
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 17 January 2022
Sec. Genomics of Plants and the Phytoecosystem
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.818880
This article is part of the Research Topic Biotechnological and Genomic Approaches for Enhancing Agronomic Performance of Crops View all 19 articles
 Qasim Raza1*
Qasim Raza1* Awais Riaz1
Awais Riaz1 Rana Muhammad Atif2,3,4Babar Hussain5,6Iqrar Ahmad Rana3,7Zulfiqar Ali8Hikmet Budak9Ibrahim A. Alaraidh10*
Rana Muhammad Atif2,3,4Babar Hussain5,6Iqrar Ahmad Rana3,7Zulfiqar Ali8Hikmet Budak9Ibrahim A. Alaraidh10*MADS-box gene family members play multifarious roles in regulating the growth and development of crop plants and hold enormous promise for bolstering grain yield potential under changing global environments. Bread wheat (Triticum aestivum L.) is a key stable food crop around the globe. Until now, the available information concerning MADS-box genes in the wheat genome has been insufficient. Here, a comprehensive genome-wide analysis identified 300 high confidence MADS-box genes from the publicly available reference genome of wheat. Comparative phylogenetic analyses with Arabidopsis and rice MADS-box genes classified the wheat genes into 16 distinct subfamilies. Gene duplications were mainly identified in subfamilies containing unbalanced homeologs, pointing towards a potential mechanism for gene family expansion. Moreover, a more rapid evolution was inferred for M-type genes, as compared with MIKC-type genes, indicating their significance in understanding the evolutionary history of the wheat genome. We speculate that subfamily-specific distal telomeric duplications in unbalanced homeologs facilitate the rapid adaptation of wheat to changing environments. Furthermore, our in-silico expression data strongly proposed MADS-box genes as active guardians of plants against pathogen insurgency and harsh environmental conditions. In conclusion, we provide an entire complement of MADS-box genes identified in the wheat genome that could accelerate functional genomics efforts and possibly facilitate bridging gaps between genotype-to-phenotype relationships through fine-tuning of agronomically important traits.
In the fight for global food security and safety, bread wheat represents one of the largest contributing grain crops. It is one of the most produced, stored, and consumed food crops worldwide and a major source of energy and nutrients in the developing countries (Igrejas and Branlard, 2020). However, following the green revolution, improvement of wheat grain production has been hindered by various bottlenecks. This includes, but is not limited to, the non-availability of a reliable and fully annotated reference genome (Brenchley et al., 2012; Lukaszewski et al., 2014; Chapman et al., 2015; Clavijo et al., 2017; Zimin et al., 2017). The slow progress in harnessing a fully annotated reference genome was mainly due to the genome’s allohexaploid nature, which comprises three closely related, but independently maintained, sub-genomes, known as A, B, and D. This, in combination with a high frequency of repetitive sequences, particularly hindered progress. Recently, an alliance of geneticists combined resources over 13 years, to produce a fully annotated sequence of the wheat genome, which had a resultant size of ∼17 Gbps. This is the largest known genome among crop plants (Appels et al., 2018). To add to this, the high-quality of the reference genome, together with large scale RNA-seq data and expression repositories (Borrill et al., 2016; Ramírez-González et al., 2018), provide a rich resource for studying evolutionary dynamics and functional characterization of important gene families, which in turn could facilitate crop improvement efforts.
MADS-box transcription factors (TFs) are a well-documented group of genes known for playing vital roles in regulating the growth and development of several important plant species (Smaczniak et al., 2012; Ali et al., 2019). These genes influence diverse biological functions, including cell development, signal transduction, biotic and abiotic stress responses, vegetative organs development, control of flowering and anthesis time, formation of meristems and flower organs, ovule development, embryo development, dehiscence zone formation, and ripening of fruits and seeds (Colombo et al., 1995; Rounsley et al., 1995; Kuo et al., 1997; Riechmann and Meyerowitz, 1997; Alvarez-Buylla et al., 2000; Samach et al., 2000; Saedler et al., 2001; Moore et al., 2002; Messenguy and Dubois, 2003; Pařenicová et al., 2003; Smaczniak et al., 2012; Ali et al., 2019). Given their importance, identification, and characterization of MADS-box genes in agriculturally important species are critical for crop improvement and fine-tuning of specific traits through genetic exploitations.
All MADS-box genes contain a highly conserved MADS (M) domain of approximately 58–60 amino acids. The M domain enables DNA binding and is in the N-terminal region of the protein (Yanofsky et al., 1990). Members of this gene family are classified into M/type I and MIKC/type II super clades, which were generated after an ancient gene duplication event that occurred before the separation of animal and plant lineages (Becker and Theißen, 2003). M-type genes have simple intron-exon structures (zero or one intron), only contain a single M domain, and encode Serum Response Factor (SRF)-like proteins. These can be subcategorized into four clades (Mα, Mβ, Mγ, and Mδ). However, the Mδ clade genes closely resemble those within the MIKC* group, as previously reported (De Bodt et al., 2003).
In comparison, MIKC-type genes have four domains: 1) a highly conserved DNA binding M domain, 2) a less conserved Intervening (I) domain of ∼ 30 AA involved in dimer formation, 3) a moderately conserved Keratin (K) domain of ∼ 70 AA which regulates heterodimerization of MADS proteins, and lastly 4) a highly inconstant C-terminal region which contributes to transcriptional regulation and higher-order protein complex formation (Henschel et al., 2002; Díaz-Riquelme et al., 2009). MIKC-type genes encode Myocyte Enhancer Factor 2 (MEF2)-like proteins and are categorized into MIKCc and MIKC* clades (Kaufmann et al., 2005). MIKC*-type genes have duplicated K domains and relatively longer I domains than MIKCc -types (Duan et al., 2014). MIKCc-type genes can be further categorized into several subclades based on their phylogenetic relationships in flowering plants (Gramzow and Theißen, 2015).
Like what has been observed in other model and crop plant species, in wheat MADS-box genes are known to confer drought tolerance through regulation of drought tolerance genes and micro-RNAs (Budak et al., 2015). In addition to this, TaMADS51, TaMADS4, TaMADS5, TaMADS6, and TaMADS18 all showed up-regulation, where TaMADAGL17, TaMADAGL2, TaMADWM31C, and TaMADS14 were downregulated, under phosphorous (P) starvation, and further functional analysis confirmed their role in P-deficient stress responses (Shi et al., 2016). In another study, several MADS-box genes were activated and differentially expressed after inoculation of wheat spikes with fusarium head blight (FHB) (Kugler et al., 2013). Furthermore, a simple yet elegant ABCDE floral organ identity model explained the complex genetic interactions among MIKC-type wheat MADS-box genes for determining the fate of floral organs (Ali et al., 2019). These significant roles of MADS-box genes in biotic and abiotic stresses, fertilizer response, and flower development highlight the necessity of their comprehensive identification and characterization in the bread wheat genome.
Genome-wide identification and characterization of important gene families are becoming main stream research approaches during recent years (Ahmed et al., 2021; Aleem et al., 2022). Previously, comprehensive genome-wide analyses of MADS-box genes have been carried out in diverse plant species. However, the available information about wheat MADS-box genes is comparatively sparse. For example, Ma et al. (2017) reported the first genome-wide analysis of MADS-box genes using only a draft version of the wheat genome (TGACv1) which contains a lesser number of genes. Likewise, Schilling et al. (2020) studied only MIKC-type MADS-box genes from an updated genome version (Ref. Seq 1.0). Moreover, a significant number of low confidence and/or truncated protein-coding pseudogenes were also included in the before-mentioned studies.
To bridge the gaps in knowledge, in this study we comprehensively identify the entire complement of high confidence, full-length, protein-coding MADS-box genes present in the publicly available reference genome of bread wheat (IWGSC RefSeq v1.1). Through a combination of different search approaches, 300 high confidence, non-redundant, full-length MADS-box genes were identified. Comparative phylogenetic analyses with model plant MADS-box genes further classified these into at least 16 Arabidopsis and/or grass specific subfamilies. Moreover, homeologous and duplicated genes were identified to study probable gene family expansion and evolution mechanisms. Furthermore, expression patterns of identified MADS-box genes were studied under several biotic and abiotic stress conditions. In this way, we provide a comprehensive resource of wheat MADS-box genes which have the potential to facilitate molecular breeders in fine-tuning important traits for further improvement.
The publicly available genome version of wheat (IWGSC Ref. Seq v1.1) was accessed through the ensemble plants database (Bolser et al., 2017) and searched using protein family database (Pfam) identifiers of MADS (PF00319) and K (PF01486) domains. A total of 281 MADS-domain and 131 K-domain encoding genes were identified. Additionally, a query-based search using “MADS” yielded 300 genes. Together, these searches identified a total of 712 genes (Supplementary Table S1), among which 300 were found to be high confidence and non-redundant and considered for further analyses. Detailed information of these 300 genes was retrieved from the ensemble plants database and is provided in Supplementary Table S2. Moreover, amino acid sequences for the identified genes were uploaded into the NCBI conserved domain database (Marchler-Bauer et al., 2015) for validation of putative protein domains (Supplementary Table S3). Gene identifiers were used as gene names for subsequent analyses; however, alternative names used in previous studies (Schilling et al., 2020) were also provided in Supplementary Table S4.
Differentiation between type I (M-type) and type II (MIKC-type) MADS-box proteins were achieved by separate MAFFT alignments using only the MADS domain (L-INS-i algorithm) (Katoh and Standley, 2013; Katoh et al., 2018) between Arabidopsis (Pařenicová et al., 2003) and wheat, and rice (Arora et al., 2007) and wheat MADS-box proteins. Subsequently, maximum likelihood (ML) phylogenies were inferred using IQ-TREE (Nguyen et al., 2015) by choosing JTT + F + G4 best fit substitution model according to the Bayesian information criterion (BIC) (Kalyaanamoorthy et al., 2017). The consistency of the ML trees was validated by setting an Ultrafast bootstrap value of 1,000 (Minh et al., 2013; Hoang et al., 2018). The final phylogenetic trees were visualized with MEGA7 (Kumar et al., 2016).
Subfamily classifications were accomplished by separate MAFFT alignments within M-type (L-INS-i algorithm) and MIKC-type (E-INS-i algorithm) MADS-box protein sequences of Arabidopsis, rice, and wheat (Katoh and Standley, 2013; Katoh et al., 2018). The full-length alignments were subjected to the Gap Strip/Squeeze v2.1.0 tool (www.hiv.lanl.gov/content/sequence/GAPSTREEZE/gap.html) for masking the individual residues by removing the gaps with default parameters. Then, masked alignments of M-type and MIKC-type proteins were independently subjected to the IQ-TREE software (Nguyen et al., 2015) for generating ML phylogenetic trees as described above. Subfamily names were given by following subfamily classifications in Arabidopsis and/or major grass species (Gramzow and Theißen, 2015) (Supplementary Table S2 and Supplementary Table S4).
Putative homeologs were recognized based on strong phylogenetic relationships (Ultrafast bootstrap value >90) within different sub-families. Classifications reported in previous studies were also considered (Appels et al., 2018). The homeologs status of thirty-one genes could not be determined due to lower Ultrafast bootstrap values (Supplementary Table S5).
Coding sequences (CDS) of all wheat MADS-box genes were retrieved from the ensemble plants database and blasted against each other using Sequence Demarcation Tool V1.2 (Muhire et al., 2014) for the identification of sequence identities. Gene pairs with ≥ 90% identity (E value < 1e−10) and non-homeologous status were considered as duplicated (Ning et al., 2017) (Supplementary Table S6). If the duplicated homologous gene pair was located on the same chromosome it was defined as tandem duplication. Otherwise, when homologous gene pairs were located on different chromosomes it was defined as segmental duplication. The CDS sequences of duplicated genes were MAFFT aligned, masked, and subjected to the Synonymous Non-Synonymous Analysis Program V2.1.1 (www.hiv.lanl.gov/content/sequence/SNAP/SNAP.html) to compute the synonymous (Ks) and non-synonymous (Ka) substitution rates. To find out which type of codon selection operated during evolution, the ratio of Ka/Ks was also calculated. The approximate divergence time between duplicated gene pairs was calculated by using formulae T = Ks/2r × 10−6 assuming a substitution rate (r) of 6.5 × 10−9 substitutions/synonymous site/year (El Baidouri et al., 2017) (Supplementary Table S6).
Expression data under all available abiotic and biotic stress conditions were retrieved from the expVIP Wheat Expression Browser (Borrill et al., 2016; Ramírez-González et al., 2018) as Log2 TPM (processed expression value in transcripts per million) obtained via RNA-seq analysis. Detailed information about expression levels of individual genes in tested tissues/growth stages and stresses/diseases are provided in the supplementary information (Supplementary Table S7). TBtools (Chen et al., 2020) was used to generate a heatmap from the obtained expression data. Expression based clustering of genes was achieved by following the K-means clustering method (K = 10, iterations = 1,000, runs = 5) (Supplementary Table S8).
In this study, conserved domains and query search-based approaches identified a total of 300 high confidence and non-redundant MADS-box genes from the publicly available wheat genome (Figure 1A, Supplementary Table S2). The NCBI-CDD batch search further revealed that 167 (∼55.7%) encode MADS-box proteins, 125 (∼41.7%) encode MADS and K-box proteins, and only 8 (∼2.6%) encode K-box domain-containing proteins (Supplementary Table S3). The protein lengths, molecular weight, and isoelectric points of MADS domain-containing proteins ranged from 58 to 450 amino acids, 6.483–48.304 kD, and 4.420–12.123 pI, respectively (Supplementary Table S2). This data suggests that different MADS-box genes may function within different environments.
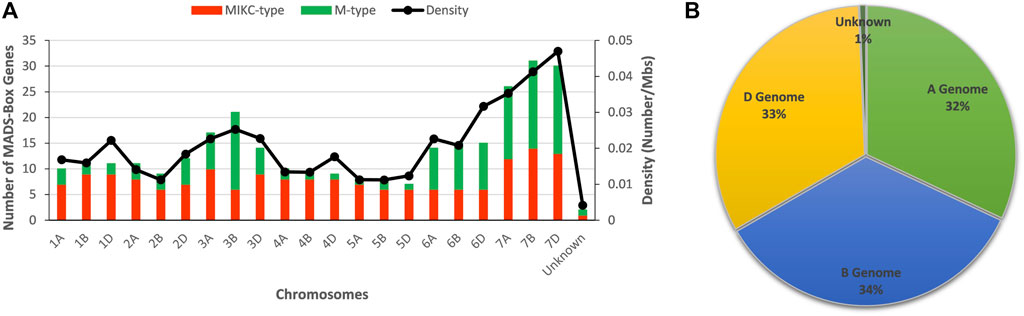
FIGURE 1. Distribution pattern of MADS-box genes on wheat chromosomes and sub-genomes. (A) Number of MADS-box genes and their density on individual wheat chromosomes. (B) Percent contribution of each sub-genome to MADS-box genes galore.
Comparative phylogenetic analyses between Arabidopsis and wheat MADS-box genes (Supplementary Figure S1), as well as between rice and wheat (Supplementary Figure S2), distinguished wheat genes into M- and MIKC-types (Supplementary Table S2). In wheat, 128 (∼43%) MADS-box genes exhibited high sequence similarity with Arabidopsis and rice M-type (type-I) genes, whereas 172 (∼57%) revealed more homology with MIKC-type (type-II) genes. In general, MADS-box genes were equally distributed among the 21 wheat chromosomes, apart from the three homeologous chromosomes (7A, 7B and 7D), which harboured a significantly higher number of genes and displayed peak gene density values (Figure 1A). This observation could be explained by the higher prevalence of duplicated gene pairs on these chromosomes (Appels et al., 2018). However, M-type genes were randomly distributed among chromosomes and were predominantly located on three homeologous chromosomes of 3rd, 6th, and 7th linkage groups (Figure 1A and Supplementary Figure S3). In comparison, MIKC-type genes were equally distributed on all chromosomes. Furthermore, the percent contributions of A, B, and D genomes were also comparable (Figure 1B). As expected, MIKC-type genes were the largest group of MADS-box genes in wheat.
Separate ML phylogenies among Arabidopsis, rice, and wheat MADS-box genes exposed 14 MIKC-type and 3 M-type major subfamilies. The 172 MIKC-type genes were unevenly dispersed into AP1 (9), AP3 (6), PI (6), AG/STK (12), SEP (28), AGL6 (3), AGL12 (6), AGL17 (31), Bsister (19), MIKC* (27), OsMADS32 (3), SOC1 (13), and SVP (9) subfamilies (Figure 2A, Supplementary Table S2). As expected, monocot (OsMADS32) and eudicot (FLC) specific gene subfamilies were also witnessed. Likewise, 128 M-type genes were randomly distributed into Mα (53), Mβ (28), and Mγ (47) subfamilies (Figure 2B).
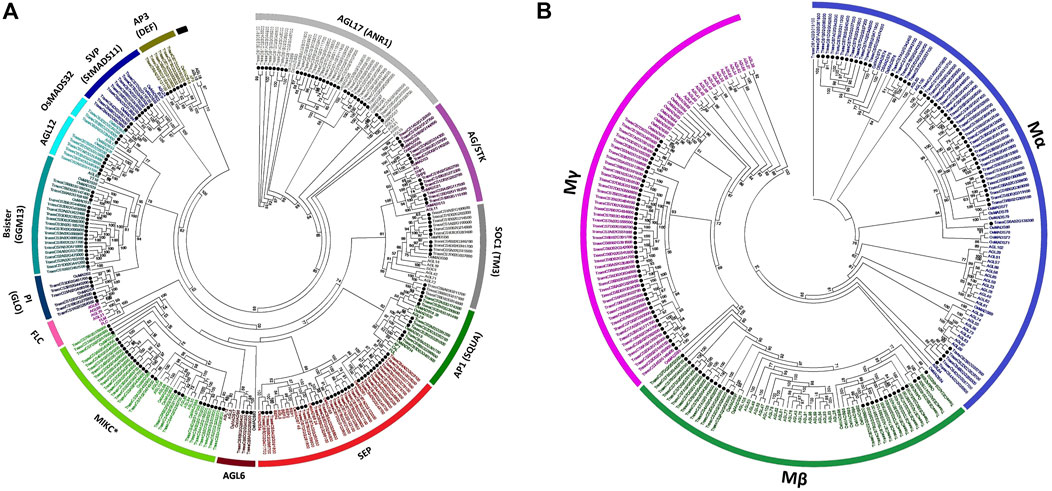
FIGURE 2. Comparative phylogenetic analysis-based subfamily classifications of wheat MADS-box genes. Arabidopsis, rice, and wheat MADS-box proteins were MAFFT aligned (Katoh and Standley, 2013; Katoh et al., 2018), maximum likelihood phylogenies inferred using IQ-TREE software (Nguyen et al., 2015) and final trees visualized with MEGA7 (Kumar et al., 2016). Separate phylogenetic trees were inferred among Arabidopsis, rice, and wheat MIKC-type (A) and M-type (B) proteins. Wheat MADS-box genes are highlighted with solid black circles. Only bootstrap values ≥ 50%, as calculated from 1,000 replicates, could be displayed on the tree nodes. Subfamily-specific colouring was adopted for differentiating the different subfamilies. Subfamily names (outer bands) were given by following the subfamily classifications in Arabidopsis and/or major grass species (Gramzow and Theißen, 2015). Alternate gene names used in previous studies are also provided in Supplementary Table S4.
In general, Arabidopsis, rice and wheat MADS-box gene subfamilies roughly followed the species-specific phylogenetic clades. Triads of wheat homeologous genes exhibited close relationships with one or more rice genes, with Arabidopsis genes representing a sister group association to grass genes (e.g., the AP1, AP3, PI, AG/STK, AGL6, AGL12, OsMADS32, SOC1, and SVP subfamilies; Figure 2A). Whereas the subfamily phylogenies were more complex in the case of AGL17, Bsister, MIKC*, SEP, and all M-type MADS-box genes, probably due to multiple duplication events during the polyploidization of wheat genome.
FLOWERING LOCUS C (FLC)-like genes have been confirmed to regulate flowering time and vernalization responses in plants (Distelfeld et al., 2009; Andrés and Coupland, 2012) and their wheat and rice counterparts have been reported based on sequence homology and phylogenetic tree reconstructions (Ruelens et al., 2013; Schilling et al., 2020). However, in this study, despite employing updated resources and tools (see Materials and Method section), none of the wheat and rice MADS-box genes fell into the Arabidopsis specific FLC-clade (Figure 2A). All the wheat genes which were previously classified as FLC-like (Supplementary Table S4) were grouped with MIKC*-like MADS-box genes. Furthermore, we compared the amino acid sequences of Arabidopsis specific FLC genes and found that these were significantly different from MIKC*-like genes (Figure 3). The most significant differences were detected in MADS domain region at 30th, 34th, and 50th positions where glutamic acid (E), glutamine (Q), and alanine/glycine/serine (A/G/S) residues of FLC genes were substituted with lysine (K), glutamic acid (E), and proline (P), respectively. Collectively, these results indicate that wheat and rice genomes might lack FLC clade.
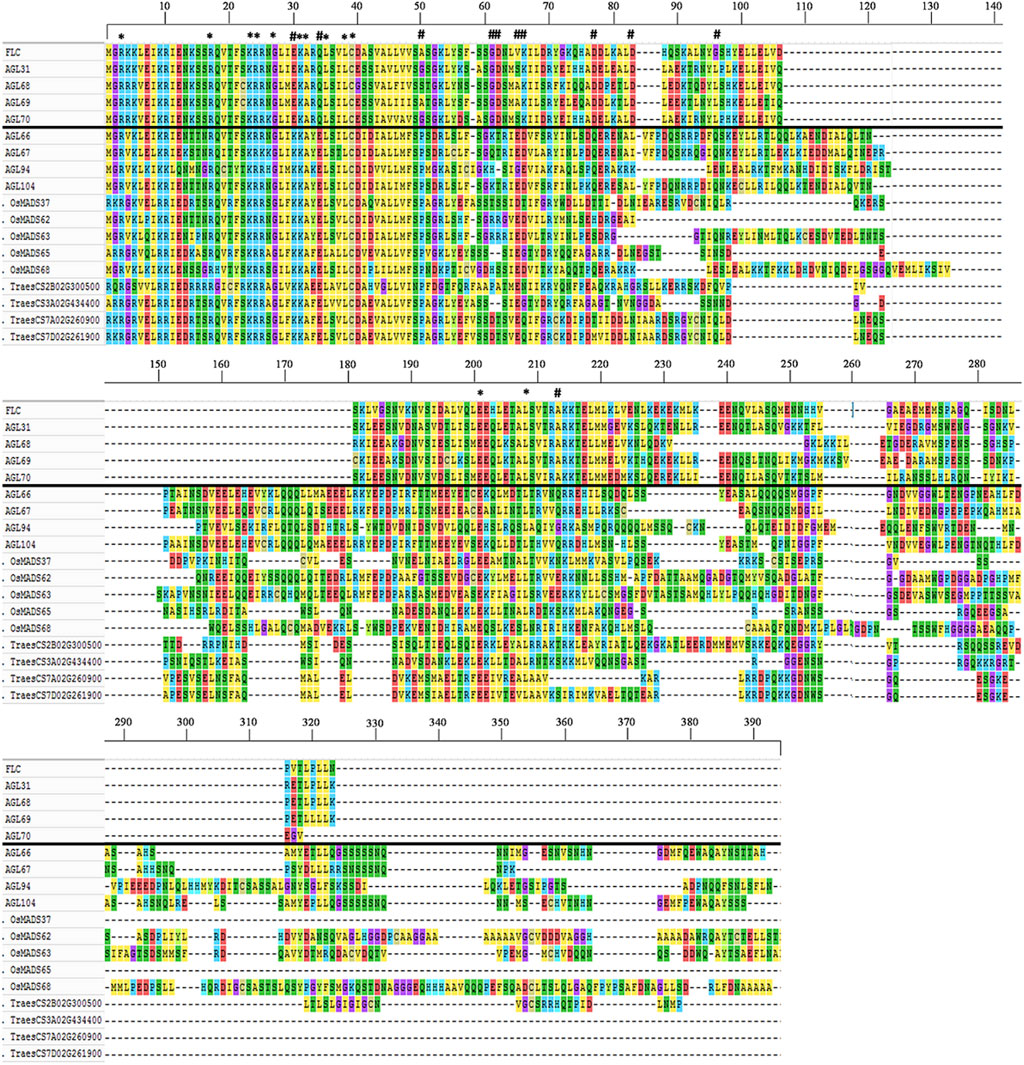
FIGURE 3. Structural differentiation between FLC- and MIKC*-like MADS-box genes. Amino acid sequences of representative genes from both subfamilies were aligned using Clustal Omega (Sievers et al., 2011) with default parameters and multiple sequence alignment visualized with MEGA7 (Kumar et al., 2016). FLC-like genes (above solid black line) were separated from MIKC*-like genes (below solid black line). The conserved and diverged residues were indicated with star (*) and asterisk (#) symbols, respectively. Multiple sequence alignment demonstrated conservation of amino acid residues/motifs within both subfamilies, whereas diversification between subfamilies.
Overall, MADS-box genes were equally located in the interstitial and proximal regions (R2a, R2b and C) and distal telomeric (R1 and R3) ends of chromosomes (49 and 51%, respectively) (Supplementary Table S2). However, substantial differences were observed among gene locations of M-type and MIKC-type genes, as well as among subfamilies. Most of the M-type genes were in distal telomeric segments (62%), whereas MIKC-type genes were more prevalent in central chromosomal segments (57%). Generally, a larger portion of genes belonging to significantly expended subfamilies tend to be in distal telomeric ends, whereas genes of smaller subfamilies were more clustered in central chromosomal segments (Supplementary Table S2).
Gene duplications were identified through sequence similarities in coding sequences of all MADS-box genes. A total of 201 duplicated gene pairs with ≥90% sequence homology were identified, which corresponded to 123 non-redundant genes (Figure 4, Supplementary Table S6). Two genes (TraesCSU02G209900, TraesCSU02G235300) with unknown chromosomal location information were also recognized to be duplicated. However, TraesCSU02G235300 showed duplications with genes located on chromosome 3B only, strongly suggesting that it was also located on the 3B chromosome. The MIKC-type MADS-box genes were also found to be more duplicated than M-type genes (60 vs 40% of duplicated gene pairs, respectively), particularly due to expended subfamilies (e.g., AGL17, Bsister, MIKC*, SEP, and SOC1). Remarkably, duplicated gene pairs were subfamily-specific and particularly recognized in subfamilies containing unbalanced homeologs, except for the AP3 subfamily (Table 1, Supplementary Table S5 and Supplementary Table S6). Among subfamilies, Mα, AGL17, MIKC*, and SEP contained 25, 21, 14, and 12% of the total duplicated gene pairs, respectively, and the majority of these were in distal telomeric and sub-telomeric (one gene located on the telomeric segment and other on the central segment) chromosomal regions. We also observed that the majority of the duplicated gene pairs (∼51%) were in distal telomeric segments, whereas only 26 and 23% of the duplicated gene pairs were in proximal and sub-telomeric segments of chromosomes. These results could be explained by higher gene density in distal vs central chromosomal segments (Appels et al., 2018). Furthermore, segmental duplications were more prevalent than tandem duplications (61 and 37%) in 197 of the duplicated gene pairs with available chromosomal information (Supplementary Table S6). Interestingly, >37% of all tandem duplications were identified on chromosome 3B, consistent with IWGSC findings (Appels et al., 2018). Collectively, these results strongly suggest that unbalanced homeologs in distal telomeric regions derive MADS-box subfamilies expansion through segmental duplications.
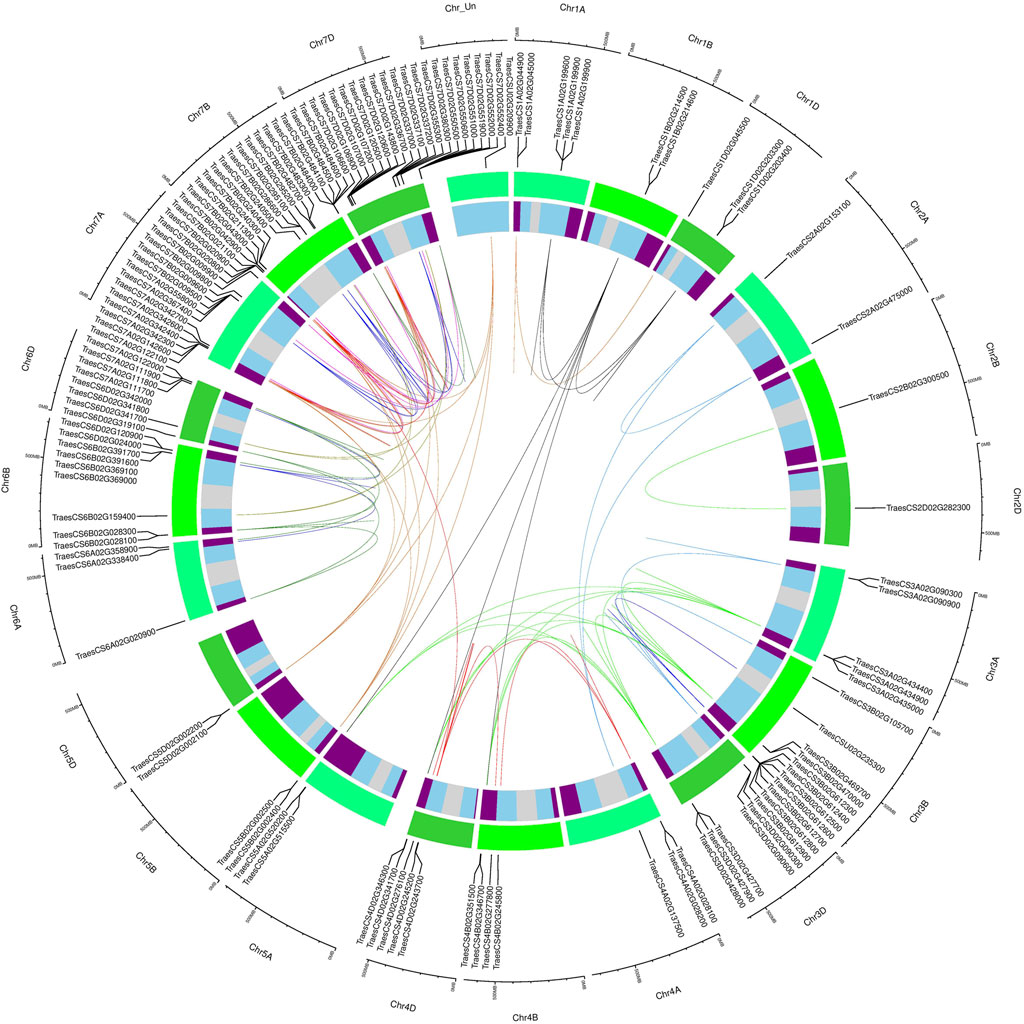
FIGURE 4. Subfamily-specific gene duplications among wheat MADS-box genes. Duplicated genes were plotted in a circular diagram using respective physical positions with shinyCircos (Yu et al., 2018). The outer track indicates three different sub-genomes (shades of green), and the inner track represents chromosomal segments (R1 and R3, purple; R2a and R2b, sky blue; C, light grey) (Appels et al., 2018). Duplicated genes were identified through sequence similarity (Supplementary Table S6; see material and method section) and linked with subfamily-specific colours as in Figures 2A,B, except for AGL17-like genes which were linked using chocolate colour. The linked duplicated genes positioned on different wheat chromosomes represent segmental duplications, whereas tandem duplications were indicated by incomplete links within the same chromosomes.

TABLE 1. A potential relationship between unbalanced homeologs and gene duplications in wheat MADS-box gene family.
To investigate evolution rates, we estimated substitutions in coding sequences and computed approximate divergence time between duplicated gene pairs (Supplementary Table S6). In M-type genes, 1st and 3rd quartiles of synonymous substitutions (Ks) were considerably narrower than MIKC-type genes, whereas non-synonymous to synonymous substitution ratios (Ka/Ks) of M-type genes were significantly higher than 1st and 3rd quartiles of MIKC-type genes (Figure 5). Furthermore, the percentage of duplication events because of positive/Darwinian selection (Ka/Ks ratio >1) was almost doubled (23%) in M-type genes as compared with MIKC-type genes (12%) (Supplementary Table S6). Additionally, the estimated divergence time of M-type genes was significantly narrower than the MIKC-type genes. The mean divergence time of M-type genes was nearly half of the mean of MIKC-type genes (Figure 5). Taken together, these data strongly indicate a rapid evolution of M-type MADS-box genes.
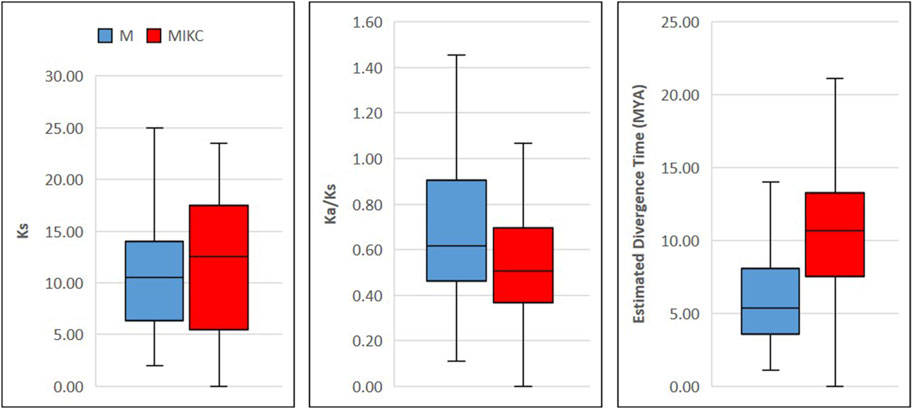
FIGURE 5. Boxplots showing evolution patterns in M- and MIKC-type wheat MADS-box genes. The coding sequences of the duplicated genes were MAFFT aligned (Katoh and Standley, 2013; Katoh et al., 2018), masked and subjected to SNAP V2.1.1 program for computing the synonymous (Ks) and non-synonymous (Ka) substitution rates. The divergence time between duplicated genes pairs was estimated in million years ago (MYA) by following El Baidouri et al. (2017) and boxplots were generated using Microsoft Excel 2019.
Extensive investigations have been carried out to study the expression patterns of MADS-box genes during the growth and development of crop plants. However, their transcriptional regulation under stressful conditions is somewhat obscure. Therefore, we analysed RNA-seq based expression data of 300 MADS-box genes under all available biotic and abiotic stress conditions in the exVIP wheat expression browser (Borrill et al., 2016; Ramírez-González et al., 2018) (Figure 6, Supplementary Table S7). Out of the total 300 genes, nearly 57% were expressed (log2 TPM 0.20–7.89) during at least one developmental stage of one or more of the stresses included in this study. Whereas the remaining 43% of genes showed no or very low expression (log2 TPM <0.0) and were subsequently considered as not expressed.
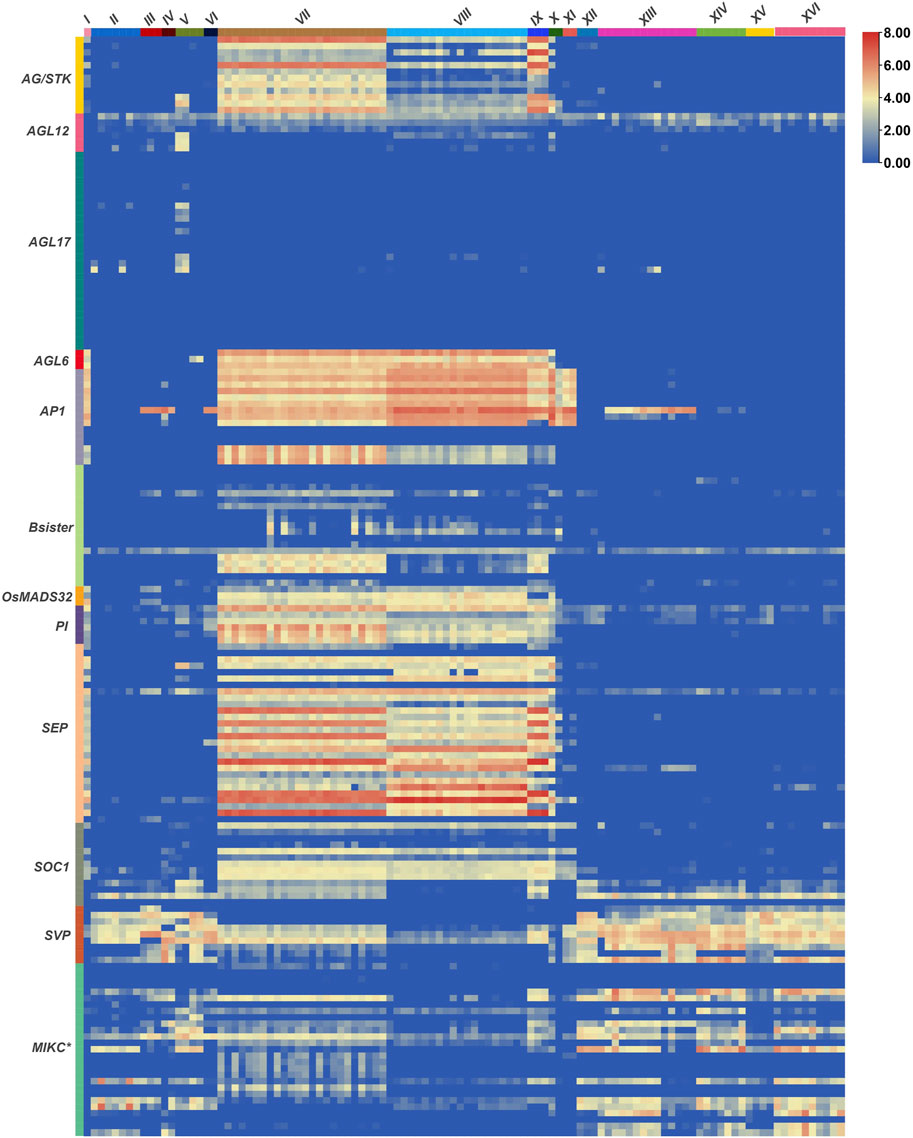
FIGURE 6. Expression patterns of wheat MADS-box genes under stressful conditions. RNA-seq based expression data of all MADS-box genes were retrieved from the exVIP wheat expression browser (Borrill et al., 2016; Ramírez-González et al., 2018) and a heatmap was generated with TBtools (Chen et al., 2020). Processed expression levels of all genes under different abiotic (I, spikes with water stress; II, drought and heat-stressed seedlings; III, seedling treated with PEG to induce drought; IV, shoots after 2 weeks of cold stress; V, phosphate starvation in roots/shoots/leaves) and biotic stresses (VI, coleoptile infection with Fusarium pseudograminearum/crown root; VII, FHB infected spikelets (0–48 h); VIII, FHB infected spikelets (30–50 h); IX, spikes inoculated with FHB and ABA/GA; X, CS spikes inoculated with FHB; XI, leaves naturally infected with Magnaporthe oryzae; XII, PAMP inoculation of seedlings; XIII, stripe rust infected seedlings; XIV, stripe rust and powdery mildew infection in seedlings; XV, Septoria tritici infected seedlings; XVI, Zymoseptoria tritici infected seedlings) (columns) and in different subfamilies (rows) are presented as Log2 transcripts per million (Log2 TPM). Detailed information about expression levels of individual genes in tested tissues/growth stages and stresses/diseases are provided in Supplementary Table S7.
Overall, MIKC-type MADS-box genes were highly expressed under all studied stresses, whereas nearly 75% of the non-expressed genes belonged to the three M-type subfamilies. Interestingly, the remaining 25% of the non-expressed genes belonged to MIKC-subfamilies containing duplicated genes, e.g., AGL17, MIKC*, and Bsister. As most of the duplicated genes were in distal telomeric and sub-telomeric segments of chromosomes, it could be expected that promoters of these genes had undergone H3K27me3 (trimethylated histone H3 lysine 27) hypermethylation which resulted in their lower or non-existent expression (Ramírez-González et al., 2018).
In general, genes of all MIKC-type subfamilies (except AGL17) were ubiquitously expressed in fusarium head blight infected spikelets/spikes (Figure 6). Nevertheless, floral homeotic genes (AP1, AP3, PI, AG/STK, AGL6, and SEP) showed the highest expression patterns. SVP, MIKC*, AGL12, and SOC1-like genes were also ubiquitously expressed in all studied abiotic and biotic stresses, strongly indicating that genes belonging to these subfamilies are active guardians of the wheat genome under stressful environments. Remarkably, all AP1-like genes demonstrated high expression in Magnaporthe oryzae infected leaves, suggesting disease-specific expression, as well as the involvement of these important subfamily genes in diverse aspects of wheat growth and development. Similarly, AGL17-like genes were expressed only in seedlings under drought, heat, and phosphate starvation conditions (Figure 6).
We also calculated expression based hierarchical clusters to analyze diversification in expression patterns present within subfamilies (Supplementary Figure S4, Supplementary Table S8). The MIKC* and SOC1-like genes were grouped into seven and six random clusters, respectively. Similarly, Bsister and SEP-like genes were separately grouped into five random clusters. By contrast, AP1 and Mγ-like genes exhibited no variation in their expression patterns, as all genes were grouped into a single random cluster. Likewise, AGL6, AGL17, AP3, OsMADS32, Mα, and Mβ-like genes displayed little diversification in expression, and genes of each subfamily were grouped into two random clusters. In comparison, many of the genes with no or very low expression (∼58%) were grouped into cluster 10 and all belonged to subfamilies with duplicated genes (Supplementary Table S8).
MADS-box gene family members have been identified across all groups of eukaryotes and are known to confer diverse biological functions. In plants, these play a central role during growth and development and are thus very important targets for crop improvement (Ali et al., 2019). Recently, significant advancements have been made in the identification and characterization of MADS-box family members in wheat, which could facilitate crop breeding efforts and aid in the development of more resilient and higher-yielding cultivars. Ma et al. (2017) reported the first genome-wide analysis of MADS-box family members and identified a total of 180 MADS-box genes in an earlier genome version (Lukaszewski et al., 2014). Their in-silico expression data provided insights into the stress associated functions of MADS-box genes. To add to this, Schilling et al. (2020) conducted a comprehensive analysis of MIKC-type MADS-box genes and identified a total of 201 genes from an updated genome version (Ref. Seq 1.0) (Appels et al., 2018). They speculated that pervasive duplications, functional conservation, and putative neofunctionalization may have contributed to the adaptation of wheat to diverse environments. These advancements, along with results presented in this study, have begun to elucidate the broad landscape of wheat MADS-box genes and provide new insights into the phylogenomics, evolution, and stress associated functions of MADS-box family members.
Bread wheat is a hexaploid species (AABBDD, 2n = 6x = 42), that originated from a series of naturally occurring hybridization events among three closely related and independently maintained sub-genomes. Its full genome size is ∼17 Gbps, among which ∼14.5 Gbps (85%) is sequenced and contains ∼107,891 coding genes (Appels et al., 2018). In this study, through a comprehensive genome-wide analysis, we identified a total of 300 high confidence MADS-box genes (Figure 1, Supplementary Table S2). To date, this is the second-highest number of MADS-box genes identified in a crop plant, closely following Brassica napus (Wu et al., 2018), which contains 307 full-lengths and/or incomplete (pseudo) MADS-box genes. Possible explanations for the abundance found in wheat may encompass the larger genome size, higher gene number, high rate of homeolog retention, and hexaploid nature of bread wheat (Appels et al., 2018). Moreover, at least 35 low confidence MIKC-type MADS-box genes were recently reported in the IWGSC reference genome (Schilling et al., 2020). If these low confidence genes are also included with the 300 MADS-box genes identified in this study, then the final number will be the highest identified in a plant genome. Furthermore, a more recent chromosome-scale assembly of the bread wheat genome revealed hundreds of new genes and thousands of additional gene copies (Alonge et al., 2020) which were substantially missing from the IWGSC reference genome explored in the current study. Collectively, all these observations strongly suggest that MADS-box genes are underestimated in wheat and their exploration might help in understanding the rapid global adaptability of wheat.
FLCs are key genes conferring vernalization requirement, which act as flowering repressors in Arabidopsis (Whittaker and Dean, 2017). High expression of FLCs repress other flowering activator genes and causes delayed flowering. In winter varieties of temperate cereals, including barley, Brachypodium, and wheat, vernalization is regulated through VERNALIZATION genes (VRN1, VRN2, VRN3) (Greenup et al., 2009). To date, strong disagreement exists in the published literature regarding the presence or absence of FLC-like genes in grasses (Paolacci et al., 2007; Zhao et al., 2011; Ruelens et al., 2013; Wei et al., 2014; Fatima et al., 2020; Schilling et al., 2020). Previously, grass genes were classified into FLC-like based on synteny and phylogenetic reconstruction methods (Ruelens et al., 2013; Schilling et al., 2020). However, despite using a more recent reference genome sequence and sophisticated phylogenetic tree reconstruction methods, we could not find FLC-clade in wheat and rice genomes (Figure 2A). Interestingly, all those genes which were previously classified as FLC-like (Ruelens et al., 2013; Schilling et al., 2020), as well as OsMADS37 and OsMADS51/65, were grouped with MIKC*-like genes (Figure 2A, Supplementary Table S4). In comparison, all other FLC homologs were grouped into a separate distinct Arabidopsis-specific FLC clade. Moreover, we also compared the multiple sequence amino acid alignments of FLC- and MIKC*-like genes and observed that these were significantly dissimilar and none of the grass genes shared sequence homology with Arabidopsis FLC paralogs (Figure 3). Altogether, these results indicate that FLC-like genes might have been lost in cereal genomes after they diverged from eudicots. Several other studies are also in agreement with current results and were unable to identify FLC-like genes in grass genomes (Arora et al., 2007; Paolacci et al., 2007; Zhao et al., 2011; Wei et al., 2014; Fatima et al., 2020). These observations suggest that vernalization pathways in monocots and dicots evolved independently and are regulated through a completely different set of genes. However, earlier reports on the identification of FLC-like genes in grasses (Ruelens et al., 2013; Sharma et al., 2017; Schilling et al., 2020) and functional conservation of ODDSOC2 with FLC in the regulation of vernalization (Greenup et al., 2010), suggest that evolution of vernalization pathways between monocots and dicots may not be fully independent. Thus, the biological question of the presence or absence of FLC-like genes in grass genomes still needs to be explored. Further research efforts in this direction might address this fundamental question, and propitious outcomes could help in breeding efforts to develop winter varieties of temperate cereals which are adapted to changing environments.
Distal telomeric chromosomal segments are evolutionary hotspots for frequent recombination events and give rise to fast-evolving genes (Glover et al., 2015; Chen N. W. G. et al., 2018). Many adaptability traits related genes that are induced in response to external stimuli are found to be predominately located in distal chromosomal segments. In comparison, genes related to housekeeping and conserved developmental functions are positioned in central chromosomal segments (Appels et al., 2018; Ramírez-González et al., 2018). In this study, the majority of the identified duplicated gene pairs were in distal telomeric (∼51%) and sub-telomeric (23%) chromosomal segments (Figure 4, Supplementary Table S6). Remarkably, all these duplications were subfamily-specific and primarily identified in larger subfamilies containing unbalanced homeologs (Mα, Mβ, Mγ, AGL17, Bsister, MIKC*, SEP, and SOC1) (Table 1). Interestingly, genes belonging to these subfamilies have been reported to regulate plants adaptability in changing environments. For example, OsMADS57 (AGL17-like gene in rice) is induced by abscisic acid, chilling, drought, and salinity stresses (Arora et al., 2007), promotes cold tolerance by interacting with a defence gene (OsWRKY94) (Chen L. et al., 2018), and modulates root to shoot nitrate translocation under deprived nitrate conditions (Huang et al., 2019). Similarly, downregulation of a SEP clade gene in pepper (CaMADS) caused more sensitivity to cold, salt, and osmotic stresses, whereas its overexpression in Arabidopsis conferred higher tolerance against these stresses (Chen et al., 2019). By contrast, almost negligible duplications were identified in a single smaller subfamily (AP3) with completely balanced homeologs (Table 1). These results strongly indicate that unbalanced homeologs of expended subfamilies have undergone subfamily-specific distal telomeric duplications, thus facilitating the rapid adaptation of bread wheat to diverse global environments. Contrarily, completely balanced homeologs of functionally conserved smaller subfamilies might have some evolutionary advantage in minimizing the developmentally detrimental gene copy number variations, due to their localization in central chromosomal segments.
Unlike MIKC-type MADS-box genes, the M-type genes have not been extensively studied and little information is available about their evolutionary origin in crop plants (Masiero et al., 2011). In this study, we observed that M-type genes were predominately located in distal telomeric chromosomal segments (Supplementary Table S2) and are evolving at a faster rate, probably due to the higher frequency of tandem gene duplications and stronger purifying selections (Figure 5, Supplementary Table S6). Moreover, lesser synonymous substitutions and higher non-synonymous to synonymous substitution ratios of M-type duplicated genes exposed their more rapid evolution as compared with MIKC-type genes (Figure 5, Supplementary Table S6). These results agree with Nam et al. (2004), who also reported faster birth-and-death evolution of M-type genes in angiosperms. This data might indicate the significance of M-type MADS-box genes in understanding the evolutionary history of the bread wheat genome.
MADS-box genes regulate diverse developmental processes, and their functions are well studied in plant morpho- and organogenesis (Smaczniak et al., 2012; Ali et al., 2019). However, several members of the MADS-box gene family are reported to be involved in the regulation of biotic and abiotic stress responses (Zhang et al., 2016; Wang Q. et al., 2018; Castelán-Muñoz et al., 2019), which point towards possible dynamic roles in stress response. In this study, we analysed the complete atlas of their expression profiles in all publicly available wheat transcriptomic data under biotic and abiotic stresses (Figure 6 and Supplementary Figure S4, Supplementary Table S7 and Supplementary Table S8). Except for AGL17, genes of all other MIKC-type subfamilies were ubiquitously expressed in FHB infected spikes/spikelets (Figure 6) mimicking their defensive roles against Fusarium infection. Previously, Yang et al. (2015) functionally characterized FgMcm1 in the causal agent (Fusarium graminearum) of barley and wheat head blight disease and demonstrated that FgMcm1 played crucial roles in cell identity and fungal development. More recently, Xu et al. (2020) also reported a close relationship between anther extrusion and field FHB resistance and pointed out that Rht and Vrn genes might have pleiotropic effects on these traits. Since FHB is a floral disease and floral organ identity is controlled by MIKC-type MADS-box genes (Ali et al., 2019), this indicates a cooperate relationship for their involvement in the pathogen response network. In future, it would be interesting to investigate how MADS-box genes are involved in fighting FHB infection.
Similarly, SVP, MIKC*, AGL12, and SOC1 subfamily genes were ubiquitously expressed under all studied stresses (Figure 6, Supplementary Table S7), pointing towards their more critical role in stress associated functions. In comparison, members of the largest MIKC group (AGL17) were expressed under phosphate starvation, and heat and drought stress conditions. Several functionally characterized genes belonging to all these subfamilies have also been reported to regulate different abiotic and/or biotic stresses in plants (Khong et al., 2015; Shi et al., 2016; Wang Z. et al., 2018; Hwang et al., 2019; Li P. et al., 2020). Interestingly, few M-type genes, especially Mα-like, were weakly expressed under different biotic stresses (Supplementary Table S7) and these expression patterns agree with Guo et al. (2013), who observed differential expression of an M-type gene in response to stripe rust infection in wheat. Collectively, these results strongly highlight MADS-box genes, especially MIKC-type, as key members of gene regulatory networks (Castelán-Muñoz et al., 2019) and their involvement in wheat response to possible pathogen insurgency and harsh environments.
Understanding the functions of candidate genes controlling agronomically important traits is critical for the acceleration of crop improvement efforts. Following the unveiling of recent wheat genetic and genomic resources (Avni et al., 2017; Luo et al., 2017; Maccaferri et al., 2019; Adamski et al., 2020), rapid testing of previous model plant discoveries and their cross-application in staple crops is paving the way towards bridging the gap between genotype and phenotype. During recent years, several wheat genes related to flowering, yield, quality, disease resistance, male sterility, and nutrient use efficiency traits have been cloned and functionally characterized [briefly reviewed by Li J. et al. (2020)], which exposed the unprecedented potential of these genetic and functional genomic resources in establishing genotype-to-phenotype relationships. These resources could also facilitate in elucidating the regulatory roles of neglected MADS-box subfamilies, such as Mα, Mβ, Mγ, and MIKC*, during wheat growth and development, and subsequently help in deciphering whether MADS-box genes confer pathogen resistance and/or abiotic stress tolerance. Furthermore, many other fundamental biological questions about their phylogenomics, evolutionary origin, and stress associated functions, could be addressed by exploiting these paramount resources.
MADS-box genes are critically important for wheat growth and development and hold enormous promise for bolstering yield potential under changing global environments. Our results speculate that the abundance of MADS-box genes in the wheat genome might be associated with its global adaptability and that subfamily-specific distal telomeric duplications in unbalanced homeologs facilitate its rapid adaptation. In addition, through comprehensive genome-wide and comparative analyses, we demonstrated that wheat and rice genomes might lack FLC-like genes, which could help molecular breeders in the identification of alternative target genes for fine-tuning of winter wheat varieties. Moreover, our in-silico expression data strongly indicated the possibility of protective roles of MADS-box genes against pathogen attacks and harsh climatic conditions. In this way, we provided an entire complement of MADS-box genes identified in the wheat genome that could accelerate functional genomics efforts and possibly facilitate in bridging genotype-to-phenotype relationships through fine-tuning of agronomically important traits.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
QR conceived and designed the study. QR, AR, RA, BH and IR retrieved the data. QR, AR and RA curated the data and performed final analyses. QR, AR and BH drafted the manuscript. QR, IR, ZA, HB and IA revised and finalized the manuscript. IA secured funding for research publication. All authors read and approved the final manuscript.
Author HB is employed by Montana BioAgriculture Incorporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
All authors are grateful to Dr Yuling Jiao, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences and the reviewers for their comments and suggestions on the manuscript. The inspiration and support from Precision Agriculture and Analytics Lab (PAAL), an affiliate of National Centre in Big Data and Cloud Computing (NCBC) is also gratefully acknowledged. A preprint version of the manuscript is publicly available at BioRxiv (Raza et al., 2020).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.818880/full#supplementary-material
Adamski, N. M., Borrill, P., Brinton, J., Harrington, S. A., Marchal, C., Bentley, A. R., et al. (2020). A Roadmap for Gene Functional Characterisation in Crops with Large Genomes: Lessons from Polyploid Wheat. Elife 9, e55646. doi:10.7554/eLife.55646
Ahmed, S., Rashid, M. A. R., Zafar, S. A., Azhar, M. T., Waqas, M., Uzair, M., et al. (2021). Genome-wide Investigation and Expression Analysis of APETALA-2 Transcription Factor Subfamily Reveals its Evolution, Expansion and Regulatory Role in Abiotic Stress Responses in Indica Rice (Oryza Sativa L. Ssp. Indica). Genomics 113, 1029–1043. doi:10.1016/J.YGENO.2020.10.037
Aleem, M., Riaz, A., Raza, Q., Aleem, M., Aslam, M., Kong, K., et al. (2022). Genome-wide Characterization and Functional Analysis of Class III Peroxidase Gene Family in Soybean Reveal Regulatory Roles of GsPOD40 in Drought Tolerance. Genomics 114, 45–60. doi:10.1016/J.YGENO.2021.11.016
Ali, Z., Raza, Q., Atif, R. M., Aslam, U., Ajmal, M., and Chung, G. (2019). Genetic and Molecular Control of floral Organ Identity in Cereals. Int. J. Mol. Sci. 20, 2743. doi:10.3390/ijms20112743
Alonge, M., Shumate, A., Puiu, D., Zimin, A. V., and Salzberg, S. L. (2020). Chromosome-Scale Assembly of the Bread Wheat Genome Reveals Thousands of Additional Gene Copies. Genetics 216, 599–608. doi:10.1534/genetics.120.303501
Alvarez-Buylla, E. R., Liljegren, S. J., Pelaz, S., Gold, S. E., Burgeff, C., Ditta, G. S., et al. (2000). MADS-box Gene Evolution beyond Flowers: Expression in Pollen, Endosperm, Guard Cells, Roots and Trichomes. Plant J. 24, 457–466. doi:10.1046/j.1365-313X.2000.00891.x
Andrés, F., and Coupland, G. (2012). The Genetic Basis of Flowering Responses to Seasonal Cues. Nat. Rev. Genet. 13, 627–639. doi:10.1038/nrg3291
Appels, R., Eversole, K., Appels, R., Eversole, K., Feuillet, C., Keller, B., et al. (2018). Shifting the Limits in Wheat Research and Breeding Using a Fully Annotated Reference Genome. Science 361, 361. doi:10.1126/science.aar7191
Arora, R., Agarwal, P., Ray, S., Singh, A. K., Singh, V. P., Tyagi, A. K., et al. (2007). MADS-box Gene Family in rice: Genome-wide Identification, Organization and Expression Profiling during Reproductive Development and Stress. BMC Genomics 8, 242. doi:10.1186/1471-2164-8-242
Avni, R., Nave, M., Barad, O., Baruch, K., Twardziok, S. O., Gundlach, H., et al. (2017). Wild Emmer Genome Architecture and Diversity Elucidate Wheat Evolution and Domestication. Science 357, 93–97. doi:10.1126/science.aan0032
Becker, A., and Theißen, G. (2003). The Major Clades of MADS-Box Genes and Their Role in the Development and Evolution of Flowering Plants. Mol. Phylogenet. Evol. 29 (3), 464–489. doi:10.1016/S1055-7903(03)00207-0
Bolser, D. M., Staines, D. M., Perry, E., and Kersey, P. J. (2017). Ensembl Plants: Integrating Tools for Visualizing, Mining, and Analyzing Plant Genomic Data. Methods Mol. Biol. 1533, 1–31. doi:10.1007/978-1-4939-6658-5_1
Borrill, P., Ramirez-Gonzalez, R., and Uauy, C. (2016). expVIP: A Customizable RNA-Seq Data Analysis and Visualization Platform. Plant Physiol. 170, 2172–2186. doi:10.1104/pp.15.01667
Brenchley, R., Spannagl, M., Pfeifer, M., Barker, G. L. A., D’Amore, R., Allen, A. M., et al. (2012). Analysis of the Bread Wheat Genome Using Whole-Genome Shotgun Sequencing. Nature 491, 705–710. doi:10.1038/nature11650
Budak, H., Hussain, B., Khan, Z., Ozturk, N. Z., and Ullah, N. (2015). From Genetics to Functional Genomics: Improvement in Drought Signaling and Tolerance in Wheat. Front. Plant Sci. 6, 1012. doi:10.3389/fpls.2015.01012
Castelán-Muñoz, N., Herrera, J., Cajero-Sánchez, W., Arrizubieta, M., Trejo, C., García-Ponce, B., et al. (2019). MADS-box Genes Are Key Components of Genetic Regulatory Networks Involved in Abiotic Stress and Plastic Developmental Responses in Plants. Front. Plant Sci. 10, 853. doi:10.3389/fpls.2019.00853
Chapman, J. A., Mascher, M., Buluç, A., Barry, K., Georganas, E., Session, A., et al. (2015). A Whole-Genome Shotgun Approach for Assembling and Anchoring the Hexaploid Bread Wheat Genome. Genome Biol. 16, 26. doi:10.1186/s13059-015-0582-8
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 13, 1194–1202. doi:10.1016/j.molp.2020.06.009
Chen, L., Zhao, Y., Xu, S., Zhang, Z., Xu, Y., Zhang, J., et al. (2018). OsMADS57 Together with OsTB1 Coordinates Transcription of its Target OsWRKY94 and D14 to Switch its Organogenesis to Defense for Cold Adaptation in rice. New Phytol. 218, 219–231. doi:10.1111/nph.14977
Chen, N. W. G., Thareau, V., Ribeiro, T., Magdelenat, G., Ashfield, T., Innes, R. W., et al. (2018). Common Bean Subtelomeres Are Hot Spots of Recombination and Favor Resistance Gene Evolution. Front. Plant Sci. 9, 1185. doi:10.3389/fpls.2018.01185
Chen, R., Ma, J., Luo, D., Hou, X., Ma, F., Zhang, Y., et al. (2019). CaMADS, a MADS-Box Transcription Factor from Pepper, Plays an Important Role in the Response to Cold, Salt, and Osmotic Stress. Plant Sci. 280, 164–174. doi:10.1016/j.plantsci.2018.11.020
Clavijo, B. J., Venturini, L., Schudoma, C., Accinelli, G. G., Kaithakottil, G., Wright, J., et al. (2017). An Improved Assembly and Annotation of the Allohexaploid Wheat Genome Identifies Complete Families of Agronomic Genes and Provides Genomic Evidence for Chromosomal Translocations. Genome Res. 27, 885–896. doi:10.1101/gr.217117.116
Colombo, L., Franken, J., Koetje, E., Van Went, J., Dons, H. J., Angenent, G. C., et al. (1995). The Petunia MADS Box Gene FBP11 Determines Ovule Identity. Plant Cell 7, 1859–1868. doi:10.1105/tpc.7.11.1859
De Bodt, S., Raes, J., Van De Peer, Y., and Theißen, G. (2003). And Then There Were many: MADS Goes Genomic. Trends Plant Sci. 8, 475–483. doi:10.1016/j.tplants.2003.09.006
Díaz-Riquelme, J., Lijavetzky, D., Martínez-Zapater, J. M., and Carmona, M. J. (2009). Genome-wide Analysis of MIKCC-type MADS Box Genes in grapevine. Plant Physiol. 149, 354–369. doi:10.1104/pp.108.131052
Distelfeld, A., Li, C., and Dubcovsky, J. (2009). Regulation of Flowering in Temperate Cereals. Curr. Opin. Plant Biol. 12, 178–184. doi:10.1016/j.pbi.2008.12.010
Duan, W., Song, X., Liu, T., Huang, Z., Ren, J., Hou, X., et al. (2014). Genome-wide Analysis of the MADS-Box Gene Family in Brassica Rapa (Chinese Cabbage). Mol. Genet. Genomics 290, 239–255. doi:10.1007/s00438-014-0912-7
El Baidouri, M., Murat, F., Veyssiere, M., Molinier, M., Flores, R., Burlot, L., et al. (2017). Reconciling the Evolutionary Origin of Bread Wheat ( Triticum aestivum ). New Phytol. 213, 1477–1486. doi:10.1111/nph.14113
Fatima, M., Zhang, X., Lin, J., Zhou, P., Zhou, D., and Ming, R. (2020). Expression Profiling of MADS-Box Gene Family Revealed its Role in Vegetative Development and Stem Ripening in S. Spontaneum. Sci. Rep. 10, 20536. doi:10.1038/s41598-020-77375-6
Glover, N. M., Daron, J., Pingault, L., Vandepoele, K., Paux, E., Feuillet, C., et al. (2015). Small-scale Gene Duplications Played a Major Role in the Recent Evolution of Wheat Chromosome 3B. Genome Biol. 16, 188. doi:10.1186/s13059-015-0754-6
Gramzow, L., and Theißen, G. (2015). Phylogenomics Reveals Surprising Sets of Essential and Dispensable Clades of MIKCc-Group MADS-Box Genes in Flowering Plants. J. Exp. Zool. (Mol. Dev. Evol. 324, 353–362. doi:10.1002/jez.b.22598
Greenup, A. G., Sasani, S., Oliver, S. N., Talbot, M. J., Dennis, E. S., Hemming, M. N., et al. (2010). ODDSOC2 Is a MADS Box floral Repressor that Is Down-Regulated by Vernalization in Temperate Cereals. Plant Physiol. 153 (3), 1062–1073. doi:10.1104/pp.109.152488
Greenup, A., Peacock, W. J., Dennis, E. S., and Trevaskis, B. (2009). The Molecular Biology of Seasonal Flowering-Responses in Arabidopsis and the Cereals. Ann. Bot. 103, 1165–1172. doi:10.1093/aob/mcp063
Guo, J., Shi, X.-X., Zhang, J.-S., Duan, Y.-H., Bai, P.-F., Guan, X.-N., et al. (2013). A Type I MADS-Box Gene Is Differentially Expressed in Wheat in Response to Infection by the Stripe Rust Fungus. Biol. Plant 57, 540–546. doi:10.1007/s10535-012-0297-6
Henschel, K., Kofuji, R., Hasebe, M., Saedler, H., Münster, T., and Theißen, G. (2002). Two Ancient Classes of MIKC-type MADS-Box Genes Are Present in the moss Physcomitrella Patens. Mol. Biol. Evol. 19, 801–814. doi:10.1093/oxfordjournals.molbev.a004137
Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q., and Vinh, L. S. (2018). UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 35, 518–522. doi:10.1093/molbev/msx281
Huang, S., Liang, Z., Chen, S., Sun, H., Fan, X., Wang, C., et al. (2019). A Transcription Factor, OsMADS57, Regulates Long-Distance Nitrate Transport and Root Elongation. Plant Physiol. 180 (2), 882–895. doi:10.1104/pp.19.00142
Hwang, K., Susila, H., Nasim, Z., Jung, J.-Y., and Ahn, J. H. (2019). Arabidopsis ABF3 and ABF4 Transcription Factors Act with the NF-YC Complex to Regulate SOC1 Expression and Mediate Drought-Accelerated Flowering. Mol. Plant 12, 489–505. doi:10.1016/j.molp.2019.01.002
Igrejas, G., and Branlard, G. (2020). “The Importance of Wheat,” in Wheat Quality for Improving Processing and Human Health. Editors G. Igrejas, T. Ikeda, and C. Guzmán (Cham, Switzerland: Springer, Cham), 1–7.
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 14, 587–589. doi:10.1038/nmeth.4285
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform 20, 1160–1166. doi:10.1093/bib/bbx108
Katoh, K., and Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780. doi:10.1093/molbev/mst010
Kaufmann, K., Melzer, R., and Theißen, G. (2005). MIKC-type MADS-Domain Proteins: Structural Modularity, Protein Interactions and Network Evolution in Land Plants. Gene 347, 183–198. doi:10.1016/j.gene.2004.12.014
Khong, G. N., Pati, P. K., Richaud, F., Parizot, B., Bidzinski, P., Mai, C. D., et al. (2015). OsMADS26 Negatively Regulates Resistance to Pathogens and Drought Tolerance in rice. Plant Physiol. 169 (4), 2935–2949. doi:10.1104/pp.15.01192
Kugler, K. G., Siegwart, G., Nussbaumer, T., Ametz, C., Spannagl, M., Steiner, B., et al. (2013). Quantitative Trait Loci-dependent Analysis of a Gene Co-expression Network Associated with Fusarium Head Blight Resistance in Bread Wheat (Triticum aestivum L.). BMC Genomics 14, 728. doi:10.1186/1471-2164-14-728
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. doi:10.1093/molbev/msw054
Kuo, M. H., Nadeau, E. T., and Grayhack, E. J. (1997). Multiple Phosphorylated Forms of the Saccharomyces cerevisiae Mcm1 Protein Include an Isoform Induced in Response to High Salt Concentrations. Mol. Cel. Biol. 17, 819–832. doi:10.1128/mcb.17.2.819
Li, J., Yang, J., Li, Y., and Ma, L. (2020). Current Strategies and Advances in Wheat Biology. Crop J. 8, 879–891. doi:10.1016/j.cj.2020.03.004
Li, P., Zhang, Q., He, D., Zhou, Y., Ni, H., Tian, D., et al. (2020). AGAMOUS-LIKE67 Cooperates with the Histone Mark Reader EBS to Modulate Seed Germination under High Temperature. Plant Physiol. 184 (1), 529–545. doi:10.1104/pp.20.00056
Lukaszewski, A. J., Alberti, A., Sharpe, A., Kilian, A., Stanca, A. M., Keller, B., et al. (2014). A Chromosome-Based Draft Sequence of the Hexaploid Bread Wheat (Triticum aestivum) Genome. Science 345, 1251788. doi:10.1126/science.1251788
Luo, M.-C., Gu, Y. Q., Puiu, D., Wang, H., Twardziok, S. O., Deal, K. R., et al. (2017). Genome Sequence of the Progenitor of the Wheat D Genome Aegilops Tauschii. Nature 551, 498–502. doi:10.1038/nature24486
Ma, J., Yang, Y., Luo, W., Yang, C., Ding, P., Liu, Y., et al. (2017). Genome-wide Identification and Analysis of the MADS-Box Gene Family in Bread Wheat (Triticum aestivum L.). PLoS One 12, e0181443. doi:10.1371/journal.pone.0181443
Maccaferri, M., Harris, N. S., Twardziok, S. O., Pasam, R. K., Gundlach, H., Spannagl, M., et al. (2019). Durum Wheat Genome Highlights Past Domestication Signatures and Future Improvement Targets. Nat. Genet. 51, 885–895. doi:10.1038/s41588-019-0381-3
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI's Conserved Domain Database. Nucleic Acids Res. 43, D222–D226. doi:10.1093/nar/gku1221
Masiero, S., Colombo, L., Grini, P. E., Schnittger, A., and Kater, M. M. (2011). The Emerging Importance of Type I MADS Box Transcription Factors for Plant Reproduction. Plant Cell 23, 865–872. doi:10.1105/tpc.110.081737
Messenguy, F., and Dubois, E. (2003). Role of MADS Box Proteins and Their Cofactors in Combinatorial Control of Gene Expression and Cell Development. Gene 316, 1–21. doi:10.1016/S0378-1119(03)00747-9
Minh, B. Q., Nguyen, M. A. T., and Von Haeseler, A. (2013). Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi:10.1093/molbev/mst024
Moore, S., Vrebalov, J., Payton, P., and Giovannoni, J. (2002). Use of Genomics Tools to Isolate Key Ripening Genes and Analyse Fruit Maturation in Tomato. J. Exp. Bot. 53, 2023–2030. doi:10.1093/jxb/erf057
Muhire, B. M., Varsani, A., and Martin, D. P. (2014). SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS One 9, e108277. doi:10.1371/journal.pone.0108277
Nam, J., Kim, J., Lee, S., An, G., Ma, H., and Nei, M. (2004). Type I MADS-Box Genes Have Experienced Faster Birth-And-Death Evolution Than Type II MADS-Box Genes in Angiosperms. Pnas 101, 1910–1915. doi:10.1073/pnas.0308430100
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 32, 268–274. doi:10.1093/molbev/msu300
Ning, P., Liu, C., Kang, J., and Lv, J. (2017). Genome-wide Analysis of WRKY Transcription Factors in Wheat (Triticum aestivum L.) and Differential Expression under Water Deficit Condition. PeerJ 5, e3232. doi:10.7717/peerj.3232
Paolacci, A. R., Tanzarella, O. A., Porceddu, E., Varotto, S., and Ciaffi, M. (2007). Molecular and Phylogenetic Analysis of MADS-Box Genes of MIKC Type and Chromosome Location of SEP-like Genes in Wheat (Triticum aestivum L.). Mol. Genet. Genomics 278, 689–708. doi:10.1007/s00438-007-0285-2
Par̆enicová, L., De Folter, S., Kieffer, M., Horner, D. S., Favalli, C., Busscher, J., et al. (2003). Molecular and Phylogenetic Analyses of the Complete MADS-Box Transcription Factor Family in Arabidopsis. Plant Cell 15, 1538–1551. doi:10.1105/tpc.011544
Ramírez-González, R. H., Borrill, P., Lang, D., Harrington, S. A., Brinton, J., Venturini, L., et al. (2018). The Transcriptional Landscape of Polyploid Wheat. Science 361, 361. doi:10.1126/science.aar6089
Raza, Q., Riaz, A., Atif, R. M., Hussain, B., Ali, Z., and Budak, H. (2020). MADS-box Genes Galore in Wheat Genome: Phylogenomics, Evolution and Stress Associated Functions. bioRxiv. doi:10.1101/2020.10.23.351635
Riechmann, J. L., and Meyerowitz, E. M. (1997). MADS Domain Proteins in Plant Development. Biol. Chem. 378 (10), 1079–1101. doi:10.1515/bchm.1997.378.10.1079
Rounsley, S. D., Ditta, G. S., and Yanofsky, M. F. (1995). Diverse Roles for MADS Box Genes in Arabidopsis Development. Plant Cell 7, 1259–1269. doi:10.1105/tpc.7.8.1259
Ruelens, P., De Maagd, R. A., Proost, S., Theißen, G., Geuten, K., and Kaufmann, K. (2013). FLOWERING LOCUS C in Monocots and the Tandem Origin of Angiosperm-specific MADS-Box Genes. Nat. Commun. 4, 2280. doi:10.1038/ncomms3280
Saedler, H., Becker, A., Winter, K. U., Kirchner, C., and Theissen, G. (2001). MADS-box Genes Are Involved in floral Development and Evolution. Acta Biochim. Pol. 48, 351–358. doi:10.18388/abp.2001_3920
Samach, A., Onouchi, H., Gold, S. E., Ditta, G. S., Schwarz-Sommer, Z., Yanofsky, M. F., et al. (2000). Distinct Roles of Constans Target Genes in Reproductive Development of Arabidopsis. Science 288, 1613–1616. doi:10.1126/science.288.5471.1613
Schilling, S., Kennedy, A., Pan, S., Jermiin, L. S., and Melzer, R. (2020). Genome‐wide Analysis of MIKC ‐type MADS ‐box Genes in Wheat: Pervasive Duplications, Functional Conservation and Putative Neofunctionalization. New Phytol. 225, 511–529. doi:10.1111/nph.16122
Sharma, N., Ruelens, P., D’Hauw, M., Maggen, T., Dochy, N., Torfs, S., et al. (2017). A Flowering Locus C Homolog Is a Vernalization-Regulated Repressor in Brachypodium and Is Cold Regulated in Wheat. Plant Physiol. 173 (2), 1301–1315. doi:10.1104/pp.16.01161
Shi, S. ya., Zhang, F. fei., Gao, S., and Xiao, K. (2016). Expression Pattern and Function Analyses of the MADS Thranscription Factor Genes in Wheat (Triticum aestivum L.) under Phosphorus-Starvation Condition. J. Integr. Agric. 15 (8), 1703–1715. doi:10.1016/S2095-3119(15)61167-4
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, Scalable Generation of High‐quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 7, 539. doi:10.1038/msb.2011.75
Smaczniak, C., Immink, R. G. H., Angenent, G. C., and Kaufmann, K. (2012). Developmental and Evolutionary Diversity of Plant MADS-Domain Factors: Insights from Recent Studies. Dev 139, 3081–3098. doi:10.1242/dev.074674
Wang, Q., Du, M., Wang, S., Liu, L., Xiao, L., Wang, L., et al. (2018). MADS-box Transcription Factor madsA Regulates Dimorphic Transition, Conidiation, and Germination of Talaromyces marneffei. Front. Microbiol. 9, 1781. doi:10.3389/fmicb.2018.01781
Wang, Z., Wang, F., Hong, Y., Yao, J., Ren, Z., Shi, H., et al. (2018). The Flowering Repressor SVP Confers Drought Resistance in Arabidopsis by Regulating Abscisic Acid Catabolism. Mol. Plant 11, 1184–1197. doi:10.1016/j.molp.2018.06.009
Wei, B., Zhang, R.-Z., Guo, J.-J., Liu, D.-M., Li, A.-L., Fan, R.-C., et al. (2014). Genome-wide Analysis of the MADS-Box Gene Family in Brachypodium Distachyon. PLoS One 9, e84781. doi:10.1371/journal.pone.0084781
Whittaker, C., and Dean, C. (2017). The FLC Locus: A Platform for Discoveries in Epigenetics and Adaptation. Annu. Rev. Cel Dev. Biol. 33, 555–575. doi:10.1146/annurev-cellbio-100616-060546
Wu, Y., Ke, Y., Wen, J., Guo, P., Ran, F., Wang, M., et al. (2018). Evolution and Expression Analyses of the MADS-Box Gene Family in Brassica Napus. PLoS One 13, e0200762. doi:10.1371/journal.pone.0200762
Xu, K., He, X., Dreisigacker, S., He, Z., and Singh, P. K. (2020). Anther Extrusion and its Association with Fusarium Head Blight in CIMMYT Wheat Germplasm. Agronomy 10, 47. doi:10.3390/agronomy10010047
Yang, C., Liu, H., Li, G., Liu, M., Yun, Y., Wang, C., et al. (2015). The MADS-Box Transcription Factor FgMcm1 Regulates Cell Identity and Fungal Development inFusarium Graminearum. Environ. Microbiol. 17, 2762–2776. doi:10.1111/1462-2920.12747
Yanofsky, M. F., Ma, H., Bowman, J. L., Drews, G. N., Feldmann, K. A., and Meyerowitz, E. M. (1990). The Protein Encoded by the Arabidopsis Homeotic Gene Agamous Resembles Transcription Factors. Nature 346, 35–39. doi:10.1038/346035a0
Yu, Y., Ouyang, Y., and Yao, W. (2018). ShinyCircos: An R/Shiny Application for Interactive Creation of Circos Plot. Bioinformatics 34, 1229–1231. doi:10.1093/bioinformatics/btx763
Zhang, Z., Li, H., Qin, G., He, C., Li, B., and Tian, S. (2016). The MADS-Box Transcription Factor Bcmads1 Is Required for Growth, Sclerotia Production and Pathogenicity of Botrytis Cinerea. Sci. Rep. 6, 33901. doi:10.1038/srep33901
Zhao, Y., Li, X., Chen, W., Peng, X., Cheng, X., Zhu, S., et al. (2011). Whole-genome Survey and Characterization of MADS-Box Gene Family in maize and Sorghum. Plant Cel Tiss Organ. Cult 105, 159–173. doi:10.1007/s11240-010-9848-8
Keywords: genome-wide analysis, in-silico expression, TaMADS’s, transcription factors, Triticum aestivum, wheat adaptability
Citation: Raza Q, Riaz A, Atif RM, Hussain B, Rana IA, Ali Z, Budak H and Alaraidh IA (2022) Genome-Wide Diversity of MADS-Box Genes in Bread Wheat is Associated with its Rapid Global Adaptability. Front. Genet. 12:818880. doi: 10.3389/fgene.2021.818880
Received: 20 November 2021; Accepted: 21 December 2021;
Published: 17 January 2022.
Edited by:
Reyazul Rouf Mir, Sher-e-Kashmir University of Agricultural Sciences and Technology, IndiaReviewed by:
Brijesh Kumar, Patanjali Research Institute, IndiaCopyright © 2022 Raza, Riaz, Atif, Hussain, Rana, Ali, Budak and Alaraidh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qasim Raza, cWFzaW1uYXphbWlAZ21haWwuY29t; Ibrahim A. Alaraidh, aWFsYXJhaWRoQGtzdS5lZHUuc2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.