 Bert van de Kooij
Bert van de Kooij Haico van Attikum*
Haico van Attikum*- Department of Human Genetics, Leiden University Medical Center, Leiden, Netherlands
Repair of DNA Double-Strand Breaks (DSBs) can be error-free or highly mutagenic, depending on which of multiple mechanistically distinct pathways repairs the break. Hence, DSB-repair pathway choice directly affects genome integrity, and it is therefore of interest to understand the parameters that direct repair towards a specific pathway. This has been intensively studied using genomic reporter constructs, in which repair of a site-specific DSB by the pathway of interest generates a quantifiable phenotype, generally the expression of a fluorescent protein. The current developments in genome editing with targetable nucleases like Cas9 have increased reporter usage and accelerated the generation of novel reporter constructs. Considering these recent advances, this review will discuss and compare the available DSB-repair pathway reporters, provide essential considerations to guide reporter choice, and give an outlook on potential future developments.
Introduction
The integrity of our genome is constantly challenged by DNA damaging lesions that arise during normal cell growth and division, and are caused by exposure to environmental mutagens and irradiation (Hoeijmakers, 2009). A particularly toxic lesion is the DNA Double-Strand Break (DSB), which separates a chromosome into two pieces and can thus cause detrimental karyotypic alterations. Detection of a DSB initiates an elaborate signaling response that halts the cell cycle, re-shapes the chromatin environment and recruits repair factors (Smeenk and van Attikum, 2013; Dantuma and van Attikum, 2016). The subsequent repair is performed by one of multiple repair pathways that are mechanistically distinct and range from error-free to highly mutagenic (Figure 1A; Scully et al., 2019).
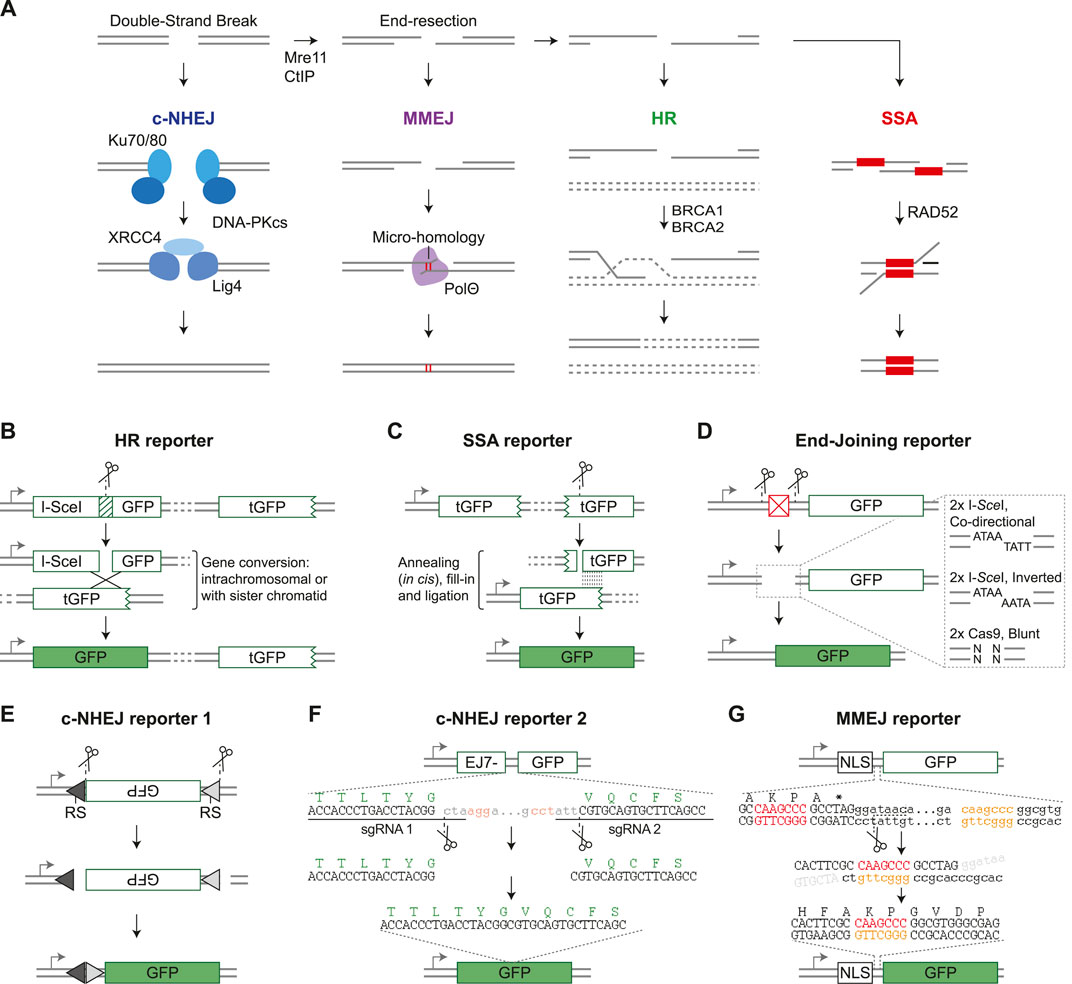
FIGURE 1. Fluorescent DSB-repair reporter systems. (A) Schematic diagram of the four double-strand break repair pathways and their main components. See main text for details. (B) DR-GFP (Pierce et al., 1999) is shown as an example HR reporter. Arrow = promoter, I-SceI GFP is a GFP gene disrupted by insertion of an I-SceI target site, tGFP = truncated GFP. (C) SA-GFP (Stark et al., 2004) is shown as an example SSA reporter. (D) Structure of a generic End-Joining reporter. Expression of GFP is prevented by a sequence element between the gene and the promoter (red box), but can be restored by nuclease-induced excision of this element followed by end-joining mediated repair of the distal DSB ends. The dashed-line box shows a zoom of the DSB ends which can be ligation compatible or non-compatible, depending on the nature of the nuclease and orientation of the target site. (E) Structure of pMX-INV (Bredemeyer et al., 2006), which is a VDJ recombination based c-NHEJ reporter specifically used in B-cells. It is cut by RAG nucleases, RS = Recombination Signal. (F) Structure of EJ7-GFP (Bhargava et al., 2018), which reports on error-free c-NHEJ. GFP amino acid sequence is depicted in green, sgRNA PAM sequences in red. Dots indicate omitted sequence. (G) EJ2-GFP (Bennardo et al., 2008) is shown as an example MMEJ reporter. Designed microhomology sequences are shown in red and orange, dots indicate omitted sequence.
In human cells the majority of DSBs are repaired by classical Non-Homologous End-Joining (c-NHEJ), which requires no or very little (≤4 nucleotides) homology at the DSB-ends to ligate them together (Figure 1A; Pannunzio et al., 2018). Repair by c-NHEJ can be either error-free, or introduce small insertions or deletions (InDels) at the break junction due to DSB end-processing by nucleases and polymerases. Alternatively, DSBs can be repaired by Homologous Recombination (HR), which is initiated by extensive nuclease-mediated resection of the DSB-ends to generate 3’ single strand overhangs (Figure 1A; Jasin and Rothstein, 2013). These overhangs invade homologous double-stranded DNA and prime polymerase-mediated extension. HR can then progress via several sub-pathways, as discussed in detail elsewhere in this special issue (Elbakry and Löbrich, 2021). In the dominant sub-pathway, the extended overhang anneals to the opposite DSB-end, and remaining single-stranded (ss) DNA gaps are closed. The genetically identical sister chromatid is the preferred repair template, and therefore HR is considered a high-fidelity repair pathway. However, other homologous sequences, either on nearby chromosomal DNA or on ectopically provided DNA, can also be used as repair template, even if homology is incomplete. In the latter case, HR can lead to sequence alterations at the repaired locus, which is known as gene conversion.
In addition to HR, DSB end-resection can prime repair by either Single-Strand Annealing (SSA) or by Micro-Homology Mediated End-joining (MMEJ), which is also known as alternative End-Joining or Theta-mediated End-joining (Figure 1A; Bhargava et al., 2016; Sallmyr and Tomkinson, 2018; Schimmel et al., 2019). During repair by both pathways, the opposite DSB-ends are joined by annealing of homologous sequence stretches, followed by nuclease-mediated removal of the non-homologous ssDNA ends. MMEJ requires short regions of microhomology (<20 nucleotides) adjacent to the DSB ends, whereas SSA depends on homologous repeats of at least 50 nucleotides long and can occur even if these are separated by distances up to 28 kilobases (Mendez-Dorantes et al., 2018; Kelso et al., 2019). Both pathways are mutagenic, but particularly SSA can result in large deletions.
Which pathway is employed to repair a given DSB is dependent on many factors including sequence context, chromatin environment, and cell cycle stage (Scully et al., 2019). Engagement of the appropriate repair pathway is essential for efficient genome maintenance, whereas an imbalance in pathway choice can have pathological consequences, including cancer development (Knijnenburg et al., 2018). This knowledge has driven studies on the fundamentals of DSB-repair for decades, but recently this research interest has grown exponentially due to the development of genome editing approaches using Cas9 and Cas9-like nucleases (Komor et al., 2017; Knott and Doudna, 2018). Understanding the determinants that direct repair of a Cas9-induced DSB towards a specific pathway is crucial to predict editing outcome, and to identify methods to control this outcome (Yeh et al., 2019). Notably, advances in genome editing techniques have not only spiked DSB-repair research, but also stimulated the development of methods to study it. This includes genomic DSB-repair reporter constructs, which have been essential tools in DSB-repair research by providing an easy and quantitative read-out for repair pathway activity (Gunn and Stark, 2012). Given these new developments, this review will discuss and compare the traditional and more recently published genomic DSB-repair reporter constructs.
Single Pathway DSB-Repair Reporters
There are numerous published methods that could be considered reporter assays because they quantitatively detect DSB-repair by a given pathway. For simplicity, this review will be limited to describing the genomic reporter constructs that are designed to detect gain or loss of expression of a marker gene, as a result of defined sequence changes associated with DSB-repair by a specific pathway. The marker gene in the reporter could be a drug resistance cassette, and we will mention a few examples of such reporters. However, the main focus of this review will be on reporters that carry a marker gene that encodes a fluorescent protein (Table 1).
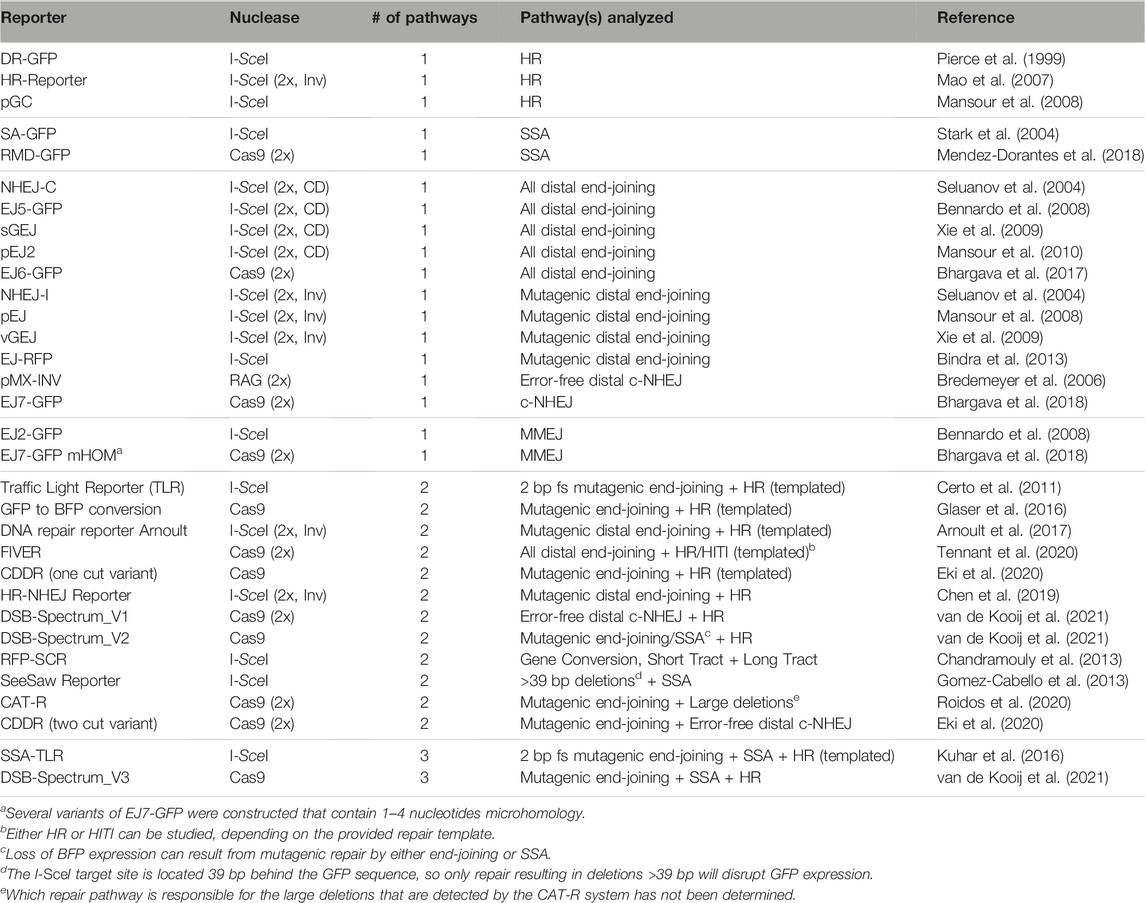
TABLE 1. Overview of fluorescent DSB-repair reporters. HR = Homologous Recombination, SSA = Single-Strand Annealing, c-NHEJ = classical Non-Homologous End-Joining, MMEJ = Microhomology-Mediated End-Joining, fs = frameshift, HITI = Homology Independent Targeted Integration, CD = Co-Directional orientation, Inv = Inverted orientation. “HR (templated)” indicates the requirement of an ectopically delivered repair template to detect repair by HR.
Early reporter systems were designed to study HR and the lay-out was based on the MAT locus of Saccharomyces cerevisiae. This locus is targeted by the HO endonuclease which can result in mating-type switching if the DSB is repaired by HR-mediated gene conversion using one of two homologous HM genes (Haber, 2012). To study this gene conversion process further, HR-reporter constructs were cloned that resembled the MAT locus, but contained marker genes like LacZ rather than the MAT gene (Rudin et al., 1989). This prototypic HR-reporter design was transferred to mammalian cells by the Jasin lab, which modified it to contain the target site for the I-SceI nuclease rather than the HO nuclease, and GFP genes rather than LacZ genes (Rouet et al., 1994; Pierce et al., 1999). The resulting HR-reporter DR-GFP consists of two non-functional GFP gene repeats; the first is disrupted by insertion of an I-SceI target site, while the second lacks a promoter and is C-terminally truncated (Figure 1B; Pierce et al., 1999). Expression of GFP, which can be measured by flow cytometry, is therefore dependent on I-SceI-induced gene conversion between the two repeats, and serves as a quantitative read-out for HR. DR-GFP is currently still widely used and inspired the design of many other fluorescent DSB-repair reporter constructs (Table 1).
Following DR-GFP, Jasin et al. developed SA-GFP, an SSA-reporter that contains a C-terminally truncated GFP gene, and a second N-terminally truncated GFP gene with an I-SceI site (Figure 1C; Stark et al., 2004). There is substantial sequence overlap between the truncated GFP genes, and annealing of these homologous sequences during SSA-repair of the I-SceI induced DSB will generate an intact GFP gene. A limited number of alternative fluorescent HR and SSA reporters has been published (Table 1), which generally follow the same design principles as DR-GFP and SA-GFP, respectively.
In contrast to HR and SSA, there is an abundance of reporters to study repair by end-joining pathways (Table 1). The majority of end-joining reporters are conceptually similar and contain an intact GFP gene that is not expressed due to an upstream inhibitory sequence element, like an out-of-frame start codon or a second gene with a stop codon (Figure 1D). DSBs are generated at two nuclease target sites flanking this inhibitory sequence, and fusion of the distal DSB-ends by end-joining repair will excise the inhibitory element and permit GFP expression. Some end-joining reporters generate ligation-compatible distal DSB-ends (see Figure 1D), and will therefore measure the collective frequency of error-free c-NHEJ, mutagenic c-NHEJ, and MMEJ. Other end-joining reporters are limited to the detection of mutagenic end-joining as their distal DSB-ends have non-compatible overhangs (Figure 1D; Table 1). In both reporter types, however, the contribution of each individual end-joining pathway to DSB-repair cannot be distinguished based on GFP expression, although it can be revealed by sequence analysis of the repair junction (Bennardo et al., 2008).
In contrast to these generic end-joining reporters, the pMX-INV reporter specifically measures c-NHEJ (Bredemeyer et al., 2006). It is based on the process of VDJ recombination that occurs in antigen-receptor genes during lymphocyte maturation (Bassing et al., 2002). The pMX-INV reporter is introduced in mouse progenitor B-cells that express viral Abl kinase and are arrested in G1 by Alb kinase inhibitors. This induces expression of the RAG nucleases that cleave adjacent to recombination signals in the reporter to excise an antisense GFP gene (Figure 1E). Subsequently, the GFP gene is inverted and re-ligated during a VDJ recombination-like reaction, which puts it behind the LTR promoter and results in GFP expression. As VDJ recombination is strictly dependent on c-NHEJ repair of the RAG-induced DSBs (Helmink and Sleckman, 2012), GFP expression from pMX-INV is a direct measure for c-NHEJ activity. More recently the EJ7-GFP reporter was developed, which also specifically quantifies repair by c-NHEJ, but resembles the other end-joining reporters rather than pMX-INV, and, unlike pMX-INV, can be used in any genetically modifiable cell-line. It contains a GFP gene with an intragenic 46 basepair (bp) spacer sequence that can be removed by targeting Cas9 precisely to the edges, and which results in an intact GFP sequence if the distal ends are joined by error-free c-NHEJ (Figure 1F; Bhargava et al., 2018).
Finally, the Stark lab published two reporters that were designed to monitor MMEJ. The EJ2-GFP reporter contains two regions with 8 nucleotides microhomology flanking an I-SceI site. Repair of the I-SceI-induced DSB by MMEJ will remove a stop codon and put GFP in frame with an upstream NLS-tag sequence (Figure 1G; Bennardo et al., 2008). Notably, sequence analysis of repair junctions from GFP positive cells revealed that 10% of the repair products contained a deletion without microhomology flanking the break, indicating that microhomology-independent DSB-repair of EJ2-GFP can also result in GFP expression. Furthermore, several EJ7-GFP variants were generated containing microhomology (Bhargava et al., 2018). One variant, with 4 nucleotides microhomology located inward from the DSB-edge, is a bona fide MMEJ reporter, as GFP expression was strongly dependent on the end-resection factor CtIP, and the MMEJ-factor PolΘ (Bhargava et al., 2018).
Multi-Pathway DSB-Repair Reporters
HR, SSA and end-joining are connected in a DSB-repair signaling network, such that loss of a pathway is compensated for by enhanced engagement of one or more of the remaining pathways (Scully et al., 2019). To study these inter-pathway dynamics, reporter systems have been developed that can quantify repair by more than one pathway (Table 1). Many multi-pathway reporters combine elements from published single-pathway reporter systems, and contain two or more genes encoding different fluorescent proteins to distinguish between the repair pathways. Rather than providing a detailed explanation of each individual multi-pathway reporter, we will discuss a few to highlight the major design concepts, and refer the reader to the references listed in Table 1 for details.
The majority of multi-pathway reporters were developed to simultaneously monitor end-joining and HR (Table 1). This was often achieved by combining a genomic end-joining reporter with an ectopic repair template. For example, the Traffic Light Reporter (TLR) contains a GFP gene disrupted by an internal I-SceI site, followed by an mCherry gene with a reading frame shifted 2 bp compared to that of GFP (Certo et al., 2011). Formation of any 2 bp frameshift-causing mutation at the I-SceI target site, predominantly generated by mutagenic c-NHEJ, will result in mCherry expression. HR can be analyzed by GFP expression, which is caused by gene conversion between the I-SceI site-containing GFP gene and a truncated GFP gene present on an ectopically provided repair template.
In several other multi-pathway reporters gene conversion causes a color-switch, i.e., a change from expression of one fluorescent protein to another. A particularly practical and widely used HR-dependent color switch is GFP to BFP conversion, or the other way around (Glaser et al., 2016). The BFP and GFP genes are completely homologous with exception of two amino acids that determine their fluorescent properties, and can therefore function as reciprocal repair templates without the need for additional homologous sequences. BFP to GFP conversion is also used as read-out for HR in the DSB-Spectrum reporters (van de Kooij et al., 2021). Conveniently, no ectopic repair template is required in these multi-pathway reporter systems because the truncated GFP repair template is located on the same construct, downstream of the BFP gene that is targeted by Cas9.
Several dual pathway reporters monitor sub-pathways rather than any of the four major DSB-repair pathways. A recent example of such a reporter is the CDDR (two cut variant), which contains a functional mCherry gene inserted into a split, and thus non-functional, GFP gene (Eki et al., 2020). Cas9 is targeted to sites flanking the mCherry gene such that distal error-free c-NHEJ reconstitutes the GFP gene, and deletes the mCherry gene. This can be distinguished from mutagenic end-joining, which results in mCherry loss without GFP expression.
Finally, whereas there are many dual-pathway reporters, only two systems can monitor three pathways simultaneously. SSA-TLR is a variant of the TLR that is flanked by truncated iRFP genes, which can be joined by SSA to form an intact gene (Kuhar et al., 2016). This system thus reports on 2 bp frameshift inducing c-NHEJ, HR with an ectopic repair template, and SSA. The more recently developed DSB-Spectrum_V3 consists of an intact BFP gene that is targeted by Cas9, and can be disrupted by end-joining mediated mutagenesis. Loss of BFP expression is thus a measure for mutagenic end-joining in general, but primarily for mutagenic c-NHEJ, as indicated by sequence analysis (van de Kooij et al., 2021). Furthermore, the reporter contains a truncated GFP gene that can be used to convert BFP to GFP as a measurement for HR (van de Kooij et al., 2021). These homologous elements can also anneal during DSB-repair by SSA, resulting in the removal of a functional mCherry gene that separates the two. As such, a single reporter can be used to simultaneously measure three DSB-repair pathways.
Considerations for Using DSB-Repair Reporters
There is substantial variation within the reporter repertoire, and one reporter might be more suited to specific research needs than the other. A first consideration is whether to use single-pathway or multi-pathway reporter systems. An advantage of single-pathway systems is their simplicity and, at least for many of them, their extensive validation by widespread use for many years. However, with similar research efforts, multi-pathway reporters generate a more comprehensive view of DSB-repair pathway activity. When studying DBS-repair factors, for instance, such reporters can immediately reveal whether a factor functions in one or multiple pathways, the latter of which can also indicate at which node in the DSB-repair network it acts.
A second consideration is to use either reporters that require ectopic HR repair templates, or repeat-containing reporters that carry the template embedded within the construct. Glaser et al. used a single-stranded oligo as template in their GFP to BFP conversion reporter (Glaser et al., 2016), which has been demonstrated to mediate gene conversion by a mechanism that diverges from canonical HR (Bothmer et al., 2017; Richardson et al., 2018). The other templated reporters use double-stranded repair templates, which might be copied in an HR-dependent process. However, unlike the sister chromatid, these ectopic repair templates are present in high copy number, exist throughout the cell cycle, and lack proximity to the broken chromosome. In repeat-containing reporters, the template is on the sister chromatid in S/G2. However, it is also present in G1 and could theoretically be used as a donor during intrachromosomal gene conversion, in contrast to HR at endogenous loci. Although these non-physiological HR-events cannot be completely excluded, early studies on DR-GFP-like reporters indicated them to be rare (Johnson and Jasin, 2000). Moreover, more recently it was shown that all detected gene conversion events with a novel repeat-containing reporter occurred in S-phase (Chen et al., 2019). Therefore, repeat-containing HR-reporters more accurately reflect HR-repair at endogenous genomic loci and may be preferred when studying fundamentals of DSB-repair, whereas templated reporters can be useful when studying genome editing approaches.
A third consideration is that some reporters, in particular end-joining reporters, might not be very specific but actually measure the collective frequency of repair by multiple pathways, as explained above. More specific detection of repair by a single pathway is in most cases preferable. However, it should be taken into account that too high specificity can come at the cost of low frequency. This is, for example, the case in the RFP-SCR reporter, which can distinguish between gene conversion sub-pathways. The frequency of cells undergoing long-tract gene conversion is consistently less than 0.1%, requiring analysis of large cell populations for reliable quantification (Chandramouly et al., 2013).
Fourth, reporters are designed to detect DSB-repair either by loss or by induction of marker gene expression. The latter confers specificity because the expression is strictly dependent on a defined change in the reporter sequence (Figure 1). This could, however, result in an underestimation of pathway usage because not all repair by the assayed pathway necessarily generates that defined sequence change. For example, mCherry expression in the TLR is dependent on generation of 2 bp frameshifts, and it therefore measures only a fraction of mutagenic c-NHEJ (Certo et al., 2011; Kuhar et al., 2016). Loss of marker gene expression, on the other hand, is theoretically less specific because it could be caused by other mutagenic repair pathways than the one measured. Nevertheless, these reporters are more inclusive because they lack the requirement for a unique mutagenic event, and their specificity can easily be validated by sequencing and genetic interrogation.
Finally, even though reporter constructs have proven extremely useful they do have limitations. First, the DSB is generated in a specific sequence context, which can impact repair pathway employment (Shen et al., 2018; Allen et al., 2019). Second, the DSB in reporter constructs is generated by efficient nucleases that keep cutting as long as they are expressed and the target site is intact. As a consequence, the phenotype analyzed might be the result of re-iterative rounds of error-free repair followed by one mutagenic repair event. This might result in an overestimation of the frequency of mutagenic repair. Moreover, the persistence of a constantly regenerated DSB might in specific cases affect pathway choice (Bennardo et al., 2009). Third, the nucleases will most likely cut both sister chromatids, which disables HR and makes the cell more reliant on alternative pathways like SSA or MMEJ. This should be taken into account when using, for example, SSA-reporters. It is, however, not an issue when using HR reporters, because the provided template lacks the nuclease target site. Altogether, these limitations should be considered for correct interpretation of reporter assays. However, they do not prevent the generation of insightful data, as proven by a large body of published DSB-repair literature in which results obtained with reporter assays were validated using orthogonal techniques.
Future Developments of DSB-Repair Reporters
The advent of targetable nucleases like Cas9 has spurred the construction of novel reporter systems (Table 1). It has enhanced the flexibility in reporter design because it negates the requirement for a specific I-SceI nuclease target sequence. Moreover, it has also expanded the nuclease repertoire with a variety of blunt-cutting enzymes, the staggered cutting Cas12a/Cpf1, and Cas9 nickase variants that can be targeted to either strand (Komor et al., 2017). Therefore, different DSB-ends can be generated, which has been shown to affect pathway choice (Bothmer et al., 2017; Schimmel et al., 2017). Given these enhanced design possibilities, a four-pathway reporter system measuring the frequency of all major DSB-repair pathways is within reach.
In addition to the development of new reporters, genome editing tools will facilitate targeted integration of reporter constructs. This will allow for comparison of repair pathway usage between genomic loci, for example, in hetero- and euchromatic regions, which is an active area of investigation (Caron et al., 2021; Schep et al., 2021). Interestingly, Cas9-based tools have been developed to modify the chromatin at target loci (Goell and Hilton, 2021). These could be used in combination with reporters to study the effect of specific chromatin marks on DSB-repair pathway choice. Importantly, ongoing research efforts are aimed at developing methods to rapidly activate and de-activate Cas9 (Liu et al., 2020; Marino et al., 2020), so that complete temporal control over Cas9 activity will be possible in the near future, thus eliminating the current problem of re-iterative cutting of reporters. Finally, reporters are ideal tools for pooled high-throughput screening, because repair phenotypes can easily be selected by FACS. Reporter screens have been done using siRNAs, but these screens have been hampered by the strong tendency of siRNAs to silence Rad51 expression as an off-target effect (Adamson et al., 2012; Howard et al., 2015). CRISPR-based screens are generally less affected by off-target editing, and the first insightful CRISPRi reporter screens have already been published (Richardson et al., 2018; Wienert et al., 2020).
In conclusion, DSB-repair reporters have evolved from designated constructs to study HR, to complex multicolor tools that can measure repair by two or three pathways in one assay. This evolution is expected to continue, driven by Cas9-technology, ensuring that reporters will remain an essential element in the DSB-repair toolkit, as they have been for multiple decades.
Author Contributions
Both authors have made an equal contribution to this work and approved it for publication.
Funding
Research in the HvA lab is supported by the European Research Council National (ERC-CoG-617485) and the NWO (VI.C.182.052). BvdK is a recipient of a Dutch Cancer Society fellowship (BUIT 2015-7546), and supported by funding from the Leiden University Medical Center under the regulation for MSCA-IF Seal of Excellence awardees.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Prof. Dr. M. B. Yaffe (MIT, Cambridge, MA, United States), Dr. J. Schimmel (LUMC, Leiden, Netherlands) and Dr. M. Rother-Krawitowska (LUMC, Leiden, Netherlands) for critical reading of the manuscript. We apologize to all researchers who generated reporter constructs that we inadvertently missed, or omitted due to space limitations.
References
Adamson, B., Smogorzewska, A., Sigoillot, F. D., King, R. W., and Elledge, S. J. (2012). A Genome-wide Homologous Recombination Screen Identifies the RNA-Binding Protein RBMX as a Component of the DNA-Damage Response. Nat. Cell Biol. 14, 318–328. doi:10.1038/ncb2426
Allen, F., Crepaldi, L., Alsinet, C., Strong, A. J., Kleshchevnikov, V., De Angeli, P., et al. (2019). Predicting the Mutations Generated by Repair of Cas9-Induced Double-Strand Breaks. Nat. Biotechnol. 37, 64–72. doi:10.1038/nbt.4317
Arnoult, N., Correia, A., Ma, J., Merlo, A., Garcia-Gomez, S., Maric, M., et al. (2017). Regulation of DNA Repair Pathway Choice in S and G2 Phases by the NHEJ Inhibitor CYREN. Nature. 549, 548–552. doi:10.1038/nature24023
Bassing, C. H., Swat, W., and Alt, F. W. (2002). The Mechanism and Regulation of Chromosomal V(D)J Recombination. Cell 109, S45–S55. doi:10.1016/S0092-8674(02)00675-X
Bennardo, N., Cheng, A., Huang, N., and Stark, J. M. (2008). Alternative-NHEJ Is a Mechanistically Distinct Pathway of Mammalian Chromosome Break Repair. Plos Genet. 4, e1000110. doi:10.1371/journal.pgen.1000110
Bennardo, N., Gunn, A., Cheng, A., Hasty, P., and Stark, J. M. (2009). Limiting the Persistence of a Chromosome Break Diminishes its Mutagenic Potential. Plos Genet. 5, e1000683. doi:10.1371/journal.pgen.1000683
Bhargava, R., Onyango, D. O., and Stark, J. M. (2016). Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 32, 566–575. doi:10.1016/j.tig.2016.06.007
Bhargava, R., Carson, C. R., Lee, G., and Stark, J. M. (2017). Contribution of Canonical Nonhomologous End Joining to Chromosomal Rearrangements is Enhanced by ATM Kinase Deficiency. Proc. Natl. Acad. Sci. U.S.A. 114, 728–733. doi:10.1073/pnas.1612204114
Bhargava, R., Sandhu, M., Muk, S., Lee, G., Vaidehi, N., and Stark, J. M. (2018). C-NHEJ without Indels Is Robust and Requires Synergistic Function of Distinct XLF Domains. Nat. Commun. 9, 2484. doi:10.1038/s41467-018-04867-5
Bindra, R. S., Goglia, A. G., Jasin, M., and Powell, S. N. (2013). Development of an Assay to Measure Mutagenic Non-Homologous End-Joining Repair Activity in Mammalian Cells. Nucleic. Acids. Res. 41, e115. doi:10.1093/nar/gkt255
Bothmer, A., Phadke, T., Barrera, L. A., Margulies, C. M., Lee, C. S., Buquicchio, F., et al. (2017). Characterization of the Interplay between DNA Repair and CRISPR/Cas9-induced DNA Lesions at an Endogenous Locus. Nat. Commun. 8, 13905. doi:10.1038/ncomms13905
Bredemeyer, A. L., Sharma, G. G., Huang, C.-Y., Helmink, B. A., Walker, L. M., Khor, K. C., et al. (2006). ATM Stabilizes DNA Double-Strand-Break Complexes during V(D)J Recombination. Nature 442, 466–470. doi:10.1038/nature04866
Caron, P., Pobega, E., and Polo, S. E. (2021). DNA Double-Strand Break Repair: All Roads Lead to HeterochROMAtin Marks. Front. Genet. 12, 1589. doi:10.3389/fgene.2021.730696
Certo, M. T., Ryu, B. Y., Annis, J. E., Garibov, M., Jarjour, J., Rawlings, D. J., et al. (2011). Tracking Genome Engineering Outcome at Individual DNA Breakpoints. Nat. Methods 8, 671–676. doi:10.1038/nmeth.1648
Chandramouly, G., Kwok, A., Huang, B., Willis, N. A., Xie, A., and Scully, R. (2013). BRCA1 and CtIP Suppress Long-Tract Gene Conversion between Sister Chromatids. Nat. Commun. 4, 2404. doi:10.1038/ncomms3404
Chen, Y., Zhang, H., Xu, Z., Tang, H., Geng, A., Cai, B., et al. (2019). A PARP1-BRG1-SIRT1 axis Promotes HR Repair by Reducing Nucleosome Density at DNA Damage Sites. Nucleic Acids Res. 47, 8563–8580. doi:10.1093/nar/gkz592
Dantuma, N. P., and Attikum, H. (2016). Spatiotemporal Regulation of Posttranslational Modifications in the DNA Damage Response. EMBO J. 35, 6–23. doi:10.15252/embj.201592595
Eki, R., She, J., Parlak, M., Benamar, M., Du, K.-P., Kumar, P., et al. (2020). A Robust CRISPR-Cas9-Based Fluorescent Reporter Assay for the Detection and Quantification of DNA Double-Strand Break Repair. Nucleic Acids Res. 48, E126. doi:10.1093/nar/gkaa897
Elbakry, A., and Löbrich, M. (2021). Homologous Recombination Subpathways: A Tangle to Resolve. Front. Genet. 12, 1402. doi:10.3389/fgene.2021.723847
Glaser, A., McColl, B., and Vadolas, J. (2016). GFP to BFP Conversion: A Versatile Assay for the Quantification of CRISPR/Cas9-mediated Genome Editing. Mol. Ther. - Nucleic Acids 5, e334. doi:10.1038/mtna.2016.48
Goell, J. H., and Hilton, I. B. (2021). CRISPR/Cas-Based Epigenome Editing: Advances, Applications, and Clinical Utility. Trends Biotechnol. 39, 678–691. doi:10.1016/j.tibtech.2020.10.012
Gomez-Cabello, D., Jimeno, S., Fernández-Ávila, M. J., and Huertas, P. (2013). New Tools to Study DNA Double-Strand Break Repair Pathway Choice. PLoS One. 8, e77206. doi:10.1371/journal.pone.0077206
Gunn, A., and Stark, J. M. (2012). “I-SceI-based Assays to Examine Distinct Repair Outcomes of Mammalian Chromosomal Double Strand Breaks,” in Methods in Molecular Biology. Editor L. Bjergbæk (Totowa, NJ: Humana Press), 379–391. doi:10.1007/978-1-61779-998-3_27
Haber, J. E. (2012). Mating-type Genes and MAT Switching in Saccharomyces cerevisiae. Genetics 191, 33–64. doi:10.1534/genetics.111.134577
Helmink, B. A., and Sleckman, B. P. (2012). The Response to and Repair of RAG-Mediated DNA Double-Strand Breaks. Annu. Rev. Immunol. 30, 175–202. doi:10.1146/annurev-immunol-030409-101320
Hoeijmakers, J. H. J. (2009). DNA Damage, Aging, and Cancer. N. Engl. J. Med. 361, 1475–1485. doi:10.1056/nejmra0804615
Howard, S. M., Yanez, D. A., and Stark, J. M. (2015). DNA Damage Response Factors from Diverse Pathways, Including DNA Crosslink Repair, Mediate Alternative End Joining. Plos Genet. 11, e1004943. doi:10.1371/journal.pgen.1004943
Jasin, M., and Rothstein, R. (2013). Repair of Strand Breaks by Homologous Recombination. Cold Spring Harbor Perspect. Biol. 5, a012740. doi:10.1101/cshperspect.a012740
Johnson, R. D., and Jasin, M. (2000). Sister Chromatid Gene Conversion Is a Prominent Double-Strand Break Repair Pathway in Mammalian Cells. EMBO J. 19, 3398–3407. doi:10.1093/emboj/19.13.3398
Kelso, A. A., Lopezcolorado, F. W., Bhargava, R., and Stark, J. M. (2019). Distinct Roles of RAD52 and POLQ in Chromosomal Break Repair and Replication Stress Response. Plos Genet. 15, e1008319. doi:10.1371/journal.pgen.1008319
Knijnenburg, T. A., Wang, L., Zimmermann, M. T., Chambwe, N., Gao, G. F., Cherniack, A. D., et al. (2018). Genomic and Molecular Landscape of DNA Damage Repair Deficiency across the Cancer Genome Atlas. Cell Rep 23, 239–e6. e6. doi:10.1016/j.celrep.2018.03.076
Knott, G. J., and Doudna, J. A. (2018). CRISPR-cas Guides the Future of Genetic Engineering. Science 361, 866–869. doi:10.1126/science.aat5011
Komor, A. C., Badran, A. H., and Liu, D. R. (2017). CRISPR-based Technologies for the Manipulation of Eukaryotic Genomes. Cell 168, 20–36. doi:10.1016/j.cell.2016.10.044
Kuhar, R., Gwiazda, K. S., Humbert, O., Mandt, T., Pangallo, J., Brault, M., et al. (2016). Novel Fluorescent Genome Editing Reporters for Monitoring DNA Repair Pathway Utilization at Endonuclease-Induced Breaks. Nucleic Acids Res. 42, e4. doi:10.1093/nar/gkt872
Liu, Y., Zou, R. S., He, S., Nihongaki, Y., Li, X., Razavi, S., et al. (2020). Very Fast CRISPR on Demand. Science 368, 1265–1269. doi:10.1126/science.aay8204
Mansour, W. Y., Rhein, T., and Dahm-Daphi, J. (2010). The Alternative End-Joining Pathway for Repair of DNA Double-Strand Breaks Requires PARP1 But is Not Dependent Upon Microhomologies. Nucleic. Acids Res. 38, 6065–6077. doi:10.1093/nar/gkq387
Mansour, W. Y., Schumacher, S., Rosskopf, R., Rhein, T., Rhein, F., Gatzemeier, F., et al. (2008). Hierarchy of Nonhomologous End-Joining, Single-Strand Annealing and Gene Conversion at Site-Directed DNA Double-Strand Breaks. Nucleic. Acids Res. 36, 4088–4098. doi:10.1093/nar/gkn347
Marino, N. D., Pinilla-Redondo, R., Csörgő, B., and Bondy-Denomy, J. (2020). Anti-CRISPR Protein Applications: Natural Brakes for CRISPR-Cas Technologies. Nat. Methods 17, 471–479. doi:10.1038/s41592-020-0771-6
Mao, Z., Seluanov, A., Jiang, Y., and Gorbunova, V. (2007). TRF2 is Required for Repair of Nontelomeric DNA Double-Strand Breaks by Homologous Recombination. Proc. Natl. Acad. Sci. U.S.A. 104, 13068–13073. doi:10.1073/pnas.0702410104
Mendez-Dorantes, C., Bhargava, R., and Stark, J. M. (2018). Repeat-mediated Deletions Can Be Induced by a Chromosomal Break Far from a Repeat, but Multiple Pathways Suppress Such Rearrangements. Genes Dev. 32, 524–536. doi:10.1101/gad.311084.117
Pannunzio, N. R., Watanabe, G., and Lieber, M. R. (2018). Nonhomologous DNA End-Joining for Repair of DNA Double-Strand Breaks. J. Biol. Chem. 293, 10512–10523. doi:10.1074/jbc.TM117.000374
Pierce, A. J., Johnson, R. D., Thompson, L. H., and Jasin, M. (1999). XRCC3 Promotes Homology-Directed Repair of DNA Damage in Mammalian Cells. Genes Dev. 13, 2633–2638. doi:10.1101/gad.13.20.2633
Richardson, C. D., Kazane, K. R., Feng, S. J., Zelin, E., Bray, N. L., Schäfer, A. J., et al. (2018). CRISPR-Cas9 Genome Editing in Human Cells Occurs via the Fanconi Anemia Pathway. Nat. Genet. 50, 1132–1139. doi:10.1038/s41588-018-0174-0
Roidos, P., Sungalee, S., Benfatto, S., Serçin, Ö., Stütz, A. M., Abdollahi, A., et al. (2020). A Scalable CRISPR/Cas9-Based Fluorescent Reporter Assay to Study DNA Double-Strand Break Repair Choice. Nat. Commun. 11, 4077. doi:10.1038/s41467-020-17962-3
Rouet, P., Smih, F., and Jasin, M. (1994). Expression of a Site-specific Endonuclease Stimulates Homologous Recombination in Mammalian Cells. Proc. Natl. Acad. Sci. 91, 6064–6068. doi:10.1073/pnas.91.13.6064
Rudin, N., Sugarman, E., and Haber, J. E. (1989). Genetic and Physical Analysis of Double-Strand Break Repair and Recombination in Saccharomyces cerevisiae. Genetics 122, 519–534. doi:10.1093/genetics/122.3.519
Sallmyr, A., and Tomkinson, A. E. (2018). Repair of DNA Double-Strand Breaks by Mammalian Alternative End-Joining Pathways. J. Biol. Chem. 293, 10536–10546. doi:10.1074/jbc.TM117.000375
Schep, R., Brinkman, E. K., Leemans, C., Vergara, X., van der Weide, R. H., Morris, B., et al. (2021). Impact of Chromatin Context on Cas9-Induced DNA Double-Strand Break Repair Pathway Balance. Mol. Cell 81, 2216–2230. e10. doi:10.1016/j.molcel.2021.03.032
Schimmel, J., Kool, H., Schendel, R., and Tijsterman, M. (2017). Mutational Signatures of Non‐homologous and Polymerase Theta‐mediated End‐joining in Embryonic Stem Cells. EMBO J. 36, 3634–3649. doi:10.15252/embj.201796948
Schimmel, J., van Schendel, R., den Dunnen, J. T., and Tijsterman, M. (2019). Templated Insertions: A Smoking Gun for Polymerase Theta-Mediated End Joining. Trends Genet. 35, 632–644. doi:10.1016/j.tig.2019.06.001
Scully, R., Panday, A., Elango, R., and Willis, N. A. (2019). DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 20, 698–714. doi:10.1038/s41580-019-0152-0
Seluanov, A., Mittelman, D., Pereira-Smith, O. M., Wilson, J. H., and Gorbunova, V. (2004). DNA End Joining be Comes Less Efficient and More Error-Prone During Cellular Senescence. Proc. Natl. Acad. Sci. U.S.A. 101, 7624–7629.
Shen, M. W., Arbab, M., Hsu, J. Y., Worstell, D., Culbertson, S. J., Krabbe, O., et al. (2018). Predictable and Precise Template-free CRISPR Editing of Pathogenic Variants. Nature 563, 646–651. doi:10.1038/s41586-018-0686-x
Smeenk, G., and van Attikum, H. (2013). The Chromatin Response to DNA Breaks: Leaving a Mark on Genome Integrity. Annu. Rev. Biochem. 82, 55–80. doi:10.1146/annurev-biochem-061809-174504
Stark, J. M., Pierce, A. J., Oh, J., Pastink, A., and Jasin, M. (2004). Genetic Steps of Mammalian Homologous Repair with Distinct Mutagenic Consequences. Mol. Cell. Biol. 24, 9305–9316. doi:10.1128/mcb.24.21.9305-9316.2004
Tennant, P. A., Foster, R. G., Dodd, D. O., Sou, I. F., McPhie, F., Younger, N., et al. (2020). Fluorescent in vivo Editing Reporter (FIVER): A Novel Multispectral Reporter of in vivo Genome Editing. bioRxiv. 2020.07.14.200170. doi:10.1101/2020.07.14.200170
van de Kooij, B., Kruswick, A., van Attikum, H., and Yaffe, M. B. (2021). Multi-Pathway DNA Double-Strand Break Repair Reporters Reveal Extensive Cross-Talk between End-Joining, Single Strand Annealing, and Homologous Recombination. bioRxiv 2021, 473809. doi:10.1101/2021.12.22.473809
Wienert, B., Nguyen, D. N., Guenther, A., Feng, S. J., Locke, M. N., Wyman, S. K., et al. (2020). Timed Inhibition of CDC7 Increases CRISPR-Cas9 Mediated Templated Repair. Nat. Commun. 11, 2109. doi:10.1038/s41467-020-15845-1
Xie, A., Kwok, A., and Scully, R. (2009). Role of Mammalian Mre11 in Classical and Alternative Nonhomologous End Joining. Nat. Struct. Mol. Biol. 16, 814–818. doi:10.1038/nsmb.1640
Keywords: genomic reporter constructs, double-strand break repair pathway choice, homologous recombination, end-joining, single-strand annealing
Citation: van de Kooij B and van Attikum H (2022) Genomic Reporter Constructs to Monitor Pathway-Specific Repair of DNA Double-Strand Breaks. Front. Genet. 12:809832. doi: 10.3389/fgene.2021.809832
Received: 05 November 2021; Accepted: 23 December 2021;
Published: 14 February 2022.
Edited by:
Judit Jimenez Sainz, Yale University, United StatesReviewed by:
Arvind Panday, Harvard Medical School, United StatesRagini Bhargava, University of Pittsburgh, United States
Copyright © 2022 van de Kooij and van Attikum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haico van Attikum, SC52YW4uQXR0aWt1bUBsdW1jLm5s