglobulins to develop new drugs and research related diseases. Compared with utilizing high-dimension features to identify immunoglobulins, this research aimed to examine a method to classify immunoglobulins and non-immunoglobulins using two features, FC* and GC*. Classification of 228 samples (109 immunoglobulin samples and 119 non-immunoglobulin samples) revealed that the overall accuracy was 80.7% in 10-fold cross-validation using the J48 classifier implemented in Weka software. The FC* feature identified in this study was found in the immunoglobulin subtype domain, which demonstrated that this extracted feature could represent functional and structural properties of immunoglobulins for forecasting.
1 Introduction
Immunoglobulins, or antibodies, are a group of proteins secreted by B lymphocytes that recognize invading antigens and bind to antigens with high affinity and specificity to neutralize toxic substances. In general, antibodies are composed of two identical polypeptide chains, each with a light chain and a heavy chain (Narciso et al., 2011). They can be divided functionally into variable (V) domains, which bind to antigens, and constant (C) domains, which activate, complement, or bind to Fc receptors (Schroeder and Cavacini, 2010). To predict the structure of immunoglobulins, (Lepore et al., 2017) developed the PIGSPro Server, an updated version of the popular PIGS Server.
Immunoglobulins have a pivotal role in disease regulation. Therefore, human and nonhuman polyclonal immunoglobulins have been used in therapeutics for many years. Five monoclonal immunoglobulins ranked in the top 10 blockbuster biotherapeutics drugs (Norman et al., 2020). Patients with primary immune deficiencies greatly benefit from the intravenous or subcutaneous administration of human immunoglobulin preparations (Perez et al., 2017). The advanced development of medicine is urged by its finite supply, which requires more identification of valuable therapeutic immunoglobulins. However, biochemical experiments are time-consuming with enzymes to fragment immunoglobulin molecules (Schroeder and Cavacini, 2010) or X-ray crystallography to obtain accurate structures (Narciso et al., 2011).
Machine learning can identify desired proteins from a large number of sequences within a short time to guide the experimental discovery process (Guo et al., 2020; Liu et al., 2020; Song G. et al., 2021; Cheng et al., 2021; Deng et al., 2021; Dong et al., 2021; Guo et al., 2021; Tang et al., 2021; Yu et al., 2021; Zhao et al., 2021). Over the past decades, researchers have developed many machine learning–based techniques for protein sequence analysis (Zhai et al., 2020; Zeng et al., 2020; Chen et al., 2021; Li et al., 2021). The bioinformatics approach of identifying immunoglobulins is to convert protein sequences into numerical vectors to reveal the internal structures of proteins. The critical aspects of protein identification can be listed as follows: feature extraction, feature selection, and machine learning. Feature extraction methods include n-gram feature type: amino acid composition (AAC), Dipeptides (Dip), Tripeptides, where frequencies of n-length peptides are used as feature vectors (Ding et al., 2011; Gautam et al., 2013; Diener et al., 2016; Rahman et al., 2018; Liu et al., 2019; Lv et al., 2019; Fu et al., 2020; Wang H. et al., 2021; Wang J. et al., 2021; Zhai et al., 2020; Shao and Liu, 2021; Yang et al., 2021; Zhang et al., 2021). In addition, pseudo–amino acid composition (PseAAC) is also a widely adopted feature extraction method, including physicochemical properties between residues (Hansen et al., 2008; Sanders et al., 2011; Gautam et al., 2013; Chen et al., 2016; Diener et al., 2016; Khan et al., 2020; Awais et al., 2021; Naseer et al., 2021).
Many feature types and complex classification methods may generate redundant information (Song B. et al., 2021). Therefore, some studies began to eliminate redundant parts to improve the predictive performance of classification models. This process is also called feature selection. MRMD (Zou et al., 2016; Ao et al., 2020; Li et al., 2020a; Li et al., 2020b; Meng et al., 2020) and ANOVA (Anderson, 2001; Lv et al., 2019) are standard feature selection methods. For optimal feature identification, (Feng et al., 2021) uses the PCA and MCE methods to make the features orthogonal and obtain the core feature set with the minimum 10-dimensional attributes for PPR gene identification and realized 97.9% accuracy. (Li et al., 2020b) used a 19-dimensional feature model to classify anticancer peptide sequences. (Ao et al., 2020) used a 10-dimensional feature model to classify antioxidant proteins and realized 90.44% accuracy. (Meng et al., 2020) used a 6-dimensional feature model to classify cell wall lytic enzymes.
However, very few tools have been developed for immunoglobulin identification. (Tang et al., 2016) used the pseudo amino acid composition (PseAAC) feature extraction approach to realize over 96% prediction accuracy in their pioneering work on immunoglobulin identification. (Gong et al., 2021) used the CC–PSSM and monoTriKGap feature extraction, MRMD feature selection, and single dimension reduction methods to realize 92.1% immunoglobulin identification accuracy by two-dimensional features. However, the link between optimal features and functional structures of immunoglobulins remains to be investigated.
To obtain a diverse feature set, this study integrated 188-D physicochemical properties, auto-cross covariance (ACC) information, and dipeptide compositions of reduced amino acids. Dimensions were reduced using the max-relevance-max-distance (MRMD) method and the single dimension reduction method. The RF and J48 classifiers implemented in Weka software were used to identify immunoglobulins. Finally, two features can correctly predict immunoglobulins, FC* and GC*. The entire modeling process is illustrated in Figure 1. The FC* feature identified in this study was found in immunoglobulin subtype domain IPR003599, which demonstrated that this extracted feature could represent functional and structural properties of immunoglobulins for forecasting.
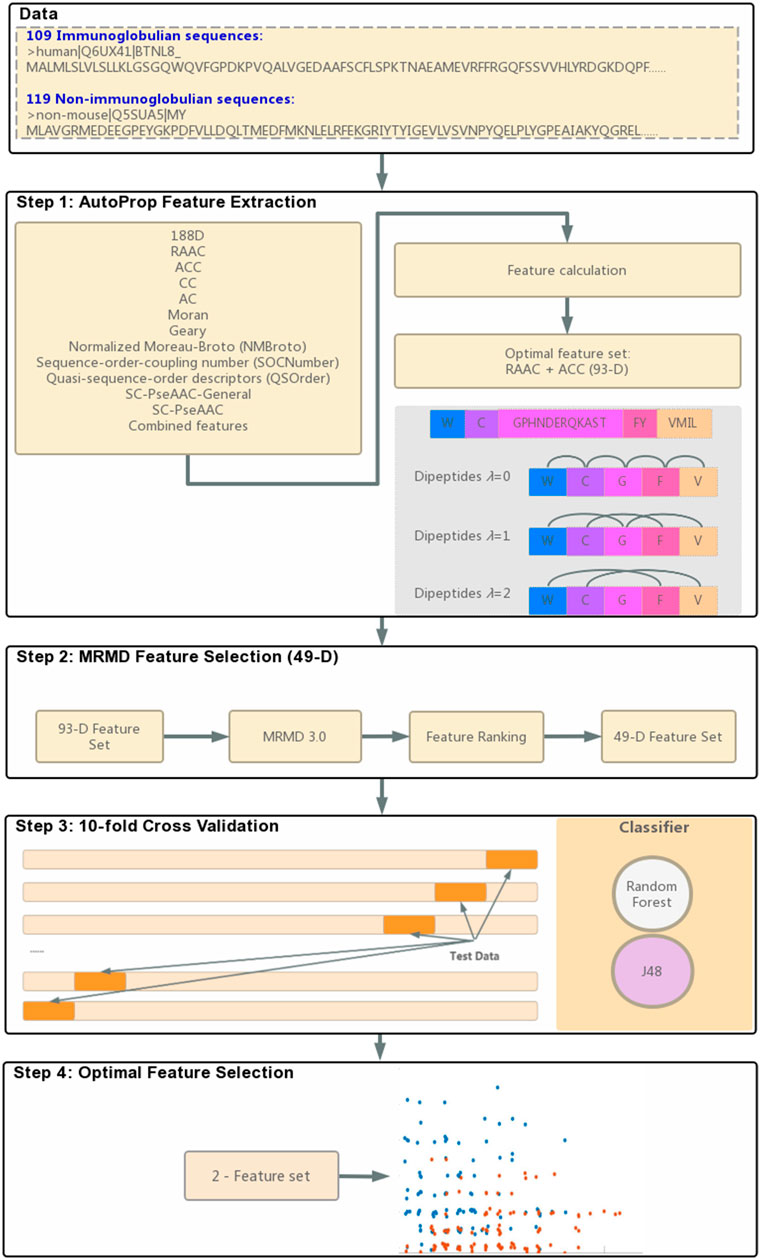
FIGURE 1. Flowchart of identifying immunoglobulins.
2 Materials and Methods
2.1 Datasets
Data for this study were collected by (Tang et al., 2016), which contain 228 samples (109 immunoglobulin samples and 119 non-immunoglobulin samples) extracted from the Universal Protein Resource (UniProt).
2.2 RAAC
Polypeptide chains fold to tertiary structures based on the physicochemical properties of residues (Tang et al., 2016). Analyzing the occurrence frequency of residue compositions cannot visualize three-dimensional protein structures. The reduced amino acid cluster (RAAC) method, replacing protein sequences with less than 20 amino acid alphabets based on a specific reducing scheme, can reduce sequence complexity. With removing non-essential information, functionally conserved regions will be displayed more clearly. Recent work presented 3D protein structures of ectonucleotide pyrophosphatase with a 1D view using the RAAC method (Solis, 2015; Zheng et al., 2019).
There are many choices of reduced schemes, and different decisions could produce distinctive protein classification results. For example, the RAACBook web server provided 74 types of reduced amino acid alphabets derived from over 1,000 published articles in PubMed (Zheng et al., 2019). Bins within the scheme are related to the chemical properties of amino acids. Dayhoff classes (AGPST, DENQ, HKR, ILMV, FWY, and C) are most used. Also, S and T are frequently together, and so are K and R, D, and E (Susko and Roger, 2007).
We used the AutoProp (Feng et al., 2020) to screen out the optimal reduced scheme of the immunoglobulin and non-immunoglobulin sequences. GPHNDERQKAST, FY, VMIL, C, and W (Figure 1 Step 1) were used. Under this reduced scheme, the 20 amino acid alphabets were represented by five alphabets: G, F, V, C, and W. For instance, any amino acid that is a G, P, H, N, D, E, R, Q, K, A, S, or T is then treated as character G. For any amino acid F and Y, it is then treated as character F, and so forth.
2.3 Feature Extraction
A sequence can be represented by sequential form and discrete form. Homolog sequences can be compared with the BLAST or FASTA program benchmark datasets for traditional sequence comparison methods. However, the similarity-based way is unsuitable for distantly related sequences (Wei et al., 2014; Chen et al., 2016; Jin et al., 2019; Manavalan et al., 2019; Hong et al., 2020; Tang et al., 2020; Wang et al., 2020; Ding et al., 2021a; Ding et al., 2021b; Huang et al., 2021; Shao et al., 2021). By converting amino acid codes to a series of discrete numerical vectors, the discrete form can overcome this drawback and be used by machine learning for protein classification. Sometimes, proteins can be classified according to fewer features, while BLAST cannot.
Different numerical values of protein codes mean different feature descriptors. Feature descriptors provided by AutoProp include 188D, ACC, PseAAC, and another nine methods (Figure 1 Step 1). Also, AutoProp provides combined features between those methods. The built-in classifiers will then calculate the accuracy percentage of each feature and decide the optimal feature.
For our data, the optimal feature is the combined features of RAAC and ACC. RAAC features also represent dipeptides of reduced amino acid, like CV, C*V (λ-gap = 1), and C**V (λ-gap = 2). The following formula was used to calculate the values of those features:
where λ = 0,1,2, and
ACC means the autocross covariance (ACC) transformation and contains auto covariance (AC) and cross-covariance (CC) and is introduced to transform protein sequences into fixed-length vectors (Feng et al., 2020). With its ability to identify sequence homologies, ACC has been successfully used for protein family classification and protein interaction prediction (Dong et al., 2009).
2.4 MRMD
The main disadvantage of the sequence word frequency vector is that they are usually huge. Therefore, dimension reduction, also called feature selection, is chosen for protein classification. The MRMD method, which is the max-relevance-max-distance–based dimensionality reduction method, is more considered for relationships among features and stability of feature selection. Cross-validation and the ROC curve are usually used to evaluate classification accuracy. The MRMD method can reduce feature dimensions with few accuracy drops (Zou et al., 2016; He et al., 2020; Tao et al., 2020).
2.5 Performance Measurement
We used three metrics to evaluate model performance. Indicators include sensitivity (SE), specificity (SP), and Accuracy (Jiang et al., 2013; Wang X. et al., 2021). Calculation methods are described as follows:
where TN, TP, FN, and FP refer to the numbers of correctly predicted non-immunoglobulin proteins, correctly predicted immunoglobulin proteins, incorrectly predicted non-immunoglobulin proteins, and incorrectly predicted immunoglobulin proteins, respectively. Sensitivity (SE) is also known as recall, and it measures the percentage that positive samples can be expected correctly over all the samples. SP indicators measure the probability of negative samples classified as non-immunoglobulins, and Accuracy is used to evaluate the overall performance of a prediction model.
3 Results and Discussion
3.1 Classification Results Under Different Features
Props returned 93D best features, and the frequency of dipeptides (λ-gap = 0, 1, 2) is saved in features 1–75, followed by 18 ACC features. The classification accuracy was 92.1% in the RF classifier and 10-fold cross-validation using Weka software. The MRMD method further reduced the dimension to 49D, and accuracy was 91.7% using the same classifier. It can be seen that MRMD reduces nearly half of the feature dimension, but the accuracy is only dropped by 0.4% (Figure 2). After continuous attempts to reduce features, the optimal two features (GC* and FC*) are finally obtained; the classification accuracy was 80.3% using the J48 classifier in Weka.
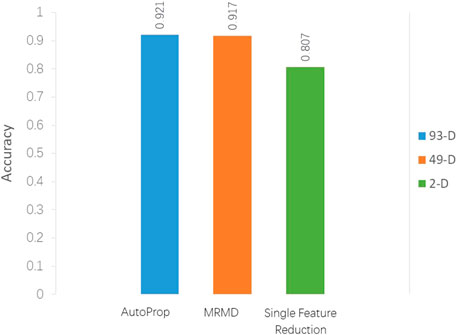
FIGURE 2. Classification accuracy comparison between models with different feature selection methods.
3.2 2D Features Scatter Distribution
Figure 3 shows the scatter plot of GC* and FC* features. What stands out in Figure 3 is that immunoglobulin and non-immunoglobulin samples can be distinguished. Immunoglobulins are scattered on the upper left with higher FC* values, and non-immunoglobulins are found in the lower right with higher GC* values. For 118 out of 119 non-immunoglobulin samples, the FC* value is equal to or less than 5. Among these, the FC* value of 49 samples is zero. The GC* value for immunoglobulin samples is less than or equal to 12.
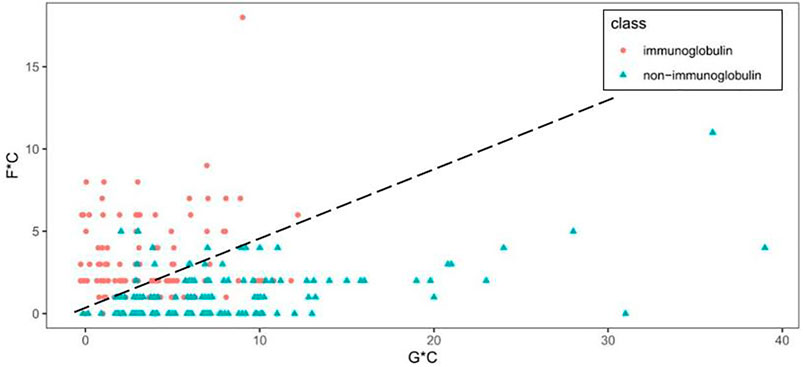
FIGURE 3. Scatter plot of GC* and FC* features.
3.3 Interpretation of Feature FC*
We noticed 49 out of 119 non-immunoglobulin samples had an FC* value of zero, whereas only four immunoglobulin samples had an FC* value of zero. Using motif search website MEME Suite 5.4.1 (Bailey and Elkan, 1994; Bailey et al., 2009) and running 109 immunoglobulin sequences, results showed that 107 out of 109 immunoglobulin samples had a motif, “ISNVTREDAGTYTC” (Figure 4). Based on the reduced scheme, Y was treated as F.

FIGURE 4. Motif discovered among immunoglobulin sequences using the MEME tool; the height of the letter indicates its relative frequency at the given position within the motif.
Immunoglobulin sequences were subjected to InterProScan (Zdobnov and Apweiler 2001) to understand the motif structure better to map protein domains. Results showed that the finding motif belonged to immunoglobulin subtype domain IPR003599.
Also, secondary structure predictions of the motif using JPred (Drozdetskiy et al., 2015) predict that the shared motif comprises alpha helices and beta sheets separated by disordered regions (Figure 5).
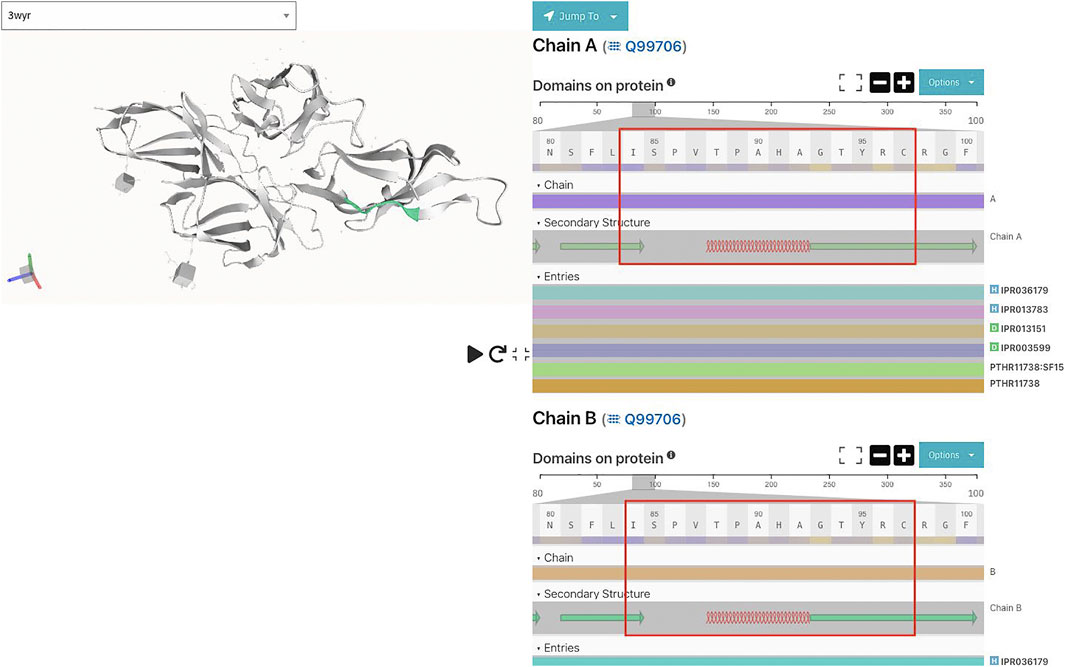
FIGURE 5. Shared motif and its secondary structure (from PDB entry 3wyr) using InterproScan.
4 Conclusion
The present research aimed to examine a method to classify immunoglobulins and non-immunoglobulins using two features, GC* and FC*. Classification of 228 samples (109 immunoglobulin samples and 119 non-immunoglobulin samples) revealed that the overall accuracy was 80.7% in the J48 classifier and 10-fold cross-validation using Weka software. The FC* feature identified in this study was found in immunoglobulin subtype domain IPR003599, which demonstrated that this extracted feature could represent functional and structural properties of immunoglobulins for forecasting.
Data Availability Statement
Publicly available datasets were analyzed in this study. These data can be found here: DOI: 10.1039/c5mb00883b.
Author Contributions
Conceptualization, HeW and GT; data collection or analysis, HaW, JZ and YD; validation, HaW; writing—original draft preparation, HaW; writing—review and editing, HeW and GT. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by the Special Science Foundation of Quzhou (2021D004).
Conflict of Interest
Authors JZ and GT were employed by the company Geneis (Beijing) Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Anderson, M. J. (2001). A New Method for Non-parametric Multivariate Analysis of Variance. Austral Ecol. 26 (1), 32–46. doi:10.1111/j.1442-9993.2001.01070.pp.x
Ao, C., Zhou, W., Gao, L., Dong, B., and Yu, L. (2020). Prediction of Antioxidant Proteins Using Hybrid Feature Representation Method and Random forest. Genomics 112 (6), 4666–4674. doi:10.1016/j.ygeno.2020.08.016
Awais, M., Hussain, W., Rasool, N., and Khan, Y. D. (2021). iTSP-PseAAC: Identifying Tumor Suppressor Proteins by Using Fully Connected Neural Network and PseAAC. Cbio 16 (5), 700–709. doi:10.2174/1574893615666210108094431
Bailey, T. L., and Elkan, C. (1994). Fitting a Mixture Model by Expectation Maximization to Discover Motifs in Biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36.
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: Tools for Motif Discovery and Searching. Nucleic Acids Res. 37, W202–W208. doi:10.1093/nar/gkp335
Chen, X. X., Tang, H., Li, W. C., Wu, H., Chen, W., Ding, H., et al. (2016). Identification of Bacterial Cell Wall Lyases via Pseudo Amino Acid Composition. Biomed. Res. Int. 2016, 1654623. doi:10.1155/2016/1654623
Chen, Y., Ma, T., Yang, X., Wang, J., Song, B., and Zeng, X., (2021). MUFFIN: Multi-Scale Feature Fusion for Drug–Drug Interaction Prediction. Bioinformatics 37 (17), 2651–2658. doi:10.1093/bioinformatics/btab169
Cheng, Y., Gong, Y., Liu, Y., Song, B., and Zou, Q. (2021). Molecular Design in Drug Discovery: a Comprehensive Review of Deep Generative Models. Brief Bioinform 22 (6). doi:10.1093/bib/bbab344
Deng, L., Huang, Y., Liu, X., and Liu, H., (2021). Graph2MDA: a Multi-Modal Variational Graph Embedding Model for Predicting Microbe-Drug Associations. Bioinform. btab792. doi:10.1093/bioinformatics/btab792
Diener, C., Garza Ramos Martínez, G., Moreno Blas, D., Castillo González, D. A., Corzo, G., Castro-Obregon, S., et al. (2016). Effective Design of Multifunctional Peptides by Combining Compatible Functions. Plos Comput. Biol. 12 (4), e1004786. doi:10.1371/journal.pcbi.1004786
Ding, H., Liu, L., Guo, F.-B., Huang, J., and Lin, H. (2011). Identify Golgi Protein Types with Modified Mahalanobis Discriminant Algorithm and Pseudo Amino Acid Composition. Ppl 18 (1), 58–63. doi:10.2174/092986611794328708
Ding, Y., Yang, C., Tang, J., and Guo, F. (2021a). Identification of Protein-Nucleotide Binding Residues via Graph Regularized K-Local Hyperplane Distance Nearest Neighbor Model. Berlin, Germany: Applied Intelligence.
Ding, Y., Tang, J., and Guo, F. (2021b). Protein Crystallization Identification via Fuzzy Model on Linear Neighborhood Representation. Ieee/acm Trans. Comput. Biol. Bioinf. 18 (5), 1986–1995. doi:10.1109/tcbb.2019.2954826
Dong, J., Zhao, M., Liu, Y., Su, Y., and Zeng, X., (2021). Deep Learning in Retrosynthesis Planning: Datasets, Models and Tools. Brief. Bioinform. bbab391. doi:10.1093/bib/bbab391
Dong, Q., Zhou, S., and Guan, J. (2009). A New Taxonomy-Based Protein Fold Recognition Approach Based on Autocross-Covariance Transformation. Bioinformatics 25 (20), 2655–2662. doi:10.1093/bioinformatics/btp500
Drozdetskiy, A., Cole, C., Procter, J., and Barton, G. J. (2015). JPred4: a Protein Secondary Structure Prediction Server. Nucleic Acids Res. 43 (W1), W389–W394. doi:10.1093/nar/gkv332
Feng, C., Ma, Z., Yang, D., Li, X., Zhang, J., and Li, Y. (2020). A Method for Prediction of Thermophilic Protein Based on Reduced Amino Acids and Mixed Features. Front. Bioeng. Biotechnol. 8, 285. doi:10.3389/fbioe.2020.00285
Feng, C., Zou, Q., and Wang, D. (2021). Using a Low Correlation High Orthogonality Feature Set and Machine Learning Methods to Identify Plant Pentatricopeptide Repeat Coding Gene/protein. Neurocomputing 424, 246–254. doi:10.1016/j.neucom.2020.02.079
Fu, X., Cai, L., Zeng, X., and Zou, Q. (2020). StackCPPred: a Stacking and Pairwise Energy Content-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency. Bioinformatics 36 (10), 3028–3034. doi:10.1093/bioinformatics/btaa131
Gautam, A., Chaudhary, K., Kumar, R., Sharma, A., Kapoor, P., Tyagi, A., et al. (2013). In Silico approaches for Designing Highly Effective Cell Penetrating Peptides. J. Transl Med. 11, 74. doi:10.1186/1479-5876-11-74
Gong, Y., Peng, D., Liao, B., and Zou, Q., (2021). Accurate Prediction and Key Feature Recognition of Immunoglobulin. Appl. Sciences-Basel 11 (15), 6894. doi:10.3390/app11156894
Guo, Y., Yan, K., Lv, H., and Liu, B. (2021). PreTP-EL: Prediction of Therapeutic Peptides Based on Ensemble Learning. Brief. Bioinform. 22 (6), bbab358. doi:10.1093/bib/bbab358
Guo, Z., Wang, P., Liu, Z., and Zhao, Y. (2020). Discrimination of Thermophilic Proteins and Non-thermophilic Proteins Using Feature Dimension Reduction. Front. Bioeng. Biotechnol. 8, 584807. doi:10.3389/fbioe.2020.584807
Hansen, M., Kilk, K., and Langel, U. (2008). Predicting Cell-Penetrating Peptides. Adv. Drug Deliv. Rev. 60 (4-5), 572–579. doi:10.1016/j.addr.2007.09.003
He, S., Guo, F., Zou, Q., and Ding, H., (2020). MRMD2.0: A Python Tool for Machine Learning with Feature Ranking and Reduction. Curr. Bioinformatics 15 (10), 1213–1221. doi:10.2174/1574893615999200503030350
Hong, Z., Zeng, X., Wei, L., and Liu, X. (2020). Identifying Enhancer-Promoter Interactions with Neural Network Based on Pre-trained DNA Vectors and Attention Mechanism. Bioinformatics 36 (4), 1037–1043. doi:10.1093/bioinformatics/btz694
Huang, S., He, X., Wang, G., and Bao, E., (2021). AlignGraph2: Similar Genome-Assisted Reassembly Pipeline for PacBio Long Reads. Brief Bioinform 22 (5), bbab022. doi:10.1093/bib/bbab022
Jiang, Q., Wang, G., Jin, S., Li, Y., and Wang, Y. (2013). Predicting Human microRNA-Disease Associations Based on Support Vector Machine. Ijdmb 8 (3), 282–293. doi:10.1504/ijdmb.2013.056078
Jin, Q., Meng, Z., Pham, T. D., Chen, Q., Wei, L., and Su, R. (2019). DUNet: A Deformable Network for Retinal Vessel Segmentation. Knowledge-Based Syst. 178, 149–162. doi:10.1016/j.knosys.2019.04.025
Khan, Y. D., Alzahrani, E., Alghamdi, W., and Ullah, M. Z., (2020). Sequence-based Identification of Allergen Proteins Developed by Integration of PseAAC and Statistical Moments via 5-Step Rule. Curr. Bioinformatics 15 (9), 1046–1055. doi:10.2174/1574893615999200424085947
Lepore, R., Olimpieri, P. P., Messih, M. A., and Tramontano, A. (2017). PIGSPro: Prediction of immunoGlobulin Structures V2. Nucleic Acids Res. 45 (W1), W17–W23. doi:10.1093/nar/gkx334
Li, H.-L., Pang, Y.-H., and Liu, B. (2021). BioSeq-BLM: a Platform for Analyzing DNA, RNA and Protein Sequences Based on Biological Language Models. Nucleic Acids Res. 49, e129. doi:10.1093/nar/gkab829
Liu, B., Gao, X., and Zhang, H. (2019). BioSeq-Analysis2.0: an Updated Platform for Analyzing DNA, RNA and Protein Sequences at Sequence Level and Residue Level Based on Machine Learning Approaches. Nucleic Acids Res. 47 (20), e127. doi:10.1093/nar/gkz740
Li, Q., Dong, B., Wang, D., and Wang, S. (2020a). Identification of Secreted Proteins from Malaria Protozoa with Few Features. Ieee Access 8, 89793–89801. doi:10.1109/access.2020.2994206
Li, Q., Zhou, W., Wang, D., Wang, S., and Li, Q. (2020b). Prediction of Anticancer Peptides Using a Low-Dimensional Feature Model. Front. Bioeng. Biotechnol. 8, 892. doi:10.3389/fbioe.2020.00892
Liu, Y., Wang, G., Wang, Y., and Huang, Y., (2020). A Deep Learning Approach for Filtering Structural Variants in Short Read Sequencing Data. Brief Bioinform 22 (4). doi:10.1093/bib/bbaa370
Lv, Z., Jin, S., Ding, H., and Zou, Q. (2019). A Random Forest Sub-golgi Protein Classifier Optimized via Dipeptide and Amino Acid Composition Features. Front. Bioeng. Biotechnol. 7, 215. doi:10.3389/fbioe.2019.00215
Manavalan, B., Basith, S., Shin, T. H., Wei, L., and Lee, G. (2019). Meta-4mCpred: A Sequence-Based Meta-Predictor for Accurate DNA 4mC Site Prediction Using Effective Feature Representation. Mol. Ther. - Nucleic Acids 16, 733–744. doi:10.1016/j.omtn.2019.04.019
Meng, C., Wu, J., Guo, F., Dong, B., and Xu, L. (2020). CWLy-Pred: A Novel Cell wall Lytic Enzyme Identifier Based on an Improved MRMD Feature Selection Method. Genomics 112 (6), 4715–4721. doi:10.1016/j.ygeno.2020.08.015
Narciso, J. E. T., Uy, I. D. C., Cabang, A. B., Chavez, J. F. C., Pablo, J. L. B., Padilla-Concepcion, G. P., et al. (2011). Analysis of the Antibody Structure Based on High-Resolution Crystallographic Studies. New Biotechnol. 28 (5), 435–447. doi:10.1016/j.nbt.2011.03.012
Naseer, S., Hussain, W., Khan, Y. D., and Rasool, N. (2021). NPalmitoylDeep-Pseaac: A Predictor of N-Palmitoylation Sites in Proteins Using Deep Representations of Proteins and PseAAC via Modified 5-Steps Rule. Cbio 16 (2), 294–305. doi:10.2174/1574893615999200605142828
Norman, R. A., Ambrosetti, F., Bonvin, A. M. J. J., Colwell, L. J., Kelm, S., Kumar, S., et al. (2020). Computational Approaches to Therapeutic Antibody Design: Established Methods and Emerging Trends. Brief. Bioinform. 21 (5), 1549–1567. doi:10.1093/bib/bbz095
Perez, E. E., Orange, J. S., Bonilla, F., Chinen, J., Chinn, I. K., Dorsey, M., et al. (2017). Update on the Use of Immunoglobulin in Human Disease: A Review of Evidence. J. Allergy Clin. Immunol. 139 (3), S1–S46. doi:10.1016/j.jaci.2016.09.023
Rahman, M. S., Rahman, M. K., Kaykobad, M., and Rahman, M. S. (2018). isGPT: An Optimized Model to Identify Sub-golgi Protein Types Using SVM and Random Forest Based Feature Selection. Artif. Intelligence Med. 84, 90–100. doi:10.1016/j.artmed.2017.11.003
Sanders, W. S., Johnston, C. I., Bridges, S. M., Burgess, S. C., and Willeford, K. O. (2011). Prediction of Cell Penetrating Peptides by Support Vector Machines. Plos Comput. Biol. 7 (7), e1002101. doi:10.1371/journal.pcbi.1002101
Schroeder, H. W., and Cavacini, L. (2010). Structure and Function of Immunoglobulins. J. Allergy Clin. Immunol. 125 (2), S41–S52. doi:10.1016/j.jaci.2009.09.046
Shao, J., and Liu, B. (2021). ProtFold-DFG: Protein Fold Recognition by Combining Directed Fusion Graph and PageRank Algorithm. Brief Bioinform 22 (3), bbaa192. doi:10.1093/bib/bbaa192
Shao, J., Yan, K., and Liu, B. (2021). FoldRec-C2C: Protein Fold Recognition by Combining Cluster-To-Cluster Model and Protein Similarity Network. Brief Bioinform 22 (3), bbaa144. doi:10.1093/bib/bbaa144
Solis, A. D. (2015). Amino Acid Alphabet Reduction Preserves Fold Information Contained in Contact Interactions in Proteins. Proteins 83 (12), 2198–2216. doi:10.1002/prot.24936
Song, G., Wang, G., Luo, X., Cheng, Y., Song, Q., Wan, J., et al. (2021). An All-To-All Approach to the Identification of Sequence-specific Readers for Epigenetic DNA Modifications on Cytosine. Nat. Commun. 12 (1), 795. doi:10.1038/s41467-021-20950-w
Song, B., Li, F., Liu, Y., and Zeng, X. (2021). Deep Learning Methods for Biomedical Named Entity Recognition: a Survey and Qualitative Comparison. Brief. Bioinform. 22 (6), bbab282. doi:10.1093/bib/bbab282
Susko, E., and Roger, A. J. (2007). On Reduced Amino Acid Alphabets for Phylogenetic Inference. Mol. Biol. Evol. 24 (9), 2139–2150. doi:10.1093/molbev/msm144
Tang, H., Chen, W., and Lin, H. (2016). Identification of Immunoglobulins Using Chou's Pseudo Amino Acid Composition with Feature Selection Technique. Mol. Biosyst. 12 (4), 1269–1275. doi:10.1039/c5mb00883b
Tang, Y.-J., Pang, Y.-H., and Liu, B. (2020). IDP-Seq2Seq: Identification of Intrinsically Disordered Regions Based on Sequence to Sequence Learning. Bioinformaitcs 36 (21), 5177–5186. doi:10.1093/bioinformatics/btaa667
Tang, Y.-J., Pang, Y.-H., and Liu, B., 2021, DeepIDP-2L: Protein Intrinsically Disordered Region Prediction by Combining Convolutional Attention Network and Hierarchical Attention Network. Bioinformatics 2021, btab810. doi:10.1093/bioinformatics/btab810
Tao, Z., Li, Y., Teng, Z., and Zhao, Y. (2020). A Method for Identifying Vesicle Transport Proteins Based on LibSVM and MRMD. Comput. Math. Methods Med. 2020, 8926750. doi:10.1155/2020/8926750
Wang, H., Ding, Y., Tang, J., and Guo, F. (2020). Identification of Membrane Protein Types via Multivariate Information Fusion with Hilbert-Schmidt Independence Criterion. Neurocomputing 383, 257–269. doi:10.1016/j.neucom.2019.11.103
Wang, H., Tang, J., Ding, Y., and Guo, F. (2021). Exploring Associations of Non-coding RNAs in Human Diseases via Three-Matrix Factorization with Hypergraph-Regular Terms on center Kernel Alignment. Brief. Bioinformatics 22 (5), bbaa409. doi:10.1093/bib/bbaa409
Wang, J., Liu, X., Shen, S., and Deng, L. (2021). DeepDDS: Deep Graph Neural Network with Attention Mechanism to Predict Synergistic Drug Combinations. Brief. Bioinform. bbab390. doi:10.1093/bib/bbab390
Wang, X., Liu, J., Yang, Y., and Wang, G. (2021). The Stacking Strategy-Based Hybrid Framework for Identifying Non-coding RNAs. Brief Bioinform 22 (5). doi:10.1093/bib/bbab023
Wei, L., Liao, M., Gao, Y., Ji, R., He, Z., and Zou, Q. (2014). Improved and Promising Identification of Human MicroRNAs by Incorporating a High-Quality Negative Set. Ieee/acm Trans. Comput. Biol. Bioinf. 11 (1), 192–201. doi:10.1109/tcbb.2013.146
Yang, C., Ding, Y., Meng, Q., Tang, J., and Guo, F. (2021). Granular Multiple Kernel Learning for Identifying RNA-Binding Protein Residues via Integrating Sequence and Structure Information. Neural Comput. Appl. 33 (17), 11387–11399. doi:10.1007/s00521-020-05573-4
Yu, L., Su, Y., Liu, Y., and Zeng, X. (2021). Review of Unsupervised Pretraining Strategies for Molecules Representation. Brief. Funct. Genomics 20 (5), 323–332. doi:10.1093/bfgp/elab036
Zeng, X., Song, X., Ma, T., Pan, X., Zhou, Y., Hou, Y., et al. (2020). Repurpose Open Data to Discover Therapeutics for COVID-19 Using Deep Learning. J. Proteome Res. 19 (11), 4624–4636. doi:10.1021/acs.jproteome.0c00316
Zhai, Y., Chen, Y., Teng, Z., and Zhao, Y. (2020). Identifying Antioxidant Proteins by Using Amino Acid Composition and Protein-Protein Interactions. Front. Cel Dev. Biol. 8, 591487. doi:10.3389/fcell.2020.591487
Zhang, J., Zhang, Z., Pu, L., Tang, J., and Guo, F. (2021). AIEpred: An Ensemble Predictive Model of Classifier Chain to Identify Anti-inflammatory Peptides. Ieee/acm Trans. Comput. Biol. Bioinf. 18 (5), 1831–1840. doi:10.1109/tcbb.2020.2968419
Zhao, X., Wang, H., Li, H., Wu, Y., and Wang, G. (2021). Identifying Plant Pentatricopeptide Repeat Proteins Using a Variable Selection Method. Front. Plant Sci. 12, 506681. doi:10.3389/fpls.2021.506681
Zheng, L., Huang, S., Mu, N., Zhang, H., Zhang, J., Chang, Y., et al. (2019). RAACBook: a Web Server of Reduced Amino Acid Alphabet for Sequence-dependent Inference by Using Chou's Five-step Rule. Database-the J. Biol. Databases Curation. baz131. doi:10.1093/database/baz131
Keywords: immunoglobulin classification, machine learning, key feature extraction, MRMD, autoprop
Citation: Wan H, Zhang J, Ding Y, Wang H and Tian G (2022) Immunoglobulin Classification Based on FC* and GC* Features. Front. Genet. 12:827161. doi: 10.3389/fgene.2021.827161
Received: 01 December 2021; Accepted: 22 December 2021;
Published: 24 January 2022.
Edited by:
Quan Zou, University of Electronic Science and Technology of China, ChinaCopyright © 2022 Wan, Zhang, Ding, Wang and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hetian Wang, aGV0dzIwMjFAMTYzLmNvbQ==; Geng Tian, dGlhbmdAZ2VuZWlzLmNu