 Shuguang Han
Shuguang Han Ning Wang
Ning Wang Yuxin Guo1,3
Yuxin Guo1,3 Lei Xu
Lei Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 15 December 2021
Sec. Computational Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.810875
This article is part of the Research Topic Machine Learning Techniques on Gene Function Prediction Volume II View all 25 articles
Inspired by L1-norm minimization methods, such as basis pursuit, compressed sensing, and Lasso feature selection, in recent years, sparse representation shows up as a novel and potent data processing method and displays powerful superiority. Researchers have not only extended the sparse representation of a signal to image presentation, but also applied the sparsity of vectors to that of matrices. Moreover, sparse representation has been applied to pattern recognition with good results. Because of its multiple advantages, such as insensitivity to noise, strong robustness, less sensitivity to selected features, and no “overfitting” phenomenon, the application of sparse representation in bioinformatics should be studied further. This article reviews the development of sparse representation, and explains its applications in bioinformatics, namely the use of low-rank representation matrices to identify and study cancer molecules, low-rank sparse representations to analyze and process gene expression profiles, and an introduction to related cancers and gene expression profile database.
In recent years, inspired by L1-norm minimization methods, such as basis pursuit (Donoho and Huo, 2001), compressed sensing (Candes et al., 2004; Candes and Tao, 2005; Lustig et al., 2007), and Lasso feature selection (Tibshirani, 1996), sparse representation shows up as a novel and potent data processing method. Sparse representation has been applied to pattern recognition, for example, digit recognition, speech recognition, and face recognition, and achieved good results. Hang and Wu (2009) first introduced sparse representation to the analysis of tumor gene expression data. They applied sparse representation to classify two multi-class tumor data, compared them with the classification performance of a support vector machine (SVM), and concluded that sparse representation was superior to SVM. Sparse representation was subsequently adopted for feature selection and the classification of tumor gene expression data. Hang applied it to gene selection and obtained sound classification results (Hang, 2009). Zheng et al. (Gan et al., 2013) proposed a sparse representation classification method based on meta-samples. The method uses singular value decomposition to extract the meta-samples of various training samples, and then uses the meta-samples to linearly represent test samples and categorizes them based on representation coefficients. The test samples compare the classification performance of this method with other classic methods on multiple two-class and multi-class datasets. The experimental results demonstrated that this method is superior to a classic SVM and other methods. These results testify the application potential of sparse representation methods in tumor gene expression data analysis.
The low-rank sparse representation model based on sparse representation has also become a topic of great interest in fields such as machine vision, machine learning, and image processing, and has been applied successfully in video image processing, target recognition, task learning, and recommendation systems (Huang et al., 2017; Yu and Gao, 2019; Liu et al., 2020a; Yu et al., 2020). In low-rank sparse representation theory, a noisy or missing data matrix is decomposed into an accurate data matrix and a singular/sparse data matrix, where the accurate data matrix has low-rank characteristics, and the singular/sparse data matrix contains data noise and singular data (Tang et al., 2020). Wright et al. proposed a classification algorithm based on sparse representation (Wright et al., 2009a) that successfully applies sparse representation theory to face recognition. Meanwhile, researchers have applied the sparsity of vectors to that of matrices, and proposed low-rank matrix recovery theory (Wright et al., 2009b; Emmanuel et al., 2009) and matrix low-rank representation (Liu et al., 2010). Low-rank representation has also received extensive attention from researchers and has become another important data representation method. It has demonstrated great potential. Sparse representation has many advantages, such as insensitivity to noise, strong robustness, insensitivity to selected features, and no “overfitting” phenomenon. Therefore, the application of sparse representation in bioinformatics should be studied further.
In recent years, inspired by discriminant analysis, researchers have combined discriminative ideas with sparse representation or low-rank representation theory to extract discriminative information from samples further to improve recognition performance. Discriminant analysis is a multivariate statistical analysis method that analyzes various characteristic values of sample data, and then discriminates the category of the observed sample. For example, Fisher Linear Discrimination (FLD). The essence of the FLD is to project sample points into a low-dimensional space so that, in the projected space, the distance between sample points of the same category is small and the distance between sample points of varying categories is large.
And because gene expression profile data research plays a vital role in genetic engineering, protein design, new drug development, etc., the use of machine learning methods including deep learning to explore gene expression profile data modeling methods has led to the biological field Wide attention of researchers. At the same time, the innovation of this article are; 1) The low-rank representation (LRR) is modified, and a new type of low-rank representation model is constructed by introducing manifold regularization and class label restriction mechanism, which is used for low-rank scoring of gene features and selecting the optimal gene subset; 2) Introduce the idea of deep learning to the low-rank sparse model, and propose a deep feature representation method for gene expression profile data, and realize the classification and clustering of gene data on this basis; 3) Propose a feature selection mechanism for gene expression profile data based on low-rank graphs; 4) Establish a genetic feature correlation measurement criterion based on low-rank representation coefficients, use this criterion to obtain a new genetic feature selection method, and use Robust Principal Component Analysis (RPCA) and Maximum Interval Criterion (MMC) to build a two-step genetic feature selection method.
As sparse representation and low-rank representation have been widely applied to the analysis and research of cancer and gene expression profiles in recent years, the databases of cancer and gene expression profiles can be adopted, respectively, for the research and application of sparse representation methods. Tables 1, 2 show the specific database description.

TABLE 1. Common cancer databases.

TABLE 2. Commonly used gene expression profile database.
The development of bioinformatics is mainly divided into three stages: gene stage, genomic stage, and post-genomic stage. The first two stages mainly focus on the research of gene sequences (Yu et al., 2019; Cai et al., 2020a; Fu et al., 2020; Wang et al., 2020; Dao et al., 2021a; Dao et al., 2021b; Huang et al., 2021). In the post-genome stage, bioinformatics has entered a new development period, and its research focus has shifted from the study of gene sequences to the study of gene functions (Wang et al., 2013; Dong et al., 2020; Wang et al., 2021a; Lv et al., 2021; Yu et al., 2021). It incorporates all aspects of the process of acquiring, storing, processing, distributing, and explaining biological information, and combines various tools of applied mathematics, computer science, and biology to clarify and understand biological significance in biological data.
As a common malignant tumor, cancer is a common fatal disease worldwide because of its complex pathogenic factors, high treatment difficulty, and high risk of recurrence and metastasis. In China, deaths from cancer are always high, and it is a severe threat to the lives and health of Chinese people (Silverberg and Lubera, 1998; Chen et al., 2020). How to prevent and treat cancer effectively has become a topic of widespread concern the world over. With the development of high-throughput sequencing technology, scientists can observe the gene expression of cancer cells at the single-cell level. Feature mining methods for cancer molecules are divided into supervised and unsupervised learning, as shown in Figure 1. The supervised method generally includes two steps: 1) First obtain the cancer classification information of the research sample through known prior information or other models. For example, using marker genes, clustering methods, or SNF algorithms. 2) Based on the sample typing information obtained in the previous step, the candidate molecular characteristics are screened out in the training data set, and then these candidate molecular characteristics are classified or survival analysis in the validation data set to determine the final effective molecular characteristics. The methods often used in this step mainly include difference hypothesis testing, support vector machine algorithm, random forest and linear discriminant analysis. Another type of unsupervised method does not require the typing information of a given sample set. It is mainly based on model assumptions and related data theories. At the same time, the molecular features and samples in the data are grouped to obtain a molecular set or module, and for the “liveness” value of the sample in the new feature space, commonly used methods include bi-clustering algorithm, matrix decomposition and manifold learning. However, existing unsupervised methods (Chen et al., 2020; Zou et al., 2020) fail to distinguish different feature subspaces. Hence, they may produce errors, or even invalid results, when applied to cancer molecular feature mining. Thus, a low-rank representation learning algorithm (Chen and Yanga, 2014) is presented based on the presumption that the sample subspace exists, and samples in the same subspace can represent each other, while those in different subspaces cannot. The algorithm can accurately identify a “clustered” structure or grouping information of inherent samples in the heterogeneous data. The effectiveness of this method has been widely recognized in image processing, and it also provides new ideas and directions for establishing accurate models for mining cancer molecular characteristics. Therefore, a mathematical model based on low-rank representation can be established by combining multiple scales, including molecules, modules, functional networks, and multi-omics molecular features. This model can be studied from the three aspects described below, and a series of mathematical models that are more in line with the heterogeneous structure of data and the biological characteristics of the disease are proposed, and a fair evaluation of the validity and practicability of the model is provided using simulated cases and the application of real data, and theoretical modeling and tools for analyzing multi-scale molecular characteristics of cancer are provided. Figure 2 shows the method of applying a low-rank representation matrix to mine the molecular characteristics of cancer.
1) A dimensionality reduction method is adopted to obtain the characteristics of the molecular module specific to the cancer subtype (Cheng et al., 2018; Tang et al., 2018; Yu et al., 2018; Zhang et al., 2018; Jiang et al., 2019; Su et al., 2019; Liu et al., 2020b; Su et al., 2020). It can address nonlinear sample structure issues that the traditional dimensionality reduction method cannot identify. This is because the dimensionality reduction model fused with low-rank representation learning can process highly heterogeneous data, adaptively capture sample cluster structure and subtype-specific module features, and improve the ability to classify tumor subtypes and obtain reliable molecular modules.
2) The fusion model with molecular function information was used to analyze the characteristics of functional subnets. Makes full use of the advantages of known functional information in biological interpretability (Liu et al., 2019; Cai et al., 2020b), deeply probes into functionally abnormal biological pathways or molecular behaviors, obtains subtype-specific functional subnets, and clarifies the molecular mechanism of cancer from a functional level.
3) A fusion model with molecular function information analyzes the features of functional subnets, makes full use of the biological characteristics of the sample representation relationship consistency of multi-omics data, further explores synergistic or complementary molecular characteristic information at the system level, and provides new clue to enable the understanding of the cross-omics pathogenic factors of cancers.

FIGURE 1. Method for mining cancer molecular features.
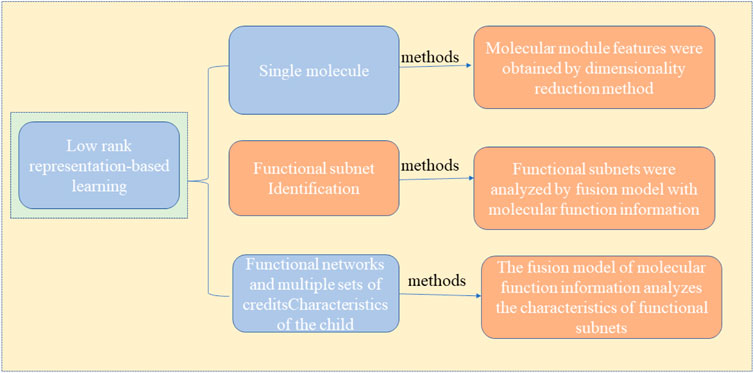
FIGURE 2. Method for mining cancer molecular features using a low-rank representation matrix.
At the same time, medical imaging is also playing an increasingly major role in helping doctors to conduct a precise diagnosis of cancer. Even medical imaging cloud and remote image center can be used for cloud reading, remote consultation, health management, disease diagnosis, image archiving and communication, etc. (Mehto and Mehra, 2016; Ma et al., 2020; Meziane, 2020; Zhang et al., 2021). Therefore, how to protect patients’ personal information in medical images, such as CT, MRI, and other medical images, so that this personal information and patients’ electronic medical records cannot be leaked has constituted a key issue for the medical industry that needs to be resolved urgently against the background of machine learning cloud computing and big data. Using medical image digital watermarking technology is an effective method to work out this problem (Hong et al, 2016; Vairaprakash and Shenbagavalli, 2017; Shen et al., 2018; Yang et al., 2018; Zhou et al., 2020). Compared with general digital watermarking technology, digital watermarking technology used in medical images, theoretically, should satisfy three characteristics: reliability, availability, and confidentiality.
Common medical image digital watermarking algorithms are divided into three categories: 1) a medical image watermarking algorithm based on non-interest area RONI (Thanki et al., 2017), which diminishes the watermark embedding capacity (Liu et al., 2016b; Gangadhar et al., 2018) and demonstrates poor robustness; 2) reversible digital watermarking; 3) classic conventional digital watermarking algorithms used to process medical images. However, these conventional watermarking algorithms demonstrate poor resistance to geometric attacks; hence, other models that can resist conventional attacks and geometric attacks effectively are necessary. Thus, the design and construction of a new medical image algorithm model based on perceptual hashing technology and neural network technology should be attempted to resolve the contradiction between the robustness and invisibility of medical image digital watermarking. Perceptual hashing mainly resolves the issue of conventional attacks and the neural network mainly resolves geometric attacks. The framework diagram is shown in Figure 3. The model process roughly uses the output vector of the hash algorithm as the input vector of the neural network, and finally obtains the output result. Perceptual hashing is a type of hashing algorithm, and its workflow has 7 main steps: 1) Reduce the size, reduce the picture to
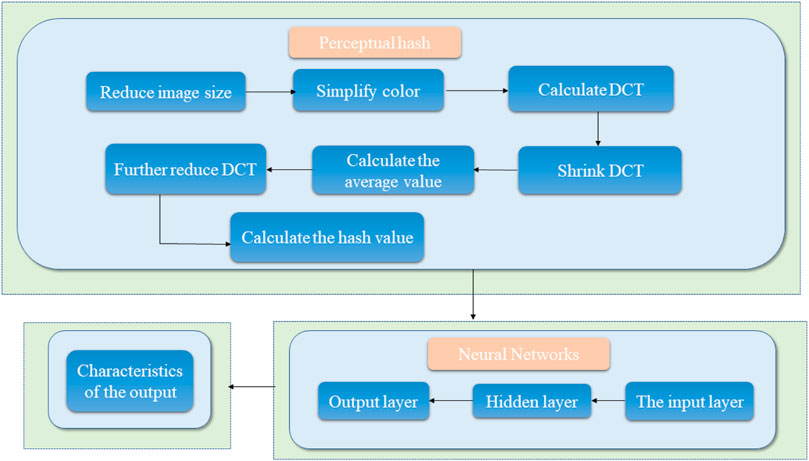
FIGURE 3. Schematic diagram of the robustness feature acquisition of medical images based on perceptual hashing and a neural network.
The robustness and invisibility of digital watermark images can be studied from the following perspectives:
1) Regarding anti-conventional attacks, research is based on the extraction of perceptual hashing medical image features in the transform domain. It is used to study the human visual system, and by combining with perceptual hashing technology, establishes a transform domain perception hash algorithm model, and locates a vector that conforms to the human visual characteristic and is robust against conventional attacks.
2) Regarding anti-geometric attacks, the extraction of medical image features based on perceptual hashing and a neural network is studied. The Osirix DICOM image library and existing medical images are adopted to construct a medical image database that is attacked using nonlinear geometry. Then, the neural network model is designed to train the 2D and 3D medical images after nonlinear geometric attacks, and find the robust feature vectors against nonlinear geometric attacks, which are used as the features of designing robust watermarking algorithms for medical images against geometric attacks.
3) Research on methods for extracting robust perceptual hashing sequences from medical images based on perceptual hashing and neural networks.
4) Regarding research on how to embed large-capacity digital watermarks in medical images, perpetual hashing sequence feature vectors that counter conventional attacks and geometric attacks are used to generate a secret key by combining with the encrypted watermark to complete the embedding and extraction of a large-capacity watermark.
The emergence of gene expression profile data helps the understanding of the pathological process of cancer cells at the molecular level. Tens of thousands of varying genes in tissue samples can be detected by gene chips, and then the gene chip expression profile data can be analyzed and processed. Thus, tumors are classified so that patients can be treated effectively. However, gene expression profiles are characterized by high dimensionality, large noise, a small number of gene samples, missing data, data redundancy, and an unbalanced distribution of class samples. Thus, advanced data modeling methods must be used to extract the classification characteristics of samples effectively from tens of thousands of gene expression profiles. With the rapid development of artificial intelligence and machine learning in speech and machine vision in recent years, the use of machine learning methods, including deep learning, to explore gene expression profile data modeling methods is destined to be a development trend in the future.
Presently, research on gene expression profiles mainly covers the following: 1) the preprocessing of gene expression profile data, 2) extraction of gene expression profile data features, 3) selection of gene expression profile data features, and 4) clustering and classification research of gene expression profile data. Common gene feature selection methods are categorized into three types: the filter method, wrapper method, and embedded method (Bolón-Canedo et al., 2014). They can also be based on low-rank scoring, low-rank representation coefficient-based gene feature correlation measurement, and a two-step method based on robust principal component analysis (RPCA) (Partridge and Jabri, 2002) and the maximum margin criterion (MMC) for feature selection. RPCA, low-rank representation (Shu et al., 2017), and matrix completion (Cao et al., 2011; Zeng et al., 2017; Liu et al., 2020c; Ran et al., 2020; Zhao et al., 2020) are three main research areas for low-rank sparse theory. As the name implies, sparse representation refers to a linear combination of fewer basic signals to express most or all of the original signal. Among them, these basic signals are called atoms, which are selected from the over-complete dictionary; and the over-complete dictionary is gathered from atoms whose number exceeds the signal dimension. Therefore, it can be seen that any signal has different sparse representations under different atom groups. For example, a
Low-rank sparse representation models have been applied in many fields (Cheng et al., 2016; Chen et al., 2017; Zhang et al., 2017; Brbic and Kopriva, 2018; Chen et al., 2018; Xie et al., 2018; Yuanyuan et al., 2018; Zeng et al., 2018; Ding et al., 2019; Shen et al., 2019; Zhang et al., 2019; Li et al., 2020; Wu and Yu, 2021), which demonstrate high superiority, particularly in terms of dimensionality reduction and subspace segmentation. Considering existing analysis methods, introduce a low-rank sparse representation model for gene expression profile data analysis, several new methods for feature selection and feature extraction of gene expression profile data based on low-rank sparse representation models are explored, and they are applied to gene expression profile clustering and classification. As shown in Figure 4, this section mainly uses the following process to study gene expression profile data based on low-rank sparse representation analysis. In typical cases, the following three specific research areas are mainly involved when studying gene expression profile data.
1) Estimation of missing points in gene expression profile data.
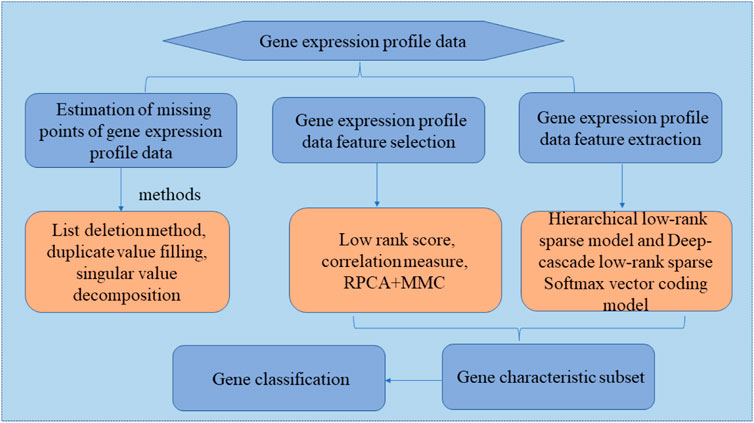
FIGURE 4. Research procedure for gene database analysis based on low-rank sparse representation.
In recent years, missing point estimation methods have included the following: 1) list deletion method; 2) duplicate value filling; 3) average value substitution method; and 4) the use of statistical methods for estimation, such as K-nearest neighbor (KNN) (Olga et al., 2001), singular value decomposition, and local least squares.
2) Feature selection for gene expression profile data.
Feature selection is a major prerequisite for the classification and clustering of gene expression profile data (Lu and Zhao, 2019; Zou et al., 2020; Qi et al., 2021a; Zulfiqar et al., 2021). Three common gene feature selection methods exist: the filter method, wrapper method, and embedded method. And Low-rank scoring, gene feature correlation measurement based on a low-rank representation coefficient, and a two-step method based on RPCA and MMC can also be used to select features. To overcome the shortcomings of traditional low-rank representation models, feature selection introduces manifold regularization constraints and class-label information constraints, sets up a manifold regularized low-rank representation model and a class-label constrained low-rank representation model, and solve the low-rank representation coefficient matrix in the two models. On this basis, two different low-rank graphs are set up, the low-rank graphs are used to score each gene feature, and a set of optimal gene feature subsets is selected according to the score.
3) Gene expression profile data feature extraction.
Common feature extraction methods can be divided into linear and nonlinear transformations. Typical linear feature extraction algorithms include sparse principal component analysis (PCA) (Min et al., 2018; Islam et al., 2020), independent component analysis (Moysés et al., 2017), and LDA. Nonlinear transformation methods primarily include neural networks, kernel methods (Qi et al., 2021b), manifold learning (Shen et al., 2017), sparse representation (Min et al., 2017), and matrix factorization methods (Wang et al., 2017; Yang et al., 2017; Yang and Hu, 2017; McCall et al., 2019). With the continuous development of machine learning and data mining, new feature extraction methods continue to arise. For example, PCA, FA, and ICA are three characteristic methods commonly used in gene expression profile data mining.
Gene expression profile data analysis has attracted widespread attention from scholars, and a series of gene expression profile analysis methods have been proposed. Classic methods such as PCA, LDA, KNN, decision-making tree method, ensemble learning, SVM, extreme learning machine, neural network, sparse representation, and gene bi-clustering method based on qualitative/quantitative measurement have been widely applied to the classification and clustering of gene expression profile data. Meanwhile, these technologies can provide techniques and comparisons for low-rank sparse representation methods. The core of the low-rank sparse representation method is low-rank sparse modeling theory. As an effective tool for large-scale data analysis, this theory has made great progress in recent years. Additionally, it has been widely used in subspace segmentation, image processing and recognition, machine vision, system modeling and control, and other large-scale data analysis.
Therefore, it has become an inevitable trend to apply low-rank sparse representation models to study them. Low-rank sparse representation models have been applied in multiple fields, particularly in dimensionality reduction and subspace segmentation. For example, in feature extraction, traditional graph-based learning algorithm feature extraction methods are constrained using a graph construction method, and the effectiveness of the extracted feature vectors is reduced. By contrast, low-rank graphs have better local and global data description capabilities. A dimensionality reduction method based on low-rank graphs is a more effective feature extraction method. Moreover, with the advancement of biological sequencing technology, scientists have been able to observe the gene expression of cancer cells at the single-cell level, and discovered that the heterogeneity of cancer tissue far exceeds previous estimates. However, so far, low-rank sparse representation models are rarely used for gene data analysis. Therefore, this article introduces low-rank sparse representation models for gene expression profile data analysis based on existing analysis methods. Discuss new methods for feature selection and feature extraction of gene expression profile data based on low-rank sparse representation model, and use it for gene expression profile clustering and classification.
At the same time, with the advancement of biological sequencing technology, scientists have been able to observe the gene expression of cancer cells at the single-cell level, and found that the heterogeneity of cancer tissues far exceeds previous estimates. The observation samples of potential strongly heterogeneous data are likely to be in multiple feature subspaces. Each subspace is composed of the same set of molecular features that represent the same cancer class (subtype), and samples from different subspaces belong to different cancer class (subtype). However, many unsupervised methods proposed before cannot distinguish different feature subspaces, so errors or even invalid results may occur when these methods are used for cancer molecular feature mining. After research, it is found that the low-rank representation learning algorithm can accurately identify the inherent sample “cluster” structure or grouping information in heterogeneous data. The algorithm assumes that the sample subspace exists, and samples in the same subspace can characterize each other. Samples in different subspaces cannot characterize each other. Moreover, the effectiveness of this algorithm has been widely recognized in the field of image processing, and it also provides us with new ideas and new directions for establishing accurate models for mining cancer molecular characteristics.
Gene expression profile data analysis has attracted widespread attention from scholars at home and abroad. Not only have they proposed a series of gene expression profile analysis methods, but they also developed a variety of gene software based on gene public databases, such as EASE network platform, pathway analysis software Gen-MAPP2 and the development of the domestically developed pathway analysis platform KOBAS, the development of these software provides a basis for the subsequent further research on gene expression profiles.
This article mainly uses low-rank sparse modeling theory to analyze experimental data. As one of the effective tools for large-scale data analysis, this theory has been widely used in different aspects in recent years. For example, sparse representation has been applied to the field of pattern recognition and has yielded fruitful results. The low-rank sparse representation model based on sparse representation has also become a research focus in machine vision, machine learning, and image processing, and has been applied successfully in video image processing, target recognition, task learning, bioinformatics (Ding et al., 2020; Hong et al., 2020; Hu et al., 2020; Lu et al., 2020; Hu et al., 2021a; Hu et al., 2021b; Wang et al., 2021b), and recommendation systems (Wei et al., 2014; Wei et al., 2017a; Wei et al., 2017b). However, further attention should be paid to low-rank representation learning. In specific applications, LRR generally uses original data as a dictionary, which requires a sufficient number of observed data samples, and only part of the data in the dictionary can be damaged. In real-world scenarios, the aforementioned assumptions may not be tenable; hence, LatLRR can be considered, and a dictionary can be constructed using observed and unobserved data.
At the same time, sparse representation also has important clinical significance. For example, data released by the National Cancer Center reveal that there are approximately 4.29 million new cancer patients in China every year, which accounts for 20% of new cases globally, and deaths have reached 2.81 million. Approximately 10,000 patients are diagnosed with cancer in China every day, that is, one patient every 7 min. Therefore, the prevention and treatment of cancers are not optimistic. It is expected that the incidence of cancers will continue to rise in the next one or two decades. The high incidence of cancer cases has resulted in severe challenges to domestic economic development and residents’ healthy life. How to prevent and treat cancer effectively has become a topic of great interest worldwide. With the advancement of high-throughput technology, biomedicine is rapidly stepping into the era of big data. Omics data represented by gene expression profiles have demonstrated particular leaps. The emergence of gene expression profile data helps people to understand the pathological process of cancer cells at the molecular level. Thousands of genes in tissue samples can be detected by gene chips, and then the tumor can be classified by analyzing and processing the gene chip expression profile data so that patients can be treated effectively. However, because of the characteristics of gene expression profile data, there are still many problems in the research field. With the rapid development of artificial intelligence and machine learning in the field of speech and machine vision, in the next few years, artificial intelligence and machine learning will play an increasingly important role in genetic biology, genomic medicine and precision medicine, especially deep learning. The rapid development has attracted widespread attention from researchers in the biomedical field, so it has become an inevitable trend to use low-rank sparse representation models to study them. An extensive application of sparse representation in bioinformatics helps to address the problem that some unsupervised algorithms cannot distinguish different feature subspaces of cancer molecules. Moreover, it is expected that, in the near future, it can provide technological references for the prevention and treatment of critical illness, and the research and development of new drugs.
However, sparse representation in bioinformatics still has varying degrees of limitations. For example: 1) Constructing a more flexible sparse representation model. In the existing sparse representation model, there is an objective function and a constraint function, the objective function is generally to minimize the energy of the noise under the assumption that the observation signal has a linear model form and contains Gaussian white noise, constraint function generally refers to sparse constraint term. On the one hand, this objective function treats the sparse components equally; on the other hand, it ignores the existence of other goals in different applications, because if you look at it from the standpoint of representation alone, it does not necessarily require the sparsest solution to be unique or the sparsest solution is not the most ideal. Therefore, it is necessary to construct a sparse representation model with multiple targets and variable regular parameters to meet the characteristics and needs of more application problems. 2) When determining the regular parameter
At the same time, sparse representation provides a powerful means in blind source separation technology, because blind source separation technology is to solve the unknown input and unknown transmission channel and output the known signal processing technology. The sparse representation technology reduces the complexity of the algorithm by separating the estimation process of the mixing matrix and the estimation process of the source signal, and improves the accuracy of the source signal separation. Therefore, sparse representation has become a popular method in the current blind source separation problem.
Conceptualization, LS and YJ; data collection or analysis, YG and LX; validation, YG and FT; writing—original draft preparation, SH and NW; writing—review and editing, NW and SH. All authors have read and agreed to the published version of the manuscript.
The work was supported by the National Key R and D Program of China (No. 2020YFB2104400), the Special Science Foundation of Quzhou (2021D004), and the research foundation of Shenzhen Polytechnic (No. 6021310016K).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors LX.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Maxine Garcia, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn/) for editing the English text of a draft of this manuscript. As well as thanks to the guidance of the tutor and the joint efforts of other authors, the success of this article is the result of everyone’s joint efforts.
Bolón-Canedo, V., Sánchez-Maroño, N., Alonso-Betanzos, A., and José, B. (2014). A Review of Microarray Datasets and Applied Feature Selection Methods. Inf. Sci. Int. J. 282, 111–135. doi:10.1016/j.ins.2014.05.042
Brbic, M., and Kopriva, I. (2018). Multi-view Low-Rank Sparse Subspace Clustering. Pattern Recognition J. Pattern Recognition Soc. 73, 247–258. doi:10.1016/j.patcog.2017.08.024
Cai, L., Ren, X., Fu, X., Peng, L., Gao, M., Zeng, X., et al. (2020). Interpretable Sequence-Based Enhancers and Their Strength Predictor. Bioinformatics 37 (8), 1060–1067. doi:10.1093/bioinformatics/btaa914
Cai, L., Wang, L., Fu, X., Xia, C., Zeng, X., and Zou, Q. (2020). ITP-pred: an Interpretable Method for Predicting, Therapeutic Peptides with Fused Features Low-Dimension Representation. Brief. Bioinform. 22 (4), bbaa367. doi:10.1093/bib/bbaa367
Candes, E. J., and Tao, T. (2005). Decoding by Linear Programming. IEEE Trans. Inform. Theor. 51 (12), 4203–4215. doi:10.1109/tit.2005.858979
Candes, E., Romberg, J., and Tao, T. (2004). Robust Uncertainty Principles: Exact Signal Reconstruction from Highly Incomplete Frequency Information.
Cao, F., Cai, M. M., and Tan, Y. (2011). Image Interpolation via Low-Rank Matrix Completion and Recovery, International Workshop on Java Technologies for Real-Time & Embedded Systems.
Chen, B., Yang, Z., and Yang, Z. (2018). An Algorithm for Low-Rank Matrix Factorization and its Applications. Neurocomputing 275, 1012–1020. doi:10.1016/j.neucom.2017.09.052
Chen, J., Mao, H., Sang, Y., and Yi, Z. (2017). Subspace Clustering Using a Symmetric Low-Rank Representation. Knowledge-Based Syst. 127, 46–57. doi:10.1016/j.knosys.2017.02.031
Chen, J., and Yanga, J. (2014). Robust Subspace Segmentation via Low-Rank Representation. IEEE Trans. Cybernetics 44 (8), 1432.
Chen, Z., Zhao, P., Li, F., Marquez-Lago, T. T., Leier, A., Revote, J., et al. (2020). iLearn: an Integrated Platform and Meta-Learner for Feature Engineering, Machine-Learning Analysis and Modeling of DNA, RNA and Protein Sequence Data. Brief. Bioinform. 21 (3), 1047–1057. doi:10.1093/bib/bbz041
Cheng, L., Hu, Y., Sun, J., Zhou, M., and Jiang, Q. (2018). DincRNA: a Comprehensive Web-Based Bioinformatics Toolkit for Exploring Disease Associations and ncRNA Function. Bioinformatics 34 (11), 1953–1956. doi:10.1093/bioinformatics/bty002
Cheng, L., Shi, H., Wang, Z., Hu, Y., Yang, H., Zhou, C., et al. (2016). IntNetLncSim: an Integrative Network Analysis Method to Infer Human lncRNA Functional Similarity. Oncotarget 7 (30), 47864–47874. doi:10.18632/oncotarget.10012
Dao, F. Y., Lv, H., Su, W., Sun, Z. J., Huang, Q. L., and Lin, H. (2021). iDHS-Deep: an Integrated Tool for Predicting DNase I Hypersensitive Sites by Deep Neural Network. Brief. Bioinformatics 22 (4), bbaa356. doi:10.1093/bib/bbab047
Dao, F. Y., Lv, H., Zhang, D., Zhang, Z. M., Liu, L., and Lin, H. (2021). DeepYY1: a Deep Learning Approach to Identify YY1-Mediated Chromatin Loops. Brief. Bioinformatics 22 (4). doi:10.1093/bib/bbaa356
Ding, Y., Tang, J., and Guo, F. (2019). Identification of Drug-Side Effect Association via Multiple Information Integration with Centered Kernel Alignment. Neurocomputing 325, 211–224. doi:10.1016/j.neucom.2018.10.028
Ding, Y., Tang, J., and Guo, F. (2020). Identification of Drug-Target Interactions via Fuzzy Bipartite Local Model. Neural Comput. Applic 32, 10303–10319. doi:10.1007/s00521-019-04569-z
Dong, L., Wang, J., and Wang, G. (2020). BYASE: a Python Library for Estimating Gene and Isoform Level Allele-specific Expression. Bioinformatics 36 (19), 4955–4956. doi:10.1093/bioinformatics/btaa636
Donoho, D. L., and Huo, X. (2001). Uncertainty Principles and Ideal Atomic Decomposition. IEEE Trans. Inform. Theor. 47 (7), 2845–2862. doi:10.1109/18.959265
Forbes, S. A., Bindal, N., Bamford, S., Cole, C., Kok, C. Y., Beare, D., et al. (2011). COSMIC: Mining Complete Cancer Genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–D950. doi:10.1093/nar/gkq929
Fu, X., Cai, L., Zeng, X., and Zou, Q. (2020). StackCPPred: a Stacking and Pairwise Energy Content-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency. Bioinformatics 36 (10), 3028–3034. doi:10.1093/bioinformatics/btaa131
Gan, B., Zheng, C. H., and Liu, J. X. (2013). “Metasample-Based Robust Sparse Representation for Tumor Classification,” in International Conference on Biomedical Engineering(ICBE).
Gangadhar, Y., Giridhar Akula, V. S., and Reddy, P. C. (2018). An Evolutionary Programming Approach for Securing Medical Images Using Watermarking Scheme in Invariant Discrete Wavelet Transformation. Biomed. Signal Process. Control. 43, 31–40. doi:10.1016/j.bspc.2018.02.007
Hang, X., and Wu, F. X. (2009). Sparse Representation for Classification of Tumors Using Gene Expression Data. J. Biomed. Biotechnol. 2009 (10), 403689. doi:10.1155/2009/403689
Hang, X. (2009). “Multiclass Gene Selection on Microarray Data Using L1-Norm Least Square Regression,” in International Joint Conference on Bioinformatics, Systems Biology and Intelligent Computing, 52–55. doi:10.1109/IJCBS.2009.76
Hong, L., Xiao, Di., Zhang, R., Zhang, Y., and Bai, S. (2016). Robust and Hierarchical Watermarking of Encrypted Images Based on Compressive Sensing. Signal. Process. Image Commun. A Publ. Eur. Assoc. Signal Process. 45, 41–51. doi:10.1016/j.image.2016.04.002
Hong, Q., Yan, R., Wang, C., and Sun, J. (2020). Memristive Circuit Implementation of Biological Nonassociative Learning Mechanism and its Applications. IEEE Trans. Biomed. Circuits Syst. 14 (5), 1036–1050. doi:10.1109/tbcas.2020.3018777
Hu, Y., Qiu, S., and Cheng, L. (2021). Integration of Multiple-Omics Data to Analyze the Population-specific Differences for Coronary Artery Disease. Comput. Math. Methods Med. 2021, 7036592. doi:10.1155/2021/7036592
Hu, Y., Sun, J. Y., Zhang, Y., Zhang, H., Gao, S., Wang, T., et al. (2021). rs1990622 Variant Associates with Alzheimer's Disease and Regulates TMEM106B Expression in Human Brain Tissues. BMC Med. 19 (1), 11. doi:10.1186/s12916-020-01883-5
Hu, Y., Zhang, H., Liu, B., Gao, S., Wang, T., Han, Z., et al. (2020). rs34331204 Regulates TSPAN13 Expression and Contributes to Alzheimer's Disease with Sex Differences. Brain 143 (11), e95. doi:10.1093/brain/awaa302
Huang, L., Li, X., Guo, P., Yao, Y., Liao, B., Zhang, W., et al. (2017). Matrix Completion with Side Information and its Applications in Predicting the Antigenicity of Influenza Viruses. Bioinformatics 33 (20), 3195–3201. doi:10.1093/bioinformatics/btx390
Huang, S., He, X., Wang, G., and Bao, E. (2021). AlignGraph2: Similar Genome-Assisted Reassembly Pipeline for PacBio Long Reads. Brief Bioinform 22 (5), bbab022. doi:10.1093/bib/bbab022
Islam, M. A., Kundu, S., and Hassan, R. (2020). Gene Therapy Approaches in an Autoimmune Demyelinating Disease: Multiple Sclerosis. Cgt 19 (6), 376–385. doi:10.2174/1566523220666200306092556
Jiang, L., Xiao, Y., Ding, Y., and Tang, J. (2019). Discovering Cancer Subtypes via an Accurate Fusion Strategy on Multiple Profile Data. Front. Genet. 10, 20. doi:10.3389/fgene.2019.00020
Li, J., Chang, M., Gao, Q., Song, X., and Gao, Z. (2020). Lung Cancer Classification and Gene Selection by Combining Affinity Propagation Clustering and Sparse Group Lasso. Cbio 15 (7), 703–712. doi:10.2174/1574893614666191017103557
Liu, C., Wei, D., Xiang, J., Ren, F., Huang, L., Lang, J., et al. (2020). An Improved Anticancer Drug-Response Prediction Based on an Ensemble Method Integrating Matrix Completion and Ridge Regression. Mol. Ther. - Nucleic Acids 21, 676–686. doi:10.1016/j.omtn.2020.07.003
Liu, G., Lin, Z., Yan, S., Sun, J., Yu, Y., and Ma, Y. (2010). Robust Recovery of Subspace Structures by Low-Rank Representation.
Liu, L., Li, Q.-Z., Jin, W., Lv, H., and Lin, H. (2019). Revealing Gene Function and Transcription Relationship by Reconstructing Gene-Level Chromatin Interaction. Comput. Struct. Biotechnol. J. 17, 195–205. doi:10.1016/j.csbj.2019.01.011
Liu, Y., Yu, Z., Chen, C., Han, Y., and Yu, B. (2020). Prediction of Protein Crotonylation Sites through LightGBM Classifier Based on SMOTE and Elastic Net. Anal. Biochem. 609, 113903. doi:10.1016/j.ab.2020.113903
Liu, Y., Qu, X., and Xin, G. (2016). A ROI-Based Reversible Data Hiding Scheme in Encrypted Medical Images. J. Vis. Commun. Image Representation 39, 51–57. doi:10.1016/j.jvcir.2016.05.008
Liu, Y., Wen, Z., and Li, M. (2020). The Power of Matrix Factorization: Methods for Deconvoluting Genetic Heterogeneous Data at Expression Level. Curr. Bioinformatics 15 (8), 841–853.
Lu, X.-X., and Zhao, S.-Z. (2019). Gene-based Therapeutic Tools in the Treatment of Cornea Disease. Cgt 19 (1), 7–19. doi:10.2174/1566523219666181213120634
Lu, X., Wang, X., Ding, L., Li, J., Gao, Y., and He, K. (2020). frDriver: A Functional Region Driver Identification for Protein Sequence. IEEE/ACM Trans. Comput. Biol. Bioinformatics 18 (5), 1773–1783. doi:10.1109/TCBB.2020.3020096
Lustig, M., Donoho, D., and Pauly, J. M. (2007). Sparse MRI: The Application of Compressed Sensing for Rapid MR Imaging. Magn. Reson. Med. 58 (6), 1182–1195. doi:10.1002/mrm.21391
Lv, H., Dao, F. Y., Zulfiqar, H., Su, W., Ding, H., Liu, L., et al. (2021). A Sequence-Based Deep Learning Approach to Predict CTCF-Mediated Chromatin Loop. Brief. Bioinformatics 22 (5), bbab031. doi:10.1093/bib/bbab031
Ma, X., Xi, B., Zhang, Y., Zhu, L., Sui, X., Tian, G., et al. (2020). A Machine Learning-Based Diagnosis of Thyroid Cancer Using Thyroid Nodules Ultrasound Images. Cbio 15 (4), 349–358. doi:10.2174/1574893614666191017091959
McCall, A. L., Stankov, S. G., Cowen, G., Cloutier, D., Zhang, Z., Yang, L., et al. (2019). Reduction of Autophagic Accumulation in Pompe Disease Mouse Model Following Gene Therapy. Cgt 19 (3), 197–207. doi:10.2174/1566523219666190621113807
Mehto, A., and Mehra, N. (2016). Adaptive Lossless Medical Image Watermarking Algorithm Based on DCT & DWT. Proced. Comp. Sci. 78, 88–94. doi:10.1016/j.procs.2016.02.015
Meziane, B. (2020). A Self-Sustained Oscillator to the Lorenz-Haken Dynamics. Physica Scripta 95 (5). doi:10.1088/1402-4896/ab6e4c
Min, C., He, X., Shao, B. D., and Ying, W. D. (2017). A Novel Gene Selection Method Based on Sparse Representation and Max-Relevance and Min-Redundancy. Comb. Chem. High Throughput Screen. 20 (999). doi:10.2174/1386207320666170126114051
Min, W., Liu, J., and Zhang, S. (2018). Edge-group Sparse PCA for Network-Guided High Dimensional Data Analysis. Bioinformatics 34 (20), 3479–3487. doi:10.1093/bioinformatics/bty362
Moysés, N., Sff, E., Thelma, S., Campana, N., Maciel, F., Azevedo, B., et al. (2017). Independent Component Analysis (ICA) Based-Clustering of Temporal RNA-Seq Data. Plos One 12 (7), e0181195. doi:10.1371/journal.pone.0181195
Olga, T., Michael, C., Gavin, S., Pat, B., Trevor, H., Robert, T., et al. (2001). Missing Value Estimation Methods for DNA Microarrays. Bioinformatics 17 (6), 520–525. doi:10.1093/bioinformatics/17.6.520
Partridge, M., and Jabri, M. (2002). , 1, 289–298. doi:10.1109/NNSP.2000.889420Robust Principal Component AnalysisNeural Networks Signal. Process. X, IEEE Signal. Process. Soc. Workshop
Qi, C., Wang, C., Zhao, L., Zhu, Z., Wang, P., Zhang, S., et al. (2021). SCovid: Single-Cell Atlases for Exposing Molecular Characteristics of COVID-19 across 10 Human Tissues. Nucleic Acids Res. doi:10.1093/nar/gkab881
Qi, R., Wu, J., Guo, F., Xu, L., and Zou, Q. (2021). A Spectral Clustering with Self-Weighted Multiple Kernel Learning Method for Single-Cell RNA-Seq Data. Brief Bioinform 22 (4), bbaa216. doi:10.1093/bib/bbaa216
Ran, W., Chen, X., Wang, B., Yang, P., Li, Y., Xiao, Y., et al. (2020). Whole-exome Sequencing of Tumor-Only Samples Reveals the Association between Somatic Alterations and Clinical Features in Pancreatic Cancer. Curr. Bioinformatics 15 (10), 1160–1167. doi:10.2174/1574893615999200626190346
Shen, C., Ding, Y., Tang, J., Xu, X., and Guo, F. (2017). An Ameliorated Prediction of Drug-Target Interactions Based on Multi-Scale Discrete Wavelet Transform and Network Features. Ijms 18 (8), 1781. doi:10.3390/ijms18081781
Shen, M., Ma, B., Zhu, L., Mijumbi, R., Du, X., and Hu, J. (2018). Cloud-Based Approximate Constrained Shortest Distance Queries over Encrypted Graphs with Privacy Protection. IEEE Trans. Inf. Forensics Security 13 (4), 940–953. doi:10.1109/TIFS.2017.2774451
Shen, Y., Tang, J., and Guo, F. (2019). Identification of Protein Subcellular Localization via Integrating Evolutionary and Physicochemical Information into Chou's General PseAAC. J. Theor. Biol. 462, 230–239. doi:10.1016/j.jtbi.2018.11.012
Shu, Z., Fan, H., Huang, P., Wu, D., Ye, F., and Wu, X. (2017). Multiple Laplacian Graph Regularised Low‐rank Representation with Application to Image Representation. Iet Image Process. 11 (6), 370–378. doi:10.1049/iet-ipr.2016.0391
Silverberg, E., and Lubera, J. A. (1998). Cancer Statistics, 1989. Ca Cancer J. Clin. 39 (1), 3–20. doi:10.3322/canjclin.39.1.3
Su, R., Hu, J., Zou, Q., Manavalan, B., and Wei, L. (2020). Empirical Comparison and Analysis of Web-Based Cell-Penetrating Peptide Prediction Tools. Brief. Bioinformatics 21 (2), 408–420. doi:10.1093/bib/bby124
Su, R., Liu, X., Wei, L., and Zou, Q. (2019). Deep-Resp-Forest: A Deep forest Model to Predict Anti-cancer Drug Response. Methods 166, 91–102. doi:10.1016/j.ymeth.2019.02.009
Tang, H., Zhao, Y.-W., Zou, P., Zhang, C.-M., Chen, R., Huang, P., et al. (2018). HBPred: a Tool to Identify Growth Hormone-Binding Proteins. Int. J. Biol. Sci. 14 (8), 957–964. doi:10.7150/ijbs.24174
Tang, X., Cai, L., Meng, Y., Xu, J., Lu, C., and Yang, J. (2020). Indicator Regularized Non-negative Matrix Factorization Method-Based Drug Repurposing for COVID-19. Front. Immunol. 11, 603615. doi:10.3389/fimmu.2020.603615
Thanki, R., Borra, S., Dwivedi, V., and Borisagar, K. (2017). A RONI Based Visible Watermarking Approach for Medical Image Authentication. J. Med. Syst. 41 (9), 143. doi:10.1007/s10916-017-0795-3
Tibshirani, R. (1996). Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. Ser. B 58 (1). doi:10.1111/j.2517-6161.1996.tb02080.x
Tomczak, K., Czerwińska, P., and Wiznerowicz, M. (2015). The Cancer Genome Atlas (TCGA): An Immeasurable Source of Knowledge. Contemp. Oncol. (Pozn) 19 (1A), A68–A77. doi:10.5114/wo.2014.47136
Vairaprakash, S., and Shenbagavalli, A., A Discrete Rajan Transform-Based Robustness Improvement Encrypted Watermark Scheme Backed by Support Vector Machine ☆. Comput. Electr. Eng. 2017, 70: 826–843. doi:10.1016/j.compeleceng.2017.12.029
Wang, D., Zhang, Z., Jiang, Y., Mao, Z., Wang, D., Lin, H., et al. (2021). DM3Loc: Multi-Label mRNA Subcellular Localization Prediction and Analysis Based on Multi-Head Self-Attention Mechanism. Nucleic Acids Res. 49 (8), e46. doi:10.1093/nar/gkab016
Wang, G., Qi, K., Zhao, Y., Li, Y., Juan, L., Teng, M., et al. (2013). Identification of Regulatory Regions of Bidirectional Genes in Cervical Cancer. BMC Med. Genomics 6 (Suppl. 1), S5. doi:10.1186/1755-8794-6-S1-S5
Wang, H., Tang, J., Ding, Y., and Guo, F. (2021). Exploring Associations of Non-coding RNAs in Human Diseases via Three-Matrix Factorization with Hypergraph-Regular Terms on center Kernel Alignment. Brief. Bioinformatics 22 (5), bbaa409. doi:10.1093/bib/bbaa409
Wang, J., Chen, S., Dong, L., and Wang, G. (2020). CHTKC: a Robust and Efficient K-Mer Counting Algorithm Based on a Lock-free Chaining Hash Table. Brief Bioinform 22.
Wang, J., Liu, J. X., Zheng, C. H., Wang, Y. X., Kong, X. Z., and Weng, C. G. (2017). A Mixed-Norm Laplacian Regularized Low-Rank Representation Method for Tumor Samples Clustering. IEEE/ACM Trans. Comput. Biol. Bioinformatics 16 (1), 172–182. doi:10.1109/TCBB.2017.2769647
Wei, L., Liao, M., Gao, Y., Ji, R., He, Z., and Zou, Q. (2014). Improved and Promising Identification of Human MicroRNAs by Incorporating a High-Quality Negative Set. Ieee/acm Trans. Comput. Biol. Bioinf. 11 (1), 192–201. doi:10.1109/tcbb.2013.146
Wei, L., Wan, S., Guo, J., and Wong, K. K. (2017). A Novel Hierarchical Selective Ensemble Classifier with Bioinformatics Application. Artif. Intelligence Med. 83, 82–90. doi:10.1016/j.artmed.2017.02.005
Wei, L., Xing, P., Zeng, J., Chen, J., Su, R., and Guo, F. (2017). Improved Prediction of Protein-Protein Interactions Using Novel Negative Samples, Features, and an Ensemble Classifier. Artif. Intelligence Med. 83, 67–74. doi:10.1016/j.artmed.2017.03.001
Wright, J., Ganesh, A., Rao, S., and Ma, Y. (2009). Robust Principal Component Analysis: Exact Recovery of Corrupted Low-Rank Matrices. IEEE.
Wright, J., Yang, A., Ganesh, A., and Sastry, S. (2009). Robust Face Recognition via Sparse Representation. IEEE Trans. Pattern Anal. Mach. Intell. 31 (2), 210–227. doi:10.1109/tpami.2008.79
Wu, X., and Yu, L. (2021). EPSOL: Sequence-Based Protein Solubility Prediction Using Multidimensional Embedding. Bioinformatics 37 (23), 4314–4350. doi:10.1093/bioinformatics/btab463
Xie, L., Yin, M., Yin, X., Liu, Y., and Yin, G. (2018). Low-Rank Sparse Preserving Projections for Dimensionality Reduction. IEEE Trans. Image Process. 27, 5261–5274. doi:10.1109/TIP.2018.2855426
Yang, G., and Hu, Z. (2017). Gene Feature Extraction Based on Nonnegative Dual Graph Regularized Latent Low-Rank Representation. Biomed. Res. Int. 2017, 1–8. doi:10.1155/2017/1096028
Yang, H., Yin, J., and Jiang, M. (2018). Perceptual Image Hashing Using Latent Low-Rank Representation and Uniform LBP. Appl. Sci. 8 (2), 317. doi:10.3390/app8020317
Yang, J., Hagen, J., Guntur, K. V., Allette, K., Schuyler, S., Ranjan, J., et al. (2017). A Next Generation Sequencing Based Approach to Identify Extracellular Vesicle Mediated mRNA Transfers between Cells. BMC Genomics 18 (1), 987. doi:10.1186/s12864-017-4359-1
Yu, L., Yao, S., Gao, L., and Zha, Y. (2019). Conserved Disease Modules Extracted from Multilayer Heterogeneous Disease and Gene Networks for Understanding Disease Mechanisms and Predicting Disease Treatments. Front. Genet. 9, 745. doi:10.3389/fgene.2018.00745
Yu, L., and Gao, L. (2019). Human Pathway-Based Disease Network. Ieee/acm Trans. Comput. Biol. Bioinf. 16 (4), 1240–1249. doi:10.1109/tcbb.2017.2774802
Yu, L., Shi, Q., Wang, S., Zheng, L., and Gao, L. (2020). Exploring Drug Treatment Patterns Based on the Action of Drug and Multilayer Network Model. Ijms 21 (14), 5014. doi:10.3390/ijms21145014
Yu, L., Wang, M., Yang, Y., Xu, F., Zhang, X., Xie, F., et al. (2021). Predicting Therapeutic Drugs for Hepatocellular Carcinoma Based on Tissue-specific Pathways. Plos Comput. Biol. 17 (2), e1008696. doi:10.1371/journal.pcbi.1008696
Yu, L., Zhao, J., and Gao, L. (2018). Predicting Potential Drugs for Breast Cancer Based on miRNA and Tissue Specificity. Int. J. Biol. Sci. 14 (8), 971–982. doi:10.7150/ijbs.23350
Yuanyuan, C., Lei, Z., and Zhang, Yi. (2018). Subspace Clustering Using a Low-Rank Constrained Autoencoder. Inf. Sci. Int. J. 424, 27–38. doi:10.1016/j.ins.2017.09.047
Zeng, X., Liao, Y., Liu, Y., and Zou, Q. (2017). Prediction and Validation of Disease Genes Using HeteSim Scores. Ieee/acm Trans. Comput. Biol. Bioinf. 14 (3), 687–695. doi:10.1109/tcbb.2016.2520947
Zeng, X., Liu, L., Lü, L., and Zou, Q. (2018). Prediction of Potential Disease-Associated microRNAs Using Structural Perturbation Method. Bioinformatics 34 (14), 2425–2432. doi:10.1093/bioinformatics/bty112
Zhang, S., Wang, Y., Gu, Y., Zhu, J., Ci, C., Guo, Z., et al. (2018). Specific Breast Cancer Prognosis‐subtype Distinctions Based onDNAmethylation Patterns. Mol. Oncol. 12 (7), 1047–1060. doi:10.1002/1878-0261.12309
Zhang, X., Zou, Q., Rodriguez-Paton, A., and Zeng, X. (2019). Meta-Path Methods for Prioritizing Candidate Disease miRNAs. Ieee/acm Trans. Comput. Biol. Bioinf. 16 (1), 283–291. doi:10.1109/tcbb.2017.2776280
Zhang, Y., Xiang, M., and Yang, B. (2017). Low-rank Preserving Embedding. Pattern Recognition 70, 112–125. doi:10.1016/j.patcog.2017.05.003
Zhang, Z., Ding, J., Xu, J., Tang, J., and Guo, F. (2021). Multi-Scale Time-Series Kernel-Based Learning Method for Brain Disease Diagnosis. IEEE J. Biomed. Health Inform. 25 (1), 209–217. doi:10.1109/jbhi.2020.2983456
Zhao, T., Hu, Y., Peng, J., and Cheng, L. (2020). DeepLGP: a Novel Deep Learning Method for Prioritizing lncRNA Target Genes. Bioinformatics 36 (16), 4466–4472. doi:10.1093/bioinformatics/btaa428
Zhou, X., Li, Z., Xie, H., Feng, T., Lu, Y., Wang, C., et al. (2020). Leukocyte Image Segmentation Based on Adaptive Histogram Thresholding and Contour Detection. Cbio 15 (3), 187–195. doi:10.2174/1574893614666190723115832
Zou, Q., Lin, G., Jiang, X., Liu, X., and Zeng, X. (2020). Sequence Clustering in Bioinformatics: an Empirical Study. Brief. Bioinform. 21 (1), 1–10.
Keywords: sparse representation, gene expression profile, machine learning, low-rank representation, cancer
Citation: Han S, Wang N, Guo Y, Tang F, Xu L, Ju Y and Shi L (2021) Application of Sparse Representation in Bioinformatics. Front. Genet. 12:810875. doi: 10.3389/fgene.2021.810875
Received: 08 November 2021; Accepted: 01 December 2021;
Published: 15 December 2021.
Edited by:
Quan Zou, University of Electronic Science and Technology of China, ChinaReviewed by:
Liang Cheng, Harbin Medical University, ChinaCopyright © 2021 Han, Wang, Guo, Tang, Xu, Ju and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Ju, eWp1QHhtdS5lZHUuY24=; Lei Shi, c2xzcGluZUAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.