
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 04 March 2022
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.816779
This article is part of the Research Topic Inherited Metabolic Diseases in Pediatrics: Clinical and Molecular Features View all 25 articles
 Shengnan Wu1
Shengnan Wu1 Linghua Shen1
Linghua Shen1 Qiong Chen1
Qiong Chen1 Chunxiu Gong2
Chunxiu Gong2 Yanling Yang3
Yanling Yang3 Haiyan Wei1
Haiyan Wei1 Bingyan Cao2*
Bingyan Cao2* Yongxing Chen1*
Yongxing Chen1*Background: Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency (HMGCS2D) is a rare autosomal recessive metabolic disorder caused by mutations of the HMGCS2 gene. To date, no more than 60 patients have been reported throughout the world.
Purpose: To analyze the clinical, biochemical, molecular, and outcome features of HMGCS2D in a case series of 10 new Chinese patients.
Methods: This retrospective study includes 10 Chinese patients diagnosed with HMGCS2D. We collected and analyzed clinical data for all patients. We also reviewed clinical data for 39 cases that had been reported previously.
Results: All of our patients had experienced their first metabolic crisis before 12 months old. The most common clinical manifestations were anorexia, dyspnea, and disturbance of consciousness (10/10), followed by vomiting (8/10), fever (7/10), cough (4/10), diarrhea, and seizures (3/10). Each patient (10/10) had a different degree of hepatomegaly and increased aminotransferase, severe metabolic acidosis, and hypofibrinogenemia. 9 patients presented with severe hypoglycemia and weak positives on qualitative tests of urinary ketone body. Patient 3 was the only one without hypoglycemia. Five patients had hypocalcemia, five patients had hyperammonemia, four patients had hyperuricemia, and three had hypertriglyceridemia. During the metabolic acidosis episode, we observed high dicarboxylic acid values in urine, and the elevated ratio of blood acetylcarnitine to free carnitine may have been an additional biochemical signature. However, all returned to normal during the interictal interval. Molecular analysis identified 15 variants in the HMGCS2 gene, of which 10 were novel (c.220G>A/p.E74K, c.407A>G/p.D136G, c.422T>A/p.V141D, c.719A>C/p.D240A, c.821G>A/p.R274H, c.39dupA/p.L14Tfs*59, c.1394delA/p.N465Tfs*10, c.788delT/p.L263Cfs*36, c.717T>G/p.Y239*, and c.1017-2A>G). Combining these with previous cases, the known mutation c.1201G>T/p.E401* has been found in 6/40 (15.0%) of mutated alleles in 21 Chinese patients from 20 families, while none have been found in other populations. We found that patients with biallelic truncation mutation appeared to show a more severe clinical condition through a literature review.
Conclusion: This study analyzed the phenotypic and genetic features of HMGCS2D in a Chinese case series. We also expanded the HMGCS2 mutational spectrum with 10 novel variants. The c.1201G>T/p.E401* mutation was the most frequent, representing 15.0% of the mutated alleles in reported unrelated Chinese patients, and thus, it may be a hot spot mutation.
Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency (mitochondrial HMG-CoA synthase deficiency; HMGCS2D; OMIM#605911) is a rare autosomal recessive metabolic disorder caused by mutations in the HMGCS2 gene (OMIM*600234). The HMGCS2 gene encodes mitochondrial HMG-CoA synthase, which catalyzes the reactions of acetyl-CoA and acetoacetyl-CoA into HMG-CoA during ketone body synthesis. Ketone bodies are an important energy supply for the brain, heart and kidneys during long-term fasting and carbohydrate deprivation. Pathogenic mutations on the HMGCS2 gene cause mitochondrial HMG-CoA synthase deficiency which disrupts the ketogenesis and blocks the energy supply to the brain during the fasting state. Patients present with intermittent vomiting, lethargy, respiratory distress, encephalopathy, and hepatomegaly, usually precipitated by an intercurrent infection or prolonged fasting (Aledo et al., 2006). Hypoketotic hypoglycemia, metabolic acidosis, increased transaminitis and dicarboxylic aciduria are common laboratory findings during the acute attack stage. Nevertheless, the clinical manifestations and biochemical abnormalities usually return to normal during the interictal period. In 1997, Thompson described the first case of mitochondrial HMGCS2D (Thompson et al., 1997). To date, no more than 60 patients carrying over 40 variants have been reported throughout the world among different ethnic groups. Among them, only 11 patients, from 10 families, are of Chinese descent.
In the present study, we report 10 new Chinese patients with HMGCS2D, and summarize their clinical, biochemical, molecular and outcome characteristics. We reviewed 39 patients with relatively complete clinical data reported for overall understanding of the molecular genetics features of Chinese patients, and to explore the correlation between genotype and clinical features and disease severity. This has not been reported in previous studies.
Ten unrelated patients (two boys and eight girls) from different Chinese families identified between 2015 and 2021 were included in this retrospective analysis. They had each been diagnosed with HMGCS2D based on clinical features (encephalopathy, and hepatomegaly, usually precipitated by an intercurrent infection or prolonged fasting), biochemical detection results (hypoketotic hypoglycemia, transaminitis, metabolic acidosis and dicarboxylic aciduria), as well as molecular analysis. We collected clinical data through the review of patients’ medical records, including the age at onset, primary clinical symptoms, medical management, and biochemical and clinical outcomes following therapy.
We performed routine blood and urine examination to assess blood gas analysis, ammonia, glucose, liver function, renal function, blood lipids and plasma electrolytes. Then, we applied tandem mass spectrometry to test serum amino acids and acylcarnitines, and analyzed the results using ChemoView software. To analyze urine organic acids, we used gas chromatography-mass spectrometry and the Inborn Errors of Metabolism Screening System. We also performed abdominal ultrasonography and cranial MRI on most patients.
We extracted genomic DNA from peripheral blood leucocytes using a QIAamp DNA Blood Midi kit (Qiagen, Hilden, Germany), and generated sequences using the Agilent Bioanalyzer. Then, we applied whole-exome sequencing for mutation screening. Additionally, we performed Sanger sequencing validation on all patients who we found to be harbouring gene mutations, as well as their parents. The pathogenicity of novel variants was evaluated according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines (Richards et al., 2015). For the novel mis-sense variants, multiple sequence alignment studies were performed to verify the amino acid conservation using the UCSC Genome Bioinformatics Database, and potential pathogenicity was analyzed using Mutation taster, PolyPhen-2, and Sorting Intolerant From Tolerant (SIFT). Swiss model (https://swissmodel.expasy.org/) was also used to predict the protein 3D structure and evaluate the impact of these novel missense variants on protein structure.
When initial metabolic decompensation occurred, all patients were admitted to the hospital and received regular supportive treatment including intravenous glucose and sodium bicarbonate. We also administered liver-protecting and carnitine therapy. We gave mechanical ventilation and continuous renal replacement therapy to any patients who were critically ill because of persistent metabolic acidosis and multiple organ dysfunction. Once the HMGCS2D diagnosis was confirmed, all patients were instructed to avoid long periods of fasting, and put on a low-fat diet.
All patients were enrolled into a simple follow-up study. During the follow-up period, each patient had several telephone survey or outpatient visits. We performed physical growth and psychomotor evaluation, as well as routine laboratory tests such as urine organic acids, plasma amino acids and acylcarnitines and other biochemical investigations, every 6–12 months. Psychomotor evaluation was based on the intellectual test results according to Gesell Developmental Schedules at the last follow-up.
For overall understanding of Chinese patients’ molecular genetics features, and to explore the correlation between genotype and clinical features and disease severity, we reviewed 39 previously reported patients with relatively complete clinical data, including the genetic analysis results. In our study, we divided the patients into 3 phenotypic groups according to their clinical severity: A deceased group, a severe group, and a mild group. The deceased group included patients who had died because of disease onset. The severe group included patients who met one of the following conditions: Recurrent attacks on multiple occasions; blood pH value lower than 7.0 in metabolic decompensation; had required mechanical ventilation or CRRT treatment in metabolic decompensation; had become cognitively disabled after disease onset. The mild group included patients with milder symptoms. According to their disease-causing variation type, we divided them into 3 genotypic groups: The A group included patients carrying 2 truncating mutations on the HMGCS2 gene; the B group included patients carrying only 1 truncating mutation on the HMGCS2 gene; and the C group included patients carrying no truncating mutations.
We performed statistical analysis in SPSS 22.0. We analyzed the correlations between different phenotypic groups and genotypic groups with a Cochran-Mantel-Haenszel test. We considered any results with p < 0.05 as having a statistically significant difference.
As summarized in Table 1, there were 8 females and 2 males in our study. All of their parents were non-consanguineous. Among the 10 cases, patient 1 had a deceased sister who had died of a sudden coma after respiratory infections at the age of 8 months, while patient 4 had an older brother who had died of Reye Syndrome at 1 year and 10 months. For patient 10, his mother had experienced spontaneous abortion in her first two pregnancies with no specific cause. None of the other patients had positive family history. Patients had experienced their first metabolic crises at ages ranging from 5 to 12 months. The most common clinical manifestations were anorexia, fever and cough, which then developed into vomiting, dyspnea and disturbance of consciousness. The time from onset to progression to dyspnea and disturbance of consciousness ranged from 2 to 7 days. During the course of disease, all patients had anorexia, dyspnea and disturbance of consciousness (10/10). The second most common symptom was vomiting (8/10), followed by fever (7/10), cough (4/10), diarrhea and seizures (3/10).
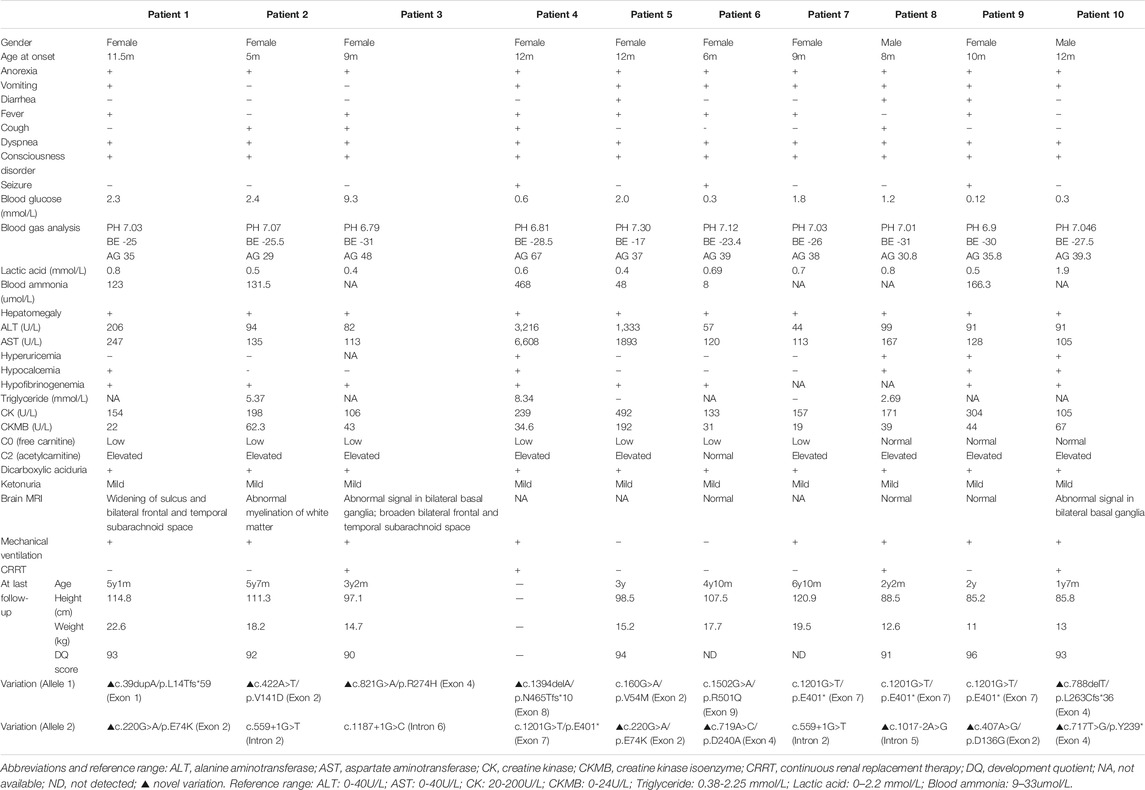
TABLE 1. Clinical information, biochemical assays, and outcome findings of 10 new patients with HMGCS2D.
Routine biochemical laboratory results at onset are shown in Table 1. Each patient had a different degree of hepatomegaly, about 2–7 cm below the right costal margin. Laboratory examinations showed severe metabolic acidosis (10/10), increased aminotransferase from mild to severe (10/10), hypofibrinogenemia (10/10), slightly increased creatine kinase isoenzyme (8/10), increased creatine kinase (3/10), and normal lactate (10/10). Among all patients, 9 presented with severe hypoglycemia and weak positives in qualitative urinary ketone body tests, while we observed no hypoglycemia in Patient 3. Five patients (Patient 1, 2, 4, 5 and 9) had hyperammonemia. Five (Patient 1, 2, 4, 9 and 10) had hypocalcemia. Three (Patient 2, 4, and 8) had hypertriglyceridemia. Eight received cranial MRI examinations, and 3 cases (Patient 1, 2 and 3) showed widening of the sulcus and bilateral frontal and temporal subarachnoid space, two (Patient 3 and 10) showed abnormal signal in the basal ganglia, one (Patient 2) showed delayed myelination in white matter, while four (Patient 6, 7, 8 and 9) were normal. During acute metabolic decompensation, eight patients (all except for Patient 5 and 6) received mechanical ventilation for respiratory failure resulting from serious metabolic acidosis. Four (Patient 3, 4, 8, and 10) had to undergo continuous renal replacement therapy (CRRT) because of serious and/or obstinate metabolic acidosis.
We performed urine organic acid analysis during the acute episode of metabolic acidosis. While we detected a massive amount of dicarboxylic acid in all patients, ketone body excretion was only mildly elevated. Plasma amino acids and acylcarnitine analysis revealed that free carnitine levels were low with a mildly increased acetylcarnitine in Patients 1–3, 5 and 7. Patients 4 and 6 had decreased free carnitine but normal acetylcarnitine, while Patients 8–10 had normal free carnitine but elevated acetylcarnitine.
We performed molecular analysis on all patients, and detected biallelic mutations in the HMGCS2 gene in all of them. In our study, we identified fifteen molecular variants in the HMGCS2 gene, ten of which were novel. Five were known mutations, including 2 missense mutations (c.160G > A/p.V54M, c.1502G > A/p.R501Q), 2 splicing mutations (c.559+1G > T, c.1187+1G > C) and one nonsense mutation (c.1201G > T/p.E401*). The ten novel potentially pathogenic variants have not been previously reported in the literature or registered in the HGMD, ClinVarMiner, ExAC or gnomAD databases. This includes the 5 missense mutations (c.220G > A/p.E74K, c.407A > G/p.D136G, c.422T > A/p.V141D, c.719A > C/p.D240A, c.821G > A/p.R274H), 3 frame-shift mutations (c.39dupA/p.L14Tfs*59, c.1394delA/p.N465Tfs*10, c.788delT/p. L263Cfs*36), one nonsense mutation (c.717T > G/p.Y239*) and one splicing mutation (c.1017-2A > G). Multiple sequence alignment studies suggested all the amino acids were conserved. Multiple lines of computational evidence supported a deleterious effect on the gene about the novel mutations. Detailed results of the 15 variants and prediction of effects of some novel mutations are shown in Table 2. In addition, we have performed a computational evaluation of the impact of the novel missense variants on protein structural using Swiss-model software. The results show that the 3D structure of HMGCS2 protein may be affected by these 5 novel mis-sense variants. Detailed results were shown in Supplementary Figure S1.
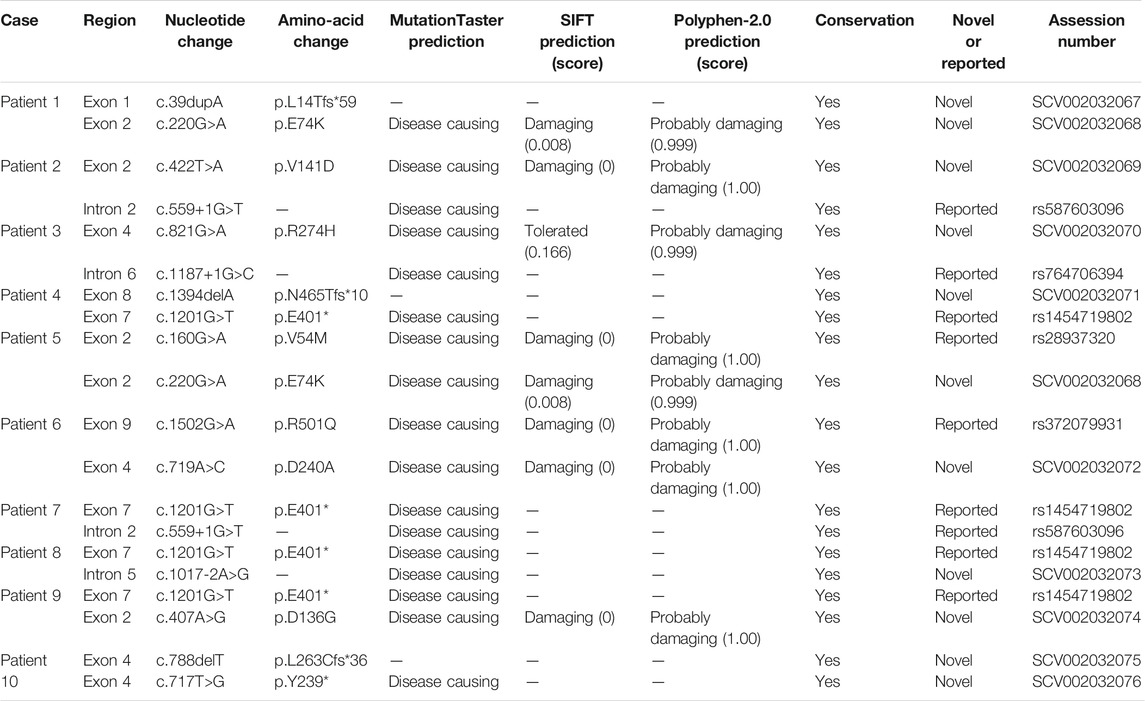
TABLE 2. Molecular analysis of HMGCS2 gene identified in 10 new Chinese patients with HMGCS2D.
Except for Patient 4 who died of multiple organ failure due to severe metabolic disorder at onset, all patients had complete recovery with symptomatic and supportive treatment. Each maintained well-being by avoiding long-term fasting and taking glucose supplements during illness. Hitherto, all 9 cases had followed up, made good progress, and reported no further episodes. The follow-up time varied from 7 months to 5 years and 3 months. As of October 2021, their ages ranged from 2 to 6 years and 9 months. During follow-up, we surveyed the routine laboratory tests including plasma amino acids and acylcarnitines, urine organic acids and other biochemical investigations. All results were within the normal range. We also summarized patients’ physical growth and developmental quotients (DQ) at the latest follow up, and found normal outcomes in all patients. Both height and weight were between mean±1SD. For the four individuals older than 4 years (Patients 1, 2, 6, and 7), all performed well in school. Additionally, DQ tests revealed that all patients examined had age-appropriate intelligence. Detailed results are shown in Table 1.
A review of previous literature has revealed that from October 1997 to January 2021, 59 patients, carrying 54 variants of the HMGCS2 gene, have been reported. Among them, only 11 individuals, from 10 families, were of Chinese descent. Including the 10 cases in our research, 27 mutations have been identified in the 21 Chinese patients from 20 families, including 15 (15/27, 55.6%) missense mutations, 5 (5/27, 18.5%) frame-shift mutations, 4 (4/27, 14.8%) nonsense mutations and 3 (3/27, 11.1%) splicing mutations. The frequency of the HMGCS2 variants in the 20 unrelated Chinese patients is shown in Table 3. The recurrent HMGCS2 mutations in the Chinese population were c.1201G > T/p.E401* (detected in 7 individuals from 6 unrelated families, mutated allele frequency 6/40, 15.0%), c.559+1G > T, c.1187+1G > C and c.1502G > A/p.R501Q (respectively detected in 3 mutated alleles, 3/40, 7.5%), c.220G > A/p.E74K (detected in 2 unrelated individuals, mutated allele frequency 2/40, 5.0%).
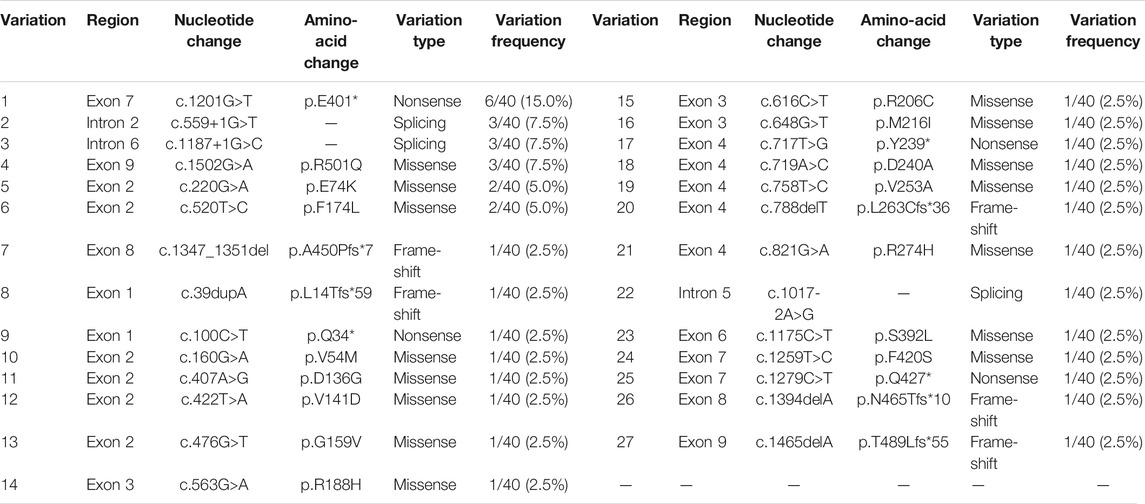
TABLE 3. The frequency of HMGCS2 variations in 20 unrelated Chinese patients (total number of mutated alleles = 40).
Including our 10 patients and the previously reported 39 patients with relatively complete clinical data, we analysed the clinical and molecular characteristics of 49 patients to explore the correlation between genotype and clinical features and disease severity. Detailed clinical data is presented in Supplementary Table S1, and the statistical analysis results are shown in Table 4. 16 patients carried biallelic truncating mutations (either nonsense, frame-shift or splicing mutation) (Group A). During acute metabolic decompensation, 4 of them (25%) died, and 7 (43.7%) had severe phenotypes. 13 patients carried monoallelic truncating mutations (Group B). Among them, one patient (7.7%) died, and 10 (76.9%) had severe phenotypes. For the 20 patients carrying biallelic non-truncating mutations (Group C), only one (5%) died, and 7 (35%) showed severe phenotypes, while 12 (60%) had mild symptoms. From another perspective, within the deceased group 66.7% (4/6) had 2 truncating mutations; within the severe group 70.9% (17/24) had at least 1 truncating mutation; and within the mild group 63.2% (12/19) had non truncating mutations. We analysed the correlation between phenotypic groups and genotypic groups with a Cochran-Mantel-Haenszel test. The results indicated that disease severity was correlated with how many truncating variations the patients carried (p < 0.05).
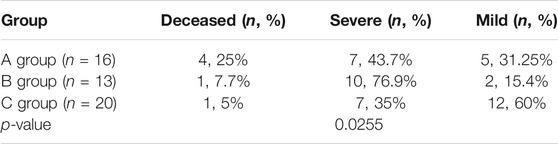
TABLE 4. Analysis of the correlation between severity of disease and number of truncating variations patients carried.
Mitochondrial HMGCS2D is a life-threatening, but treatable, inherited metabolic disorder caused by a defect in the critical enzyme that regulates ketone body formation (Aledo et al., 2006). Therefore, patients with HMGCS2D are prone to episodic metabolic decompensation triggered by fasting or stressful conditions that require fatty acid decomposition for the provision of energy. In the present study, we reported 10 new unrelated Chinese patients with HMGCS2D. All patients had suffered their first metabolic crisis before their first birthday. Prior to metabolic crisis, there had been prodromic infection of either the digestive tract or the respiratory tract in all cases. The most common clinical manifestations before admission were poor intake, fever, cough and vomiting, which progressed rapidly to life-threatening dyspnea and coma. One third of patients had seizures, although each had only one throughout the disease course. Nonetheless, this still suggests there may have been an insufficient energy supply to the brain which may have harmed the nervous system, or even caused sequelae. All of our patients presented with severe metabolic acidosis with significantly increased anion gap, hepatomegaly, increased aminotransferase, and abnormal coagulation function, but normal lactate. Although their primary clinical characteristics were similar to those previously reported, there were several points of concern.
All of our patients, with the exception of Patient 3, had hypoglycemia. According to previously reported data, 3 patients had no hypoglycemia during metabolic decompensation (Lee et al., 2019; Conlon et al., 2020; Wang et al., 2020a). The specific mechanism for this is unknown, and it may be related to compensatory glycogen decomposition and gluconeogenesis. However, the evidence suggested that hypoketotic hypoglycemia was important, but not indispensable, to HMGCS2D diagnosis. Similar to the patients observed by other researchers (Fukao et al., 2014; Conboy et al., 2018), urinary ketones in all of our patients were weakly positive (i.e., not negative). This may have interfered with the initial HMGCS2D diagnosis. Moreover, the observed ketones may have been due to the moderate amounts of ketone bodies produced by leucine catabolism pathway. In addition, in our study, four patients developed hypocalcemia during acute decompensation. Previously, this had only been reported in two patients (Wolf et al., 2003; Kilic et al., 2020). The specific pathogenesis of hypocalcemia remains unclear. We speculate the activation of coagulation and the fibrinolytic system, cell membrane damage caused by severe acidosis and tissue energy deficiency, Vitamin D deficiency and insufficient calcium intake in the acute stage could each be factors. Three patients had transient hyperuricemia, similar to the recent Turkish cases (Kilic et al., 2020). Many organic acids can stimulate the transporter, increasing the reabsorption of uric acid, which may account for the hyperuricemia (George and Minter, 2021). Moreover, Patient 3 had a concomitant disease, congenital laryngeal web, which had been diagnosed when she was 10-months-old based on clinical symptoms including recurrent cough, asthma, laryngeal stridor and hoarseness, and the results of electronic bronchoscopy. Then she accepted interventional therapy, with exceptional results. To our knowledge, this is the first time this has been reported in patients with HMGCS2D.
Detection of metabolic markers are crucial to diagnosing most inherited metabolic diseases. However, as far as we know, there are no specific biochemical markers in HMGCS2D. All patients in this study showed elevated dicarboxylic acid in their urine during decompensation. It has been reported that this can provide an indication, but cannot differentiate HMGCS2D from other fatty acid metabolic disorders (Kilic et al., 2020; Wang et al., 2020b). It has been reported that increased urine 4-hydroxy-6-methyl-2-pyrone (4-HMP) can be a specific biochemical hallmark indicator which can distinguish HMGCS2D from fatty acid oxidation defects (Pitt et al., 2015; Wang et al., 2020a). Unfortunately, in our study, only 3 patients received urine 4-HMP tests. Among them, 2 had elevated urinary 4-HMP and the other patient’s urinary 4-HMP was normal. These results indicated that using 4-HMP as a positive prediction needs further study through more patients. We also detected the blood acylcarnitine profile in all individuals. As previously reported in the literature, blood acetylcarnitine (C2) can be either normal or increased during decompensation (Aledo et al., 2006; Ramos et al., 2013; Liu et al., 2019). In our study, the blood C2 level in 8 of the patients increased, while the free carnitine (C0) level in 7 of the patients decreased. This led to elevated C2/C0 ratios in all cases. These data suggest that for clinically suspected patients, the high ratio of C2 to C0 may have been more specific and informative than the solitary high C2 or low C0 levels. Consequently, we considered that simultaneous detection of blood acylcarnitine profile and urinary organic acids was essential for HMGCS2D diagnosis. An elevated C2/C0 ratio combined with plenty of dicarboxylic acids in urine during an episode of acute hypoglycemia and metabolic acidosis may be an additional biochemical signature of HMGCS2D. This observation needs further research to identify whether there is sufficient specificity for clinical utilization.
As mentioned above, the lack of specificity in clinical and biochemical characteristics, as well as any abnormalities, may turn normal during intermissions. This complicates HMGCS2D diagnosis. On the other hand, detecting enzyme activity is invasive and complex, and the results are unstable. Therefore, at present, molecular genetic analysis of the HMGCS2 gene still consists of the recommended diagnostic method (Zschocke et al., 2002; Ramos et al., 2013). In recent years, more and more patients have been confirmed as carrying biallelic pathogenic variants on HMGCS2 gene by genetic testing. The HMGCS2 gene is located on chromosome 1p13-p12, with a length of 20.9 kb. It consists of 10 exons, encoding 508 amino acids. Since the first description of the disease in 1997 (Thompson et al., 1997), over 40 variants have been identified. Prior reports have shown that these mutations have an irregular distribution and involve all exons (Wang Q. et al., 2020). Most of them are sporadic, specific to families, and have ethnic and regional differences. The c.634A > G/p.G212R has been identified in 3 families, respectively from Poland (Conlon et al., 2020), Britain (Pitt et al., 2015) and Germany (Zschocke et al., 2002). This implies that it was a common HMGCS2D mutation in Europe. The large deletion on exon 1, denoted as c.1-?_104+?del, has only been identified in patients of Mediterranean descent (Pitt et al., 2015), while c.725-2A > C has only been reported in two families from Turkey (Kilic et al., 2020). No relevant research on phenotype-genotype correlations has been reported to date.
Our finding is unlike similar studies conducted on other populations. In our cohort, the recurrent variants were c.1201G > T/p.E401*, c.559+1G > T and c.220G > A/p.E74K. If combined with the 11 patients reported previously (Thompson et al., 1997; Ma and Yu, 2018; Liu et al., 2019; Wang et al., 2019; Wang et al., 2020a; Wang et al., 2020b; Yang et al., 2020), the most frequent mutation was c.1201G > T/p.E401*. This may result in a premature termination codon at amino acid residue 401 located in exon 7 of HMGCS2. This leads to a truncated protein, and causes the loss of gene function. We detected this mutation as compound heterozygous in 7 Chinese patients from 6 families, while no patient in any other population has been a carrier of c.1201G > T/p.E401* in prior studies. All of these observations suggested that c.1201G > T/p.E401* may be a hot-spot mutation in Chinese HMGCS2D patients. The second most common mutations were c.559+1G > T, c.1187+1G > C and c.1502G > A/p.R501Q, all accounting for 3/42 mutated alleles, repectively. All had been reported in previous Chinese HMGCS2D patients, but not in other populations. c.559+1G > T was reported to be the second described splicing mutation of the HMGCS2 gene, and minigene assay results revealed that it may influence protein structure and function (Wang et al., 2020b). c.1187+1G > C is the classical site splicing mutation, which may cause abnormal splicing resulting in loss of protein function. We detected the missense mutation c.1502G > A/p.R501Q in Patient 6 in the compound heterozygous state, and a previous study has reported a Chinese case in the homozygous state (Ma and Yu, 2018). Previous reports have also suggested that arginine at position 501 is critical to enzymatic function (Bagheri-Fam et al., 2020). Moreover, variation in amino acids in the same position, c.1502G > C/p.R501P, has also been found in two Thai patients (Rojnueangnit et al., 2020). All of these factors imply that it was a causative variant.
Furthermore, our study also identified 10 novel mutations of HMGCS2. The c.1017-2A > G was the fifth splicing mutation of HMGCS2. It occurred at a classical splicing site and may lead to abnormal formation of HMGCS2 protein, demonstrating its pathogenicity. c.717T > G/p.Y239*, c.39dupA/p.L14Tfs*59, c.788delT/p.L263Cfs*36 and c.1394delA/p.N465Tfs*10 all led to a premature termination codon respectively in exons 4, 1, 4, and 8. This led to encoded truncated peptides, thereby resulting in an enzyme defect. The 5 missense mutations each occurred in highly conserved regions. They were predicted to be damaged by Mutation taster, SIFT, and Polyphen 2. The computational evaluation using Swiss-model software showed that they may affect the 3D structure of HMGCS2 protein. Among them, c.220G > A/p.E74K was detected in two unrelated patients. We speculated that it is a common mutation in Chinese patients.
In order to explore the correlation between genotype and clinical features and disease severity, which had remained unknown according to previous studies, we analysed the clinical and molecular characteristics of 49 patients. This included our 10 patients, as well as the previously reported 39 patients with relatively complete clinical data (as shown in Supplementary Table S1). In Table 4, Group A (patients carrying biallelic truncating mutations) had the highest mortality (25%), while Group C (patients carrying biallelic non-truncating mutations) had the highest percentage of mild cases (60%). Group B (carrying monoallelic truncating mutations) had the highest percentage of severe cases (76.9%). Statistical analysis showed a correlation between disease severity and how many truncating variations patients carried. This suggests that patients with biallelic truncation mutation are likely to have more severe phenotypes. However, there was no convincing evidence indicating that individuals with biallelic missense mutation would have milder or later presentation. On the other hand, due to the limited number of patients, the frequency of compound heterozygotes, and the lack of enzymatic studies, it is difficult to assess the precise relationship between genotype and phenotype. Furthermore, inter-allelic complementation complicated the predictions of potential genotype-phenotype correlations. Therefore, more cases are needed for further exploration.
With regard to the prognosis, most of our patients had full recovery after symptomatic and supportive treatment, and maintained normal growth and development, without further episodes during follow-up. According to the literature, only one case had neurological sequelae (Conboy et al., 2018). However, there still were 6 patients who died of hypoglycemic crisis and serious metabolic acidosis during acute metabolic decompensation (Liu et al., 2019; Kilic et al., 2020; Wang et al., 2020a; Yang et al., 2020). These results suggest that HMGCS2D is a fatal, but treatable, hereditary metabolic disease. If we can diagnose it before onset, and then prevent those trigger factors like long-term fasting, and administrate oral glucose when patients have poor food intake, we may be able to prevent metabolic crises. However, patients cannot be diagnosed before disease onset through the current newborn screening program. We believe that the combination of rapid and accurate next-generation sequencing technology and biochemical screening may improve newborn HMGCS2D screening efficiency in the future (Luo et al., 2020).
In summary, mitochondrial HMGCS2 deficiency is a rare ketone synthesis disorder. We have described clinical symptoms, biochemical features, clinical outcomes, and molecular analysis of 10 new Chinese patients. We have also expanded the HMGCS2 mutational spectrum with 10 novel variants. The variation c.1201G > T/p.E401* is the most common, and may be a hot-spot mutation of the HMGCS2 gene in Chinese patients. Through a literature review, we found that patients with biallelic truncation mutation appeared to show a more severe clinical condition.
The data that support the findings of this study are submitted to ClinVar through ClinVar Submission Portal and available from the corresponding author upon reasonable request. Accession link to HMGCS2 cDNA sequence NM_005518 https://www.ncbi.nlm.nih.gov/nuccore/NM_005518.4. ClinVar accession link of new variants of HMGCS2 gene identified in this study https://www.ncbi.nlm.nih.gov/clinvar/?term= SUB10769664.
The studies involving human participants were reviewed and approved by The Research and Ethics Committee of Zhengzhou Children’s Hospital. Written informed consent to participate in this study was provided by the participants’ or legal guardian/next of kin.
YC and BC contributed to conception and design of the study. SW performed the statistical analysis, and wrote the first draft of the manuscript. HW and YY collected the clinical data. CG made many suggestions for the revised manuscript. All authors critically reviewed, revised, and approved the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank our patients and their families for their corporation, and all pediatricians who sent us patients’ information.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.816779/full#supplementary-material
Aledo, R., Mir, C., Dalton, R. N., Turner, C., Pié, J., Hegardt, F. G., et al. (2006). Refining the Diagnosis of Mitochondrial HMG-CoA Synthase Deficiency. J. Inherit. Metab. Dis. 29 (1), 207–211. doi:10.1007/s10545-006-0214-2
Bagheri-Fam, S., Chen, H., Wilson, S., Ayers, K., Hughes, J., Sloan-Bena, F., et al. (2020). The Gene Encoding the Ketogenic Enzyme HMGCS2 Displays a Unique Expression during Gonad Development in Mice. PLoS One 15 (1), e0227411. doi:10.1371/journal.pone.0227411
Conboy, E., Vairo, F., Schultz, M., Agre, K., Ridsdale, R., Deyle, D., et al. (2017). Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency: Unique Presenting Laboratory Values and a Review of Biochemical and Clinical Features. JIMD Rep. 40, 63–69. doi:10.1007/8904_2017_59
Conlon, T. A., Fitzsimons, P. E., Borovickova, I., Kirby, F., Murphy, S., Knerr, I., et al. (2020). Hypoglycemia Is Not a Defining Feature of Metabolic Crisis in Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency: Further Evidence of Specific Biochemical Markers Which May Aid Diagnosis. JIMD Rep. 55 (1), 26–31. doi:10.1002/jmd2.12146
Fukao, T., Mitchell, G., Sass, J. O., Hori, T., Orii, K., and Aoyama, Y. (2014). Ketone Body Metabolism and its Defects. J. Inherit. Metab. Dis. 37 (4), 541–551. doi:10.1007/s10545-014-9704-9
George, C., and Minter, D. A. (2021). “Hyperuricemia,” in Treasure Island (FL): In StatPearls. [Internet].
Kılıç, M., Dorum, S., Topak, A., Yazıcı, M. U., Ezgu, F. S., and Coskun, T. (2020). Expanding the Clinical Spectrum of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency with Turkish Cases Harboring Novel HMGCS2 Gene Mutations and Literature Review. Am. J. Med. Genet. 182 (7), 1608–1614. doi:10.1002/ajmg.a.61590
Lee, T., Takami, Y., Yamada, K., Kobayashi, H., Hasegawa, Y., Sasai, H., et al. (2019). A Japanese Case of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency Who Presented with Severe Metabolic Acidosis and Fatty Liver without Hypoglycemia. JIMD Rep. 48 (1), 19–25. doi:10.1002/jmd2.12051
Liu, H., Miao, J.-k., Yu, C.-w., Wan, K.-x., Zhang, J., Yuan, Z.-j., et al. (2019). Severe Clinical Manifestation of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency Associated with Two Novel Mutations: a Case Report. BMC Pediatr. 19 (1), 344. doi:10.1186/s12887-019-1747-5
Luo, X., Sun, Y., Xu, F., Guo, J., Li, L., Lin, Z., et al. (2020). A Pilot Study of Expanded Newborn Screening for 573 Genes Related to Severe Inherited Disorders in China: Results from 1,127 Newborns. Ann. Transl. Med. 8 (17), 1058. doi:10.21037/atm-20-1147
Ma, D., and Yu, D. (2018). Mitochondrial 3-Hydroxy-3-Methylglutaryl CoA Synthase Deficiency: a Case Report and Literature Review. Zhongguo Dang Dai Er Ke Za Zhi 20 (11), 930–933. doi:10.7499/j.issn.1008-8830.2018.11.010
Pitt, J. J., Peters, H., Boneh, A., Yaplito-Lee, J., Wieser, S., Hinderhofer, K., et al. (2015). Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency: Urinary Organic Acid Profiles and Expanded Spectrum of Mutations. J. Inherit. Metab. Dis. 38 (3), 459–466. doi:10.1007/s10545-014-9801-9
Ramos, M., Menao, S., Arnedo, M., Puisac, B., Gil-Rodríguez, M. C., Teresa-Rodrigo, M. E., et al. (2013). New Case of Mitochondrial HMG-CoA Synthase Deficiency. Functional Analysis of Eight Mutations. Eur. J. Med. Genet. 56 (8), 411–415. doi:10.1016/j.ejmg.2013.05.008
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Rojnueangnit, K., Maneechai, P., Thaweekul, P., Piriyanon, P., Khositseth, S., Ittiwut, C., et al. (2020). Expanding Phenotypic and Mutational Spectra of Mitochondrial HMG-CoA Synthase Deficiency. Eur. J. Med. Genet. 63 (12), 104086. doi:10.1016/j.ejmg.2020.104086
Thompson, G. N., Hsu, B. Y. L., Pitt, J. J., Treacy, E., and Stanley, C. A. (1997). Fasting Hypoketotic Coma in a Child with Deficiency of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase. N. Engl. J. Med. 337 (17), 1203–1207. doi:10.1056/NEJM199710233371704
Wang, H., Li, D., Song, C., Yang, Y., Qian, S., and Cheng, Y. (2020a). Case Report of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency. Zhonghua Shi Yong Er Ke Lin Chuang Za Zhi 35 (16), 1269–1271. doi:10.3760/cma.j.cn101070-20190621-00559
Wang, M., Gong, Y., Ma, X., Zhu, D., and Zhong, X. (2019). Clinical Features of a Chinese Infant with Mitochondrial 3-Hydroxy-3-Methylglutaryl CoA Synthase Deficiency and Review of the Literature. Lin Chuang Er Ke Za Zhi 37 (11), 858–861. doi:10.3969/j.issn.1000-3606.2019.11.015
Wang, Q., Yang, Y.-L., Liu, M., Chen, J.-J., Li, X.-q., Cao, B.-y., et al. (2020b). Clinical, Biochemical, Molecular and Therapeutic Characteristics of Four New Patients of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency. Clinica Chim. Acta 509, 83–90. doi:10.1016/j.cca.2020.04.004
Wolf, N. I., Rahman, S., Clayton, P. T., and Zschocke, J. (2003). Mitochondrial HMG-CoA Synthase Deficiency: Identification of Two Further Patients Carrying Two Novel Mutations. Eur. J. Pediatr. 162 (4), 279–280. doi:10.1007/s00431-002-1110-x
Yang, Q., Li, S., Xiong, H., Cai, Y., Shi, C., Xiao, X., et al. (2020). 3-Hydroxy-3-methylglutaryl-CoA Synthase Deficiency: a New Case and Literature Review. Zhongguo Xiao Er Ji Jiu Yi Xue Za Zhi 27, 10. doi:10.3760/cma.j.issn.1673-4912.2020.10.016
Keywords: 3-hydroxy-3-methylglutaryl-CoA synthase deficiency, hypoglycemia, ketogenesis, HMGCS2 gene, HMGCS2D
Citation: Wu S, Shen L, Chen Q, Gong C, Yang Y, Wei H, Cao B and Chen Y (2022) Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients. Front. Genet. 12:816779. doi: 10.3389/fgene.2021.816779
Received: 17 November 2021; Accepted: 31 December 2021;
Published: 04 March 2022.
Edited by:
Huiwen Zhang, Xinhua Hospital, ChinaReviewed by:
Julnar A. R. Usta, American University of Beirut, LebanonCopyright © 2022 Wu, Shen, Chen, Gong, Yang, Wei, Cao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongxing Chen, Y3l4NzVAMTI2LmNvbQ==; Bingyan Cao, Y2FvYnkxOTgyQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.