 Yulin Bai1†
Yulin Bai1† Jie Gong1†
Jie Gong1† Zhixiong Zhou1
Zhixiong Zhou1 Bijun Li1Ji Zhao1Qiaozhen Ke1,2Xiaoqing Zou1
Bijun Li1Ji Zhao1Qiaozhen Ke1,2Xiaoqing Zou1 Fei Pu1Linni Wu1Weiqiang Zheng2
Fei Pu1Linni Wu1Weiqiang Zheng2 Tao Zhou1,3
Tao Zhou1,3 Peng Xu1,3*
Peng Xu1,3*- 1State Key Laboratory of Marine Environmental Science, College of Ocean and Earth Sciences, Xiamen University, Xiamen, China
- 2State Key Laboratory of Large Yellow Croaker Breeding, Ningde Fufa Fisheries Company Limited, Ningde, China
- 3Fujian Key Laboratory of Genetics and Breeding of Marine Organisms, College of Ocean and Earth Sciences, Xiamen University, Xiamen, China
The Rock Bream (Oplegnathus fasciatus) is an economically important rocky reef fish of the Northwest Pacific Ocean. In recent years, it has been cultivated as an important edible fish in coastal areas of China. Despite its economic importance, genome-wide adaptions of domesticated O. fasciatus are largely unknown. Here we report a chromosome-level reference genome of female O. fasciatus (from the southern population in the subtropical region) using the PacBio single molecule sequencing technique (SMRT) and High-through chromosome conformation capture (Hi-C) technologies. The genome was assembled into 120 contigs with a total length of 732.95 Mb and a contig N50 length of 27.33 Mb. After chromosome-level scaffolding, 24 chromosomes with a total length of 723.22 Mb were constructed. Moreover, a total of 27,015 protein-coding genes and 5,880 ncRNAs were annotated in the reference genome. This reference genome of O. fasciatus will provide an important resource not only for basic ecological and population genetic studies but also for dissect artificial selection mechanisms in marine aquaculture.
Introduction
The Rock Bream (Oplegnathus fasciatus), inhabiting in coastal rocky reefs and feeds on invertebrates inhabiting the seabed, is an endemic marine fish in East Asia that belongs to Oplegnathidae in Perciformes (Figure 1A) (An et al., 2008). In recent years, O. fasciatus has become one of the most commercially valuable marine fishery species in China aquaculture (Xiao et al., 2019). As a sedentary species, due to habitat restrictions, O. fasciatus is broadly and fragmented distributed in the coastal areas of eastern China (Xiao et al., 2016a). According to the difference of genetic variation, the researchers identified the O. fasciatus population as northern (Jiaonan, Qingdao) and southern regions (Zhoushan, Zhejiang) of Chinese coastal waters (Xiao et al., 2016b). The different environments make them divergent in genetic structure and living habits, which provides a suitable fish model for the study of population genetic in marginal sea (Jian et al., 2017; Chen et al., 2019a). The male and female reference genome of O. fasciatus derived from the northern population has been reported and revealed its particularity in gender mechanism (Xiao et al., 2019; Xiao et al., 2020). However, a highly accurate reference genome of subtropical O. fasciatus species is still lacking, which hinders the progress of genome-scale genetic breeding and genetic studies of its temperature plasticity and adaptation at lower latitudes.
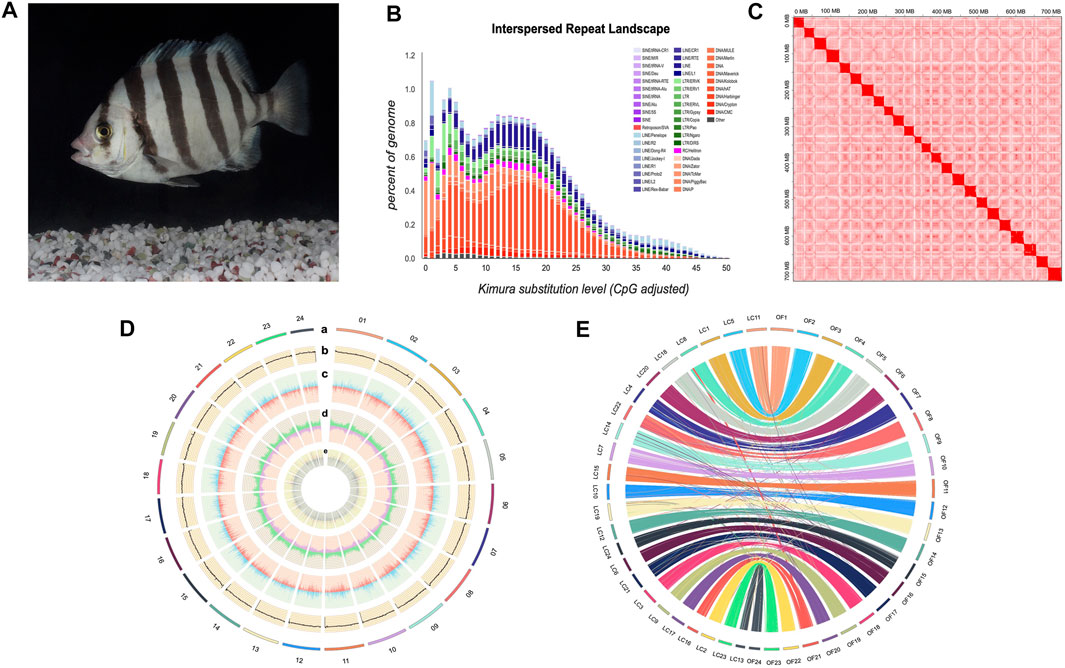
FIGURE 1. Characteristics of Oplegnathus fasciatus genome assembly. (A) A photograph of female O. fasciatus (from Zhoushan population). (B) Distribution of divergence rate for each type of TE in O. fasciatus genome. Percentages of TEs in genome (Y-axis) and Kimura divergence (X-axis). Values from 0-50 reflect older copies (right side) and new copies (left side). (C) Heat map of chromosomal interactions in the O. fasciatus genome using Hi-C data. (D) A circos plot of 24 chromosomes in O. fasciatus genome, the tracks from outside to inside are: a. Lines represent O. fasciatus chromosomes; b. GC content; c. Gene density; d. Repeat element density; e. ncRNA density. b-e. are drawn in nonoverlapping 100 kb sliding windows. (E) Circos diagram between O. fasciatus and L. crocea. Each coloured arc represents a 1 Kb fragment match between two species. We re-ordered the chromosome numbers of L. crocea for better illustration.
Recently, the genetic breeding of economic fish has attracted much attention, mainly aiming to improve growth rates and disease resistance under aquaculture conditions (Bangera et al., 2017; Gong et al., 2021; Zhao et al., 2021). Genome selection (GS) has become an efficient and popular breeding strategy, which depends on high-quality reference genome (Singh et al., 2018). It is worthwhile to invest time and money to obtain a reference genome more suitable for breeding populations to improve the prediction accuracy (Benevenuto et al., 2019). Our breeding work related to the growth traits of O. fasciatus reported recently also supports this conclusion (Gong et al., 2021). In this report, we provided a chromosome-level reference genome of O. fasciatus (from the southern population in the subtropical region) using a combination of the PacBio single molecule sequencing technique (SMRT) and high-through chromosome conformation capture (Hi-C) technologies. We assembled the genome sequences into 120 contigs with a total length of 732.95 Mb and a contig N50 length of 27.33 Mb. After chromosome-level scaffolding, 24 scaffolds were constructed corresponding to 24 chromosomes with a total length of 723.22 Mb (98.67% of the total length of all contigs). The availability of data is essential to support the population genetic studies, and will also provide an important resource for the upcoming breeding program of O. fasciatus.
Materials and Methods
Sample Collection, Library Construction and Sequencing
A healthy female O. fasciatus (from Zhoushan population) were obtained from a commercial breeding company, Ningde Fufa Aquatic Breeding (Fujian, China). All samples were collected, snap frozen in the liquid nitrogen and stored at −80°C to maintain nucleic acid integrity.
The muscle tissues were collected for DNA extraction. For the PacBio sequencing project, frozen samples were lysed in SDS digestion buffer with proteinase-K (50 µg/ml). Then, the lysates were purified using AMPure XP beads (Beckman Coulter, High Wycombe, United Kingdom) to obtain High-Molecular-Weight gDNA. Library construction and sequencing were conducted according to the manufacturer’s protocol with the PacBio RS-II platform at Novogene (Tianjin). Meanwhile, the Normal-Molecular-Weight gDNA for Illumina sequencing was extracted from the same samples using the PureLink™ Pro 96 Genomic DNA Purification Kit (Invitrogen, Shanghai, China). A pair-end library with 350 bp insert size was constructed and sequenced using the Illumina NovaSeq 6000 platform with a read length of 2 × 150 bp.
For Hi-C sequencing, The DNA was fixed by formaldehyde to maintain the conformation and the restriction enzyme (HindIII) was applied on DNA digestion, followed by repairing 5’ overhangs with biotinylated residues. After in-situ ligation of these fragments, DNA was reverse-cross linked and purified. Finally, sequencing of the Hi-C library was performed on an Illumina NovaSeq 6000 platform.
Additionally, 11 different tissues (heart, liver, spleen, intestine, kidney, skin, eye, gill, brain, blood, muscle) were collected to extract RNA for RNA sequencing (RNA-seq) following the protocols of the PureLink™ RNA Mini Kit (Invitrogen, Shanghai, China). The library was constructed using the Illumina standard protocol (San Diego, CA, United States) and sequenced on the Illumina HiSeq 6000 platform.
Data Processing and Genome Assembly
Before assembly, the PacBio data was further filtered, and reads with length less than 1500 bp or low-quality were removed. For the Illumina data, adapter sequences and low-quality reads were filtered using fastp (v. 0.23.1) software. The remaining reads were used for further assembly and estimation of genome size using the K-mer analysis of the short reads (see below).
SOAPec (v. 2.01) and GenomeScope (v. 2.0) softwares were used to analyze the K-mer frequencies in the sequencing reads to obtain genome characteristics such as genome size, heterozygosity, and repeatability.
To obtain chromosome-level whole genome assembly for O. fasciatus, we utilized a combined approach of Illumina, PacBio and Hi-C technology for the genome assembly and chromosome-level scaffolding. Then, low-quality and duplicated reads were filtered out. The O. fasciatus genome was assembled using a hybrid SMRT-Illumina-HiC strategy as follows: 1) Contigs from Continuous Long-Read (CLR) clean reads were assembled using Canu (v. 2.0) (Sergey et al., 2017) with default parameters. The high-fidelity contig sets was produced by using a combination of circular consensus CLR reads, Illumina paired-end reads with sufficient overlap to merge into single extended accurate reads; 2) Purge_Dups (v. 1.2.5) was employed to resolve redundancy in the assembly; 3) The non-redundant contig sets were reordered and scaffolded using 3D-DNA pipeline (Dudchenko et al., 2017); 4) Scaffolds were fine-tuned and discordant contigs were removed from scaffolds using Juicebox (v. 1.5) (Robinson et al., 2018). Finally, we obtained a chromosome level reference genome of O. fasciatus containing linkage group information.
Annotation of Genomic Repeats
Repetitive sequences of the O. fasciatus genome were annotated using both homology-based search and de novo methods. RepeatModeler (v. 2.0.1) and LTR_Finder (v. 1.07) were used to detect repeat sequences in the O. fasciatus genome. Combined with Repbase (v. 20,181,026; http://www.girinst.org/repbase), a repeat sequence library was constructed. RepeatMasker (v. 4.1.0) were utilized to search and classify repeats based on this library. TEclass (v. 2.1.3) was used to further annotate unclassified repeats. The built-in script buildSummary.pl from RepeatMasker (v. 4.1.0) was used to summarize Transposable Elements (TEs) annotation results. Then two scripts, calcDivergenceFromalign.pl and createRepeatLandscape.pl, were used to calculate the Kimura divergence value and draw repeated landscapes (Figure 1B). The nucleotide distances between all copies of each TE measured using the Kimura two-parameter method were compared to estimate insertion age (Schemberger et al., 2019). Tandem Repeats Finder (v. 4.09) was used to identify tandem repeats. All repetitive regions except tandem repeats were soft-masked for protein-coding gene annotation.
Protein-Coding Gene Finding and Function Annotation
The coding sequences of genetically close species, including L. crocea, L. maculatus, G. aculeatus, and P. olivaceus, were retrieved from Ensembl and NCBI. These coding sequences were provided to the software package of Braker2 (v. 2.1.5) (Bruna et al., 2021), and then the genes in the repeat-masked reference genome were annotated with the close homologous protein model. RNA-seq data were aligned to O. fasciatus contigs using Blat (v. 36 × 5) and GMAP (v. 2017-11-15), and a comprehensive transcriptome database was built using PASA (v. 2.4.1) (Haas and Salzberg, 2008). The transdecoder software (v. 5.5.0) was used to predict gene structure based on ESTs evidence, and the credible gene structure annotation information was provided to train parameters for the following de novo gene prediction software packages: Augustus (v. 3.4.0) and GeneMark (v. 4.62). Finally, evidence from the gene finders, protein homology searches and ESTs were provided to the EvidenceModeler (v 1.1.1) (Haas and Salzberg, 2008) to obtain a comprehensive and non-redundant gene set.
For gene function annotation, we used Diamond (v. 2.0.6) to search the homologous sequences from the Swiss-Prot (http://www.uniprot.org/), TremBL (http://www.uniprot.org/) and NR protein databases. We were also subjected to GO annotation and protein family annotation by InterProScan (v. 4.8) (https://www.ebi.ac.uk/interpro/). KO terms for each gene are assigned by an online website (KAAS, https://www.genome.jp/tools/kaas/). The programs tRNAScan-SE (v. 1.3.1) and RNAmmer (v. 1.2) were used to predict tRNA and rRNA, respectively. The other ncRNAs were identified by searching against the Rfam database (http://eggnogdb.embl.de/).
Internal scripts were used to calculate GC content, gene density, repetitive element density, ncRNA density, and gene components distribution based on gff3 format file in the annotation results.
The Completeness Assessment of Assembly and Annotation
Assembly completeness and accuracy were evaluated by multiple methods. First, the Illumina short reads were re-mapped to the genome using BWA (v. 0.7.17), and the mapping ratio was counted by samtools (v. 1.8; with the pattern “flagstat”). Then, we used the Benchmarking Universal Single Copy Orthologues (BUSCO) (v. 5.2.2) (Seppey et al., 2019) to test the integrality of the final assembly and the lineage dataset was actinopterygii_odb10. Similarly, the annotation integrity was evaluated based on the protein sequence sets using the protein pattern built into BUSCO software (v. 5.2.2).
Phylogenetic Analysis
Single-copy genes in O. fasciatus and 10 related species (G. morhua, L. maculatus, L. crocea, G. aculeatus, N. coriiceps, T. bimaculatus, O. latipes, C. semilaevis, B. petinirostris, and D. reio) were identified based on gene families constructed from protein sequences of all species employing OrthoFinder (v. 2.5.4) (Emms and Kelly, 2019) software with default parameters. Single-copy ortholog proteins were aligned by MUSCLE (v. 3.8.31). Subsequently, all obtained alignments were converted to their corresponding coding DNA sequences using an in-house python script. A combined continuous ultra-long sequence was constructed from all the translated coding DNA alignments for minimum evolution (ME) phylogenetic tree construction using MEGA. The divergence time is estimated using MCMCTREE (PAML package) (Yang, 1997) based on the molecular clock data of Timetree database (Hedges et al., 2006).
Results and Discussions
With a 100x sequencing depth sequencing reads from Illumina and Pacbio platforms, we assembled a high-quality chromosome-level reference genome from a female O. fasciatus. Based on the 17-kmer analysis and a dominant peak depth of 51.57, the genome size was estimated to be 751.19 Mb with the heterozygosity and repetitive sequence content were approximately 0.28 and 23.09%, respectively (Supplementary Table S1). The genome size is slightly smaller than that of the O. fasciatus derived from the northern population (previously reported; male ∼762 Mb and female ∼768.8 Mb) (Xiao et al., 2019; Xiao et al., 2020). A trimodal pattern was observed in the 17-mer frequency distribution analysis. The main peak (the second peak) is much higher than the other two sub peaks, indicating that there is a certain degree of heterozygosity and duplication in O. fasciatus genome (Supplementary Figure S1) (Manekar and Sathe, 2019).
The PacBio sequencing reads were used for de novo assembly of the genome. And the average read length and N50 of read length were 21,083 and 30,815 bp, respectively. The initial assembly yielded a total length of 1.1 Gb, comprising 2,789 contigs with a contig N50 length of 21.91 Mb. The genome assembly was larger than the estimated genome size of 751.19 Mb (see above), because some redundant sequences failed to be merged due to high heterozygosity (Zhang et al., 2012). After eliminating the redundancy, we obtained a final genome assembly of 732.95 Mb for the O. fasciatus, which is nearly equal to the estimated genome size (Table1 and Supplementary Table S1). Then, the PacBio draft assembled contigs were anchored and oriented into a chromosomal-scale assembly via the Hi-C scaffolding approach (Figure 1C). Finally, we generated a chromosome-level genome assembly of 732.99 Mb in length, with a contig N50 and scaffold N50 value of 27.33 and 31.10 Mb, respectively (Figure 1D and Table 1). The total length of assembly contained 24 chromosomes (the lengths ranged from 18.22 to 37.18 Mb) was 723.22 Mb, and the integration efficiency was 98.67% (Table 1 and Supplementary Table S2). The genome sizes of O. fasciatus (733.99 Mb) were similar with two closely related Perciformes species i.e., L. crocea (723.86 Mb) (Chen et al., 2019a) and Oplegnathus punctatus (718 Mb) (Li et al., 2021). To further verify the integrality of the assembled genome, the reads from the short-insert library were re-mapped onto the assembled genome using BWA (version 0.7.17). A total of 97.89% of the reads mapped to a reference sequence in this genome (Table 1). Additionally, we tested completeness by attempting the recovery of conserved single-copy genes from O. fasciatus genome by BUSCO (v. 5.0.0). Out of a database containing 3,640 single-copy protozoan orthologs, ∼97% were fully recovered from the assembly (Table 1 and Supplementary Table S3). The high integration efficiency (∼98.67%), mapping ratio (97.89%), and the recognition rate of single copy orthologs (∼97%) show that our assembly of O. fasciatus is of high quality, which is at the same level as some recently published high-accurate reference genomes of other marine fish (Chen et al., 2019a; Zhou et al., 2019).
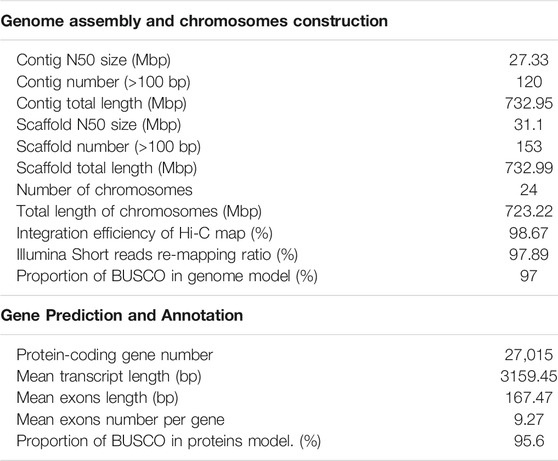
TABLE 1. Summary of the Oplegnathus fasciatus genome assembly and annotation.
There was a total of 224.97 Mb of consensus and nonredundant repetitive sequences obtained by a combination of known, novel and tandem repeats, occupying more than 30.69% of the whole genome assembly (Figure 1B and Supplementary Table S4). The de novo and homologous prediction were utilized to investigate the repeat sequences (Supplementary Table S5). TE divergence analysis suggested recent activity of DNA transposons and long terminal repeats in this genome (Figure 1B) (Wang et al., 2019). DNA transposons were the most abundant repetitive elements, spanning at least 113.54 Mb, accounting for 15.49% of the whole genome of O. fasciatus. Among them, the repetitive sequences also comprised of long interspersed elements in 44.78 Mb (LINEs; 6.11%), short interspersed nuclear elements in 1.37 Mb (SINEs; 0.19%) and long terminal repeats in 34.02 Mb (LTRs; 4.64%) (Supplementary Table S5).
A total of 27,015 nonredundant protein-coding genes were successfully yielded combining de novo, homologous searching and transcriptome-assisted predictions (Table 1). The statistics of the predicted gene models were compared to the homologous protein sequences of G. aculeatus, L. crocea, L. japonicus and P. olivaceus, which indicated closely distribution patterns in mRNA length, CDS length, exon length and exon number (Figure 2B and Supplementary Table S7). There were 17,054 complete ORFs, 27,015 complete transcripts and 250,306 exons detected in the O. fasciatus genome. The mean lengths of exon were 167.47 bp. The average exon number for each gene was 9.27 and the average length of CDS was 1551.68 bp (Supplementary Table S7). We annotated these genes against several public databases, including NR, TrEMBL, Swissprot and InterPro, resulting in 98.98, 97.94, 84.77 and 78.58% of the genes functionally assigned, respectively. Furtherly, we detected protein domains in multiple databases, and 59.76 and 59.40% of the predicted genes were annotated using GO and KEGG database, respectively. Finally, 27,015 genes were successfully functional annotated in at least one of these databases (Figure 2A and Supplementary Table S6). The number of predicted genes of O. fasciatus (27,015) through de novo prediction and homologue annotation was slightly more than others in Perciformes, such as L. crocea (23,657) (Chen et al., 2019a), L. maculatus (22,509) (Chen et al., 2019b), and previous version of O. fasciatus (24,003) from the northern population (Xiao et al., 2019). BUSCO analysis with protein pattern suggested that 97.2% (3,537) of the core conserved genes were detected in the O. fasciatus gene set, with 3,480 (95.6%) and 57 (1.6%) being identified as complete and fragmented, respectively (Table 1 and Supplementary Table S3). The results indicate that our gene structure annotation is relatively complete (Seppey et al., 2019). To verify the accuracy of the contig arrangement in 24 chromosomes, we aligned 1 K bp small fragments with 50 K bp spacing as anchors of the assembled genome against the published L. crocea genome to compare consistency between these two genomes. The 24 chromosomes we identified in the O. fasciatus genome aligned exactly against the chromosomes of the L. crocea, suggesting high continuity with the O. fasciatus genome (Figure 1E). Furthermore, four types of non-coding RNAs were identified in O. fasciatus genome, including 1,188 miRNA, 1,808 tRNAs, 1,793 rRNAs and 1,091 snRNAs (Table 1 and Supplementary Table S8).
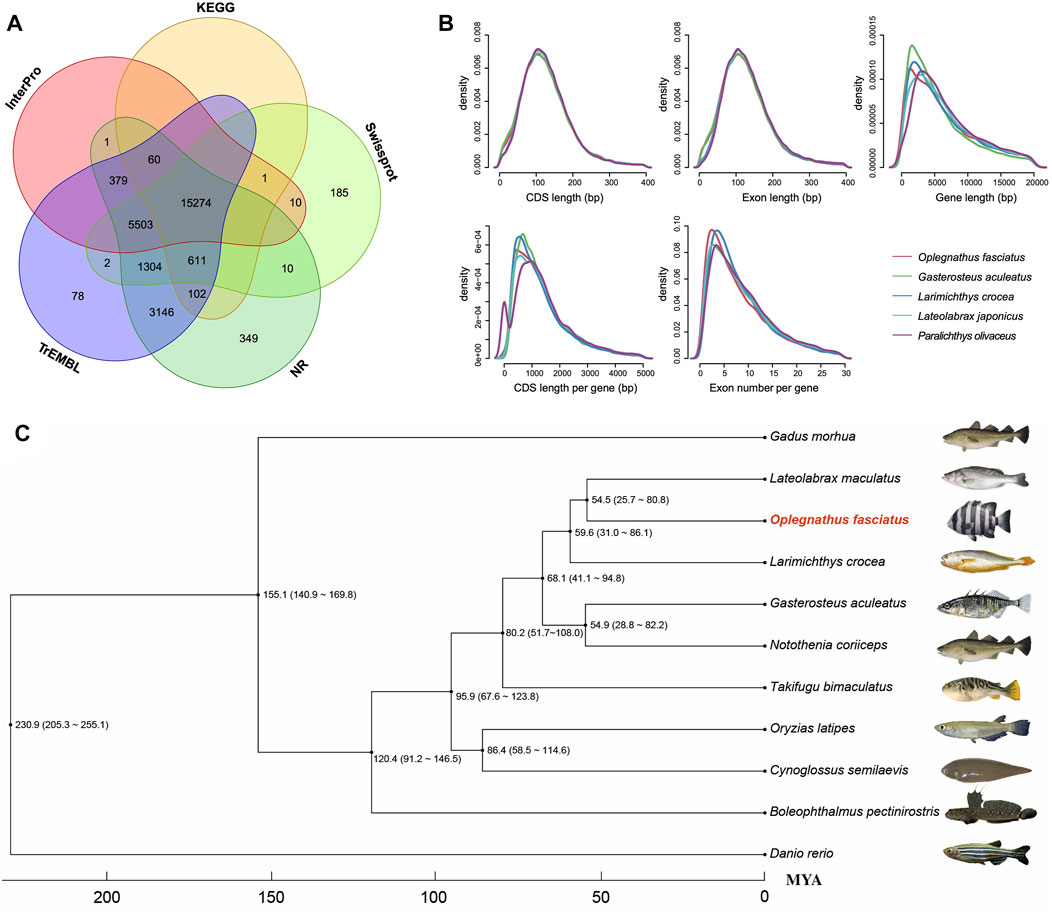
FIGURE 2. Gene annotations of the Oplegnathus fasciatus genome and phylogenetic relationships among eleven species. (A) Venn diagram of the functionally annotated protein-coding genes. (B) Gene components distribution patterns among O. fasciatus and related species (G. aculeatus, L. crocea, L. maculatus, and P. olivaceus). (C) Phylogenetic analysis among O. fasciatus and representative species (G. morhua, L. maculatus, L. crocea, G. aculeatus, N. coriiceps, T. bimaculatus, O. latipes, C. semilaevis, B. petinirostris, and D. reio).
To reveal the phylogenetic relationships among O. fasciatus and other species, a total of 2,516 single copy ortholog protein families in a 1:1:1 manner from the 10 related species (as described above) were identified and used for phylogenetic analysis (Figure 2C). Previous reports showed that O. fasciatus and L. crocea had close genetic distance and clustered in Perciformes (Eupercaria) (Xiao et al., 2019). According to our phylogenetic analysis, we further found that the divergence time between O. fasciatus and the common ancestor with L. maculatus (25.7-80.8 million years ago) was shorter than L. crocea (31.0–86.1 million years ago). In addition, the phylogenetic relationship between O. fasciatus and other fish is also consistent with previous taxonomic studies (Oh et al., 2007; Betancur-R et al., 2017), indicating that the estimation of divergence time in this study should be reasonable.
Certainly, a high-precision chromosomal genome resource is foremost to identify genetic variation underlying phenotypic traits of economic interest for aquaculture production (Chen et al., 2019a; Chen et al., 2019b; Zhou et al., 2019). The sex determination mechanism of O. fasciatus has attracted wide attention of researchers in recent years (Xiao et al., 2020). Previous cytogenetic analysis of O. fasciatus has shown that it has morphologically distinguishable sex chromosomes, and there are differences in the number of male (2n = 47; X1X2Y) and female (2n = 48; X1X1X2X2) chromosomes (Xu et al., 2019). Xiao et al. have reported the sex-related comparative genomics study of the northern population of O. fasciatus (Xiao et al., 2020), but it is still unclear whether the key genetic locus is restricted by geography. Our report also provides an important resource for further research on the mechanism of gender determination.
Conclusion
Here, we report a highly accurate chromosome-level genome assembly of O. fasciatus from the southern population in the subtropical region based on PacBio and Hi-C technologies. The genome size (732.99 Mb) is slightly smaller than that of the O. fasciatus derived from the northern population. A total of 27,015 gene structures were annotated using the strategy of multi-evidence combination. We found that the divergence time between O. fasciatus and the common ancestor with L. maculatus was shorter than L. crocea. The genome data created in this study will serve as valuable resources for species diversity and population genetic research, and will further promote the progress of genome-scale genetic breeding and genetic studies of its temperature plasticity and adaptation at lower latitudes.
Code Availability
Genome Survey and Assembly
1) SOAPec: version 2.01; -k 17 -l 11; 2) GenomeScope: version 2.0; all parameters were set as default; 3) Canu: version 2.0; all parameters were set as default; 4) Racon: version 1.4.3; all parameters were set as default; 5) NextPolish: version 1.4.0; job_type = local; task = 1212; rewrite = no; rerun = 3; sgs_options = -max_depth 100 -bwa; 6) Purge_Dups: version 1.2.5; all parameters were set as default.
Genome Annotation:
1) RepeatMasker: version 4.1.0; -no_is -nolow -norna -gff -poly -html; 2) RepeatModeler: version 2.0.1; -database genome -engine ncbi; 3) TEclass: version 2.1.3; all parameters were set as default; 4) TRF: version 4.09; 2 7 7 80 10 50, 500 -m -f -d; 5) Braker2: version 2.1.5; -gff3 –species --genome --prot_seq --bam --prg = gth --softmasking 6) Transdecoder: version 5.5.0; -t transcripts.fasta; 7) PASA: version 2.4.1; -c alignAssembly.config -C -R -g genome -t transcripts.fasta.clean -T -u transcripts.fasta --ALIGNERS blat, gmap; 8) Diamond: version 2.0.6; --outfmt 5; 9) EVidenceModeler: version 1.1.1; --gene_predictions --protein_alignments --transcript_alignments --segmentSize 100000 --overlapSize 10000 –weights weights.txt.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA778612, https://figshare.com/, https://doi.org/10.6084/m9.figshare.16950832.
Ethics Statement
The animal study was reviewed and approved by the Animal Care and Use Committee at the College of Ocean and Earth Sciences, Xiamen University.
Author Contributions
PX conceived the study. YB, JG, ZZ, BL, and QK performed bioinformatics analysis. YB, JG, JZ, and QK collected samples. YB and JG extracted DNA and RNA. FP, LW, and WZ performed the quality control. YB, TZ, and PX wrote the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by National Key R and D Program of China (2019YFE0119000), Special Foundation for Major Research Program of Fujian Province (2020NZ08003), Ningbo Science and Technology Innovation 2025 Major Project (2021Z002), the special project of local science and technology development guided by the central government (2019L3032), China Agriculture Research System (CARS-47) and Zhejiang Provincial Natural Science Foundation of China (LQ20C190008).
Conflict of Interest
WZ and QK were employed by the Ningde Fufa Fisheries Company Limited.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.811798/full#supplementary-material
Abbreviations
B. petinirostris: Blue Spotted Mudskipper, Boleophthalmus petinirostris; C. semilaevis: Half-Smooth Tongue Sole, Cynoglossus semilaevis; BUSCO, Benchmarking Universal Single Copy Orthologues; CLR, Continuous Long-Read; D. reio: Zebrafish, Danio reio; GS, Genome selection; G. aculeatus: Three-Spined Stickleback, Gasterosteus aculeatus; G. morhu: Atlantic Cod, Gadus morhua; Hi-C, High-through chromosome conformation capture; L. crocea: Large Yellow Croaker, Larimichthys crocea; L. japonicus: Chinese Seabass, Lateolabrax maculatus; PacBio SMRT, ME, Minimum Evolution; N. coriiceps: Antarctic rock cod, Notothenia coriiceps; PacBio Single molecule sequencing technique; O. fasciatus: Rock Bream, O. latipes: Japanese Rice Fish, Oryzias latipes; Oplegnathus fasciatus; P. olivaceus: Olive Flounder, Paralichthys olivaceus; T. bimaculatus: Pufferfish, Takifugu bimaculatus; TEs, Transposable Elements.
References
An, H. S., Kim, M. J., and Hong, S. W. (2008). Genetic Diversity of Rock Bream Oplegnathus fasciatus in Southern Korea. Gene Genet. 30, 451–459.
Bangera, R., Correa, K., Lhorente, J. P., Figueroa, R., and Yáñez, J. M. (2017). Genomic Predictions Can Accelerate Selection for Resistance against Piscirickettsia Salmonis in Atlantic salmon (Salmo salar). Bmc Genomics 18, 121. doi:10.1186/s12864-017-3487-y
Benevenuto, J., Ferrão, L. F. V., Amadeu, R. R., and Munoz, P. (2019). How Can a High-Quality Genome Assembly Help Plant Breeders? GigaScience 8, giz068. doi:10.1093/gigascience/giz068
Betancur-R, R., Wiley, E. O., Arratia, G., Acero, A., Bailly, N., Miya, M., et al. (2017). Phylogenetic Classification of Bony Fishes. BMC Evol. Biol. 17, 162. doi:10.1186/s12862-017-0958-3
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M., and Borodovsky, M. (2021). BRAKER2: Automatic Eukaryotic Genome Annotation with GeneMark-Ep+ and AUGUSTUS Supported by a Protein Database. NAR Genom Bioinform 3, lqaa108. doi:10.1093/nargab/lqaa108
Chen, B., Li, Y., Peng, W., Zhou, Z., and Shi, Y. (2019a). Chromosome-Level Assembly of the Chinese Seabass (Lateolabrax Maculatus) Genome. Front. Genet. 10, 275. doi:10.3389/fgene.2019.00275
Chen, B., Zhou, Z., Ke, Q., Wu, Y., Bai, H., Pu, F., et al. (2019b). The Sequencing and De Novo Assembly of the Larimichthys Crocea Genome Using PacBio and Hi-C Technologies. Sci. Data 6, 188. doi:10.1038/s41597-019-0194-3
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De Novo assembly of the Aedes aegypti Genome Using Hi-C Yields Chromosome-Length Scaffolds. Science 356, 92–95. doi:10.1126/science.aal3327
Emms, D. M., and Kelly, S. (2019). OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 20, 238. doi:10.1186/s13059-019-1832-y
Gong, J., Zhao, J., Ke, Q., Li, B., Zhou, Z., Wang, J., et al. (2021). First Genomic Prediction and Genome‐wide Association for Complex Growth‐related Traits in Rock Bream (Oplegnathus fasciatus). Evol. Appl. doi:10.1111/eva.13218
Haas, B. J., Salzberg, S. L., Zhu, W., and Pertea, M. (2008). Automated Eukaryotic Gene Structure Annotation Using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7. doi:10.1186/gb-2008-9-1-r7
Hedges, S. B., Dudley, J., and Kumar, S (2006). TimeTree: a Public Knowledge-Base of Divergence Times Among Organisms. Bioinformatics 22, 2971–2972. doi:10.1093/bioinformatics/btl505
Jian, X., Chao, B., Kunci, C., Liu, G., Jiang, Y., Luo, Q., et al. (2017). Draft Genome of the Northern Snakehead, Channa argus. GigaScience 6, 1–5. doi:10.1093/gigascience/gix011
Li, M., Zhang, R., Fan, G., Xu, W., Zhou, Q., Wang, L., et al. (2021). Reconstruction of the Origin of a Neo-Y Sex Chromosome and its Evolution in the Spotted Knifejaw, Oplegnathus Punctatus. Mol. Biol. Evol. 38, 2615–2626. doi:10.1093/molbev/msab056
Manekar, S. C., and Sathe, S. R. (2019). Estimating the K-Mer Coverage Frequencies in Genomic Datasets: A Comparative Assessment of the State-Of-The-Art. Curr. genomics 20, 2–15. doi:10.2174/1389202919666181026101326
Oh, D. J., Kim, J. Y., Lee, J. A., Yoon, W. J., Park, S. Y., and Jung, Y. H. (2007). Complete Mitochondrial Genome of the Rock Bream Oplegnathus fasciatus (Perciformes, Oplegnathidae) with Phylogenetic Considerations. Gene 392, 174–180. doi:10.1016/j.gene.2006.12.007
Robinson, J. T., Turner, D., Durand, N. C., Thorvaldsdóttir, H., Mesirov, J. P., and Aiden, E. L. (2018). Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cel Syst. 6, 256–258. doi:10.1016/j.cels.2018.01.001
Schemberger, M. O., Nascimento, V. D., Coan, R., Ramos, É., Nogaroto, V., Ziemniczak, K., et al. (2019). DNA Transposon Invasion and Microsatellite Accumulation Guide W Chromosome Differentiation in a Neotropical Fish Genome. Chromosoma 128, 547–560. doi:10.1007/s00412-019-00721-9
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 1962, 227–245. doi:10.1007/978-1-4939-9173-0_14
Sergey, K., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: Scalable and Accurate Long-Read Assembly via Adaptive K-Mer Weighting and Repeat Separation. Genome Res. 27, 722–736. doi:10.1101/gr.215087.116
Singh, K., Chhuneja, P., Gupta, O. P., Jindal, S., and Yadav, B. (2018). Shifting the Limits in Wheat Research and Breeding Using a Fully Annotated Refrence Genome. Science 361, 1–13. doi:10.1126/science.aar7191
Wang, X., Xu, W., Wei, L., Zhu, C., and Feng, C. (2019). Nanopore Sequencing and De Novo Assembly of a Black-Shelled Pacific Oyster (Crassostrea gigas) Genome. Front. Genet. 10, 1211. doi:10.3389/fgene.2019.01211
Xiao, Y., Li, J., Ren, G., Ma, D., Wang, Y., Xiao, Z. Z., et al. (2016a). Pronounced Population Genetic Differentiation in the Rock Bream Oplegnathus fasciatus Inferred from Mitochondrial DNA Sequences. Mitochondrial Dna 27, 1–8. doi:10.3109/19401736.2014.982553
Xiao, Y., Ma, D., Dai, M., Liu, Q., Xiao, Z., Li, J., et al. (2016b). The Impact of Yangtze River Discharge on the Genetic Structure of a Population of the Rock Bream, Oplegnathus fasciatus. Mar. Biol. Res. 12, 1–9. doi:10.1080/17451000.2016.1154576
Xiao, Y., Xiao, Z., Ma, D., and Liu, J. (2019). Genome Sequence of the Barred Knifejaw Oplegnathus fasciatus (Temminck & Schlegel, 1844): the First Chromosome-Level Draft Genome in the Family Oplegnathidae. GigaScience 8, giz013. doi:10.1093/gigascience/giz013
Xiao, Y., Xiao, Z., Ma, D., Zhao, C., and Herrera-Ulloa, A. (2020). Chromosome-Level Genome Reveals the Origin of Neo-Y Chromosome in the Male Barred Knifejaw Oplegnathus fasciatus. iScience 23, 101039. doi:10.1016/j.isci.2020.101039
Xu, D., Sember, A., Zhu, Q., Oliveira, E. A., Liehr, T., Abh, A. R., et al. (2019). Deciphering the Origin and Evolution of the X1X2Y System in Two Closely-Related Oplegnathus Species (Oplegnathidae and Centrarchiformes). Int. J. Mol. Sci. 20, 3571. doi:10.3390/ijms20143571
Yang, Z. (1997). PAML: a Program Package for Phylogenetic Analysis by Maximum Likelihood. Computer Appl. Biosciences Cabios 13, 555. doi:10.1093/bioinformatics/13.5.555
Zhang, G., Fang, X., Guo, X., Li, L., Luo, R., Xu, F., et al. (2012). The Oyster Genome Reveals Stress Adaptation and Complexity of Shell Formation. Nature 490, 49–54. doi:10.1038/nature11413
Zhao, J., Bai, H., Ke, Q., Li, B., Zhou, Z., Wang, H., et al. (2021). Genomic Selection for Parasitic Ciliate Cryptocaryon Irritans Resistance in Large Yellow Croaker. Aquaculture 531, 735786. doi:10.1016/j.aquaculture.2020.735786
Keywords: Oplegnathus fasciatus, genomic resources, PacBio, Hi-C, phylogenetic analysis
Citation: Bai Y, Gong J, Zhou Z, Li B, Zhao J, Ke Q, Zou X, Pu F, Wu L, Zheng W, Zhou T and Xu P (2021) Chromosome-Level Assembly of the Southern Rock Bream (Oplegnathus fasciatus) Genome Using PacBio and Hi-C Technologies. Front. Genet. 12:811798. doi: 10.3389/fgene.2021.811798
Received: 09 November 2021; Accepted: 29 November 2021;
Published: 21 December 2021.
Edited by:
Roger Huerlimann, Okinawa Institute of Science and Technology Graduate University, JapanReviewed by:
Tianxiang Gao, Zhejiang Ocean University, ChinaSyed Farhan Ahmad, Kasetsart University, Thailand
Copyright © 2021 Bai, Gong, Zhou, Li, Zhao, Ke, Zou, Pu, Wu, Zheng, Zhou and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Xu, eHVwZW5nNzdAeG11LmVkdS5jbg==
†These authors have contributed equally to this work