 Wei Wu1
Wei Wu1 Wai Kit Chu
Wai Kit Chu- 1Zhejiang Provincial Key Laboratory of Pancreatic Diseases, The First Affiliated Hospital of Zhejiang University, Hangzhou, China
- 2Department of Ophthalmology and Visual Sciences, The Chinese University of Hong Kong, Hong Kong, Hong Kong, China
Accurate replication of the entire genome is critical for cell division and propagation. Certain regions in the genome, such as fragile sites (common fragile sites, rare fragile sites, early replicating fragile sites), rDNA and telomeres, are intrinsically difficult to replicate, especially in the presence of replication stress caused by, for example, oncogene activation during tumor development. Therefore, these regions are particularly prone to deletions and chromosome rearrangements during tumorigenesis, rendering chromosome fragility. Although, the mechanism underlying their “difficult-to-replicate” nature and genomic instability is still not fully understood, accumulating evidence suggests transcription might be a major source of endogenous replication stress (RS) leading to chromosome fragility. Here, we provide an updated overview of how transcription affects chromosome fragility. Furthermore, we will use the well characterized common fragile sites (CFSs) as a model to discuss pathways involved in offsetting transcription-induced RS at these loci with a focus on the recently discovered atypical DNA synthesis repair pathway Mitotic DNA Synthesis (MiDAS).
Introduction
To proliferate, a cell needs to go through a division cycle, where it duplicates its chromosomes in S phase. The replicated chromosomes are then separated and segregated into daughter cells during mitosis. Due to the large size of human genome, cells start DNA replication from multiple origins and up to thousands of replication forks are established and coordinated to replicate the genome in a very short time period (S phase). The untimely duplication of the whole genome in S phase can lead to cells entering mitosis with under replicated DNA (URD). URD can affect sister chromatids separation and genome stability, which is a hallmark of cancer (Hanahan and Weinberg, 2011).
There are certain regions in the human genome that are inherently hard-to-replicate, posing a great challenge for the timely duplication of the entire genome. Such regions including fragile sites, rDNA and telomeres have been well documented and are the major cause of chromosome fragility (Özer and Hickson, 2018). Intriguingly, many of these regions are either characterized as containing very large genes or harboring highly transcribed genes, indicating transcription might play an important role in determining their fragility. Indeed, for very long genes that take more than one cell cycle for them to be transcribed, collisions of transcription and replication machineries are inevitable (Helmrich et al., 2011). In this article, we will first review the transcription-replication conflicts and the associated DNA:RNA hybrid called R-loop. R-loop can trigger RS leading to chromosome fragility. We will then discuss the strategies employed by cells to counteract this transcription associated RS to maintain chromosome stability.
Transcription-Mediated Replication Obstacles
During transcription, RNA polymerase (RNAP) together with multiple transcription elongation and RNA processing factors form a large complex, which is tightly bound to DNA. In addition, eukaryotic transcription needs to be coupled with other downstream events, like RNA splicing to generate mature mRNA. Thus, transcription machineries together with RNA processing factors could be obstacles for an advancing replication fork. Since DNA replication and transcription compete for the same DNA template, it is inevitable that on some occasions there will be a collision between the two machineries. Indeed, it has long been known that transcription can trigger replication-dependent genome instability that would lead to recombination and mutations (Keil and Roeder, 1984; Voelkel-Meiman et al., 1987; Chávez and Aguilera, 1997; Prado and Aguilera, 2005; Kim and Jinks-Robertson, 2012).
Depending on how transcription and replication machineries approach each other, they can either collide co-directionally or in a head-on manner. When transcription uses the leading stand of DNA replication as a template, co-directional collisions might happen; when transcription uses the lagging strand as the substrate, head-on collisions might take place. In general, it is thought that a co-directional encounter is less toxic to cells than the head-on collision. Consistent with this notion, an in vitro study suggested that a reconstituted Escherichia coli replisome can remove or bypass a co-directional RNAP and use the newly synthesized mRNA as a primer to carry on DNA synthesis (Liu et al., 1993; Pomerantz and O’Donnell, 2008). Another fact is that highly transcribed regions of rRNA, tRNA and some other essential genes are almost exclusively co-directional to fork progression in most of the studied bacteria (Rocha and Danchin, 2003; Guy and Roten, 2004). This is probably because of the evolutionary pressure for the proper replication fork progression at these regions, which is critical for genome stability and cell survival. Indeed, inverting the highly transcribed ribosomal RNA (rRNA) operons can severely affect replication fork progression at these regions and induce DNA damage responses and cell death (Srivatsan et al., 2010). Intriguingly, there is also a bias towards co-directional collision between transcription and replication in the human genome (Petryk et al., 2016). On the other hand, co-directional encounters of transcription and replication machineries can also cause DNA damages. For example, a co-directional encounter can induce replication restart with the help from helicases and replication restart proteins in Bacillus subtilis (Merrikh et al., 2011). In budding yeast, DNA polymerases tend to accumulate at highly transcribed genes in an orientation independent manner (Azvolinsky et al., 2009). Furthermore, by generating an episomal system in human cells, Hamperl et al. discovered that co-directional collisions can provoke ATM-dependent DNA damage responses (Hamperl et al., 2017).
Multiple experimental data has indicated that head-on collisions are more deleterious to fork progression than co-directional collisions and are commonly associated with DNA damage formation (Deshpande and Newlon, 1996; Prado and Aguilera, 2005; Merrikh et al., 2012). First, the tightly bound large RNAP complex can be a physical barrier that is difficult for the replisome to bypass. Second, when transcription and replication machineries move towards each other, the accumulated positive supercoiled DNA structures would slow down the progression of replication fork (Bermejo et al., 2012). Consistent with this, torsional stress reliving factors: DNA topoisomerases I and II, travel with replication forks and are required for the avoidance of transcription and replication conflicts (Tuduri et al., 2009; Bermejo et al., 2012). Lastly, head-on collisions are always associated with the formation of stable pathological nucleic acid structure: R-loop. R-loop is a three-stranded nucleic acid structure containing one DNA-RNA hybrid and one single strand of DNA. It is much more stable than double stranded DNA, and therefore can directly interfere with DNA replication leading to fork stalling or collapse (Aguilera and García-Muse, 2012; Santos-Pereira and Aguilera, 2015). Consistent with this idea, R-loops are significantly enriched at head-on regions in human cells, and introducing a head-on collision on a plasmid causes plasmid loss in a R-loop dependent manner (Hamperl et al., 2017).
Transcription at Chromosomal Fragile Loci
As mentioned above, certain regions in the human genome are intrinsically difficult to replicate and are prone to recombination and viral integration (Thorland et al., 2003; Gao et al., 2017). These regions are called chromosomal fragile loci. Accumulating evidence suggests that transcription and replication conflict is an important factor contributing to chromosomal fragility. Some of these fragile regions are discussed below.
Fragile Sites
Fragile sites are specific regions that tend to form gaps or breaks on metaphase chromosomes in the presence of RS (Sutherland, 1979). Fragile sites could potentially comprise genome stability and are highly associated with the development of various diseases, including cancer and neurological diseases. Currently, there are three known types of fragile sites:
Rare fragile sites (RFSs) are only found in less than 5% individuals in the general population, and their fragility is associated with the expansion of dinucleotide or trinucleotide repeats, which can form secondary DNA structures, affecting DNA replication and transcription (Magenis et al., 1970). Furthermore, R-loop has been found to be at RFS:FRAXA, and is associated with its silencing (Kumari et al., 2015; Bhowmick et al., 2016).
Common fragile sites (CFSs) are present in all individuals. Some of the characteristics of CFSs might explain their fragility. First, CFSs are late replicating, which might leave insufficient time for cells to clear DNA replication obstacles or to repair damaged DNA following replication perturbations (Le Beau, 1998; Hellman et al., 2000). Second, CFSs are short of replication origins (Letessier et al., 2011; Sugimoto et al., 2018; Pruitt et al., 2017). Thus, to completely duplicate CFSs, two replication forks need to travel a long distance to converge, which is challenging to replication forks, especially when cells experience RS. Recent evidence suggests CFSs fragility is tightly associated with transcription. Indeed, CFSs harboring actively transcribed long genes tend to be hotspots for copy number variants (CNVs) (Wilson et al., 2015). Interestingly, more than 80% of human CFSs and all mouse embryonic flbroblast CFSs overlap with long genes of more than 300 kb (et al., 2013). Due to their large sizes, these regions can take the entire cell cycle for them to be completely transcribed, therefore the collisions between replication and transcription are inevitable, creating stable R-loops and consequent CFSs fragility (Helmrich et al., 2011) (Figure 1). Consistent with this notion, the depletion of R-loop by RNase H1 overexpression can reduce CFSs fragility (Helmrich et al., 2011). Also, low doses of aphidicolin (APH, commonly used as a CFSs fragility inducer) induced the formation of FANCD2 protein foci, which decorate the location of CFS loci, were found to bind to the central regions of large genes in human and chicken DT40 cells (Pentzold et al., 2018). In addition, FANCD2 has also been shown to be required to promote DNA replication at CFSs by preventing R-loop formation (García-Rubio et al., 2015; Schwab et al., 2015; Madireddy et al., 2016). Our recent study has identified RTEL1 as a key factor in suppressing CFSs fragility by resolving transcription-replication conflicts caused by G-quadruplex and R-loop structures. Moreover, through DNA-RNA immunoprecipitation (DRIP) sequencing, we found both RTEL1 and low dose APH can induce genome-wide R-loop formation. Importantly, around 70% of well characterized CFSs contains at least one region with high signal intensity of R-loop, which are significantly enriched following RTEL1 depletion or APH treatment (Wu et al., 2020). Therefore, transcription and replication collisions and their associated R-loops might underline CFSs fragility.
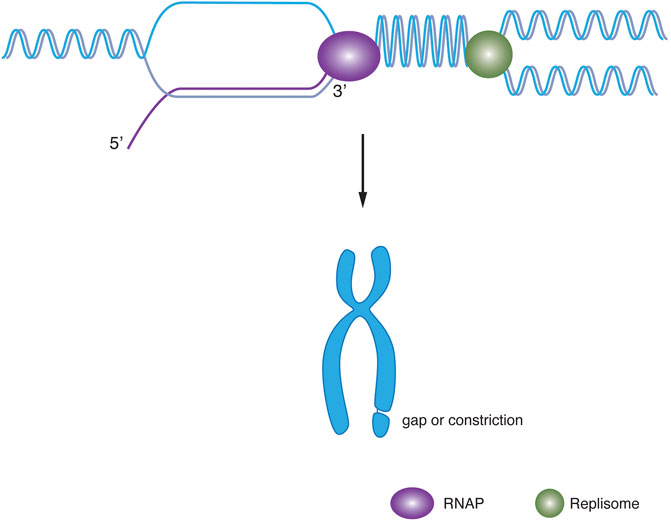
FIGURE 1. Model for head-on collision and chromosome fragility. A head-on collision of the RNA polymerase (purple, left) with replisome (green, right) results in supercoiling and R-loop leading to chromosome fragility.
Early replicating fragile sites (ERFSs) are newly discovered fragile sites that reside in highly transcribed, early replicating genes. Besides, ERFSs are very close to replication origins, which further increases the incidence of transcription and replication collisions (Barlow et al., 2013; Mortusewicz et al., 2013). Recent data suggested that transcription and replication conflicts might explain the mechanism of oncogene-induced replication stress. It has been proposed that oncogene activation forces cells exiting G1 phase and entering S phase prematurely, where transcription is not able to suppress replication origins within the body of such genes, resulting in transcription and replication conflicts. Of note, ERFSs have been mapped as hotspots for sites of oncogene-induced replication and transcription collisions (Halazonetis et al., 2008; Macheret and Halazonetis, 2018).
rDNA
Ribosomal DNA (rDNA) are regions clustered with long tandem repeats, where rRNA are transcribed. rDNA is highly transcribed by RNA polymerase I to meet the demand for the translation activity in cells. Each copy of a repeat harbors a potential replication origin (Linskens and Huberman, 1988). In yeast, it is estimated that each active rDNA gene can accommodate up to 150 RNA polymerases (French et al., 2003), which can form a physical barrier for the replication machinery (Berger et al., 1997; Coffman et al., 2005). Therefore, there are very high chances of transcription and replication collisions at rDNA (Deshpande and Newlon, 1996; Takeuchi et al., 2003; Kobayashi, 2014). In agreement with this, transcription and replication conflicts associated with R-loops are commonly found at rDNA (Chen et al., 2017; Lindström, 2018). Immunofluorescence staining analysis using a specific antibody recognizing R-loops showed the strongest signal in the nucleoli. Nuclear regions containing lots of rDNA, implying stable R-loops formation at rDNA loci. In addition, proteins involved in R-loops prevention or resolution, such as RNase H1, PIF1, and Top I are found to accumulate at rDNA region and are essential for their integrity (El Hage et al., 2010; El Hage et al., 2014; Shen et al., 2017; Tran et al., 2017). Moreover, the RNAi (RNA interference) component, Dicer, was found to be required to limit transcription and replication collisions at rDNA through promoting transcription termination (Castel et al., 2014).
Telomere
Telomeres are regions composed of thousands of TTAGGG repeats that lie at the ends of chromosomes. Usually, telomeres are bound by the shelterin complex. These nucleoprotein structures can prevent telomeres from being recognized as DNA double strand breaks (DSBs), which is critical for genome stability and cell survival (Palm and de Lange, 2008; de Lange, 2009). Although, telomeres are heterochromatic, transcription still happens from the subtelomeric regions towards the end of chromosomes. Currently, two types of telomere associated RNA have been reported, telomeric repeat-containing G-rich RNA (TERRA) and the complementary C-rich transcripts (ARIA) (Kwapisz and Morillon, 2020). Although the biological functions of ARIA remain to be explored, TERRA has been shown to invade into telomeres and induce the formation of stable R-loop structures (Azzalin et al., 2007). TERRA R-loops can be found in all eukaryotes (Rippe and Luke, 2015). As mentioned above, stable R-loops can be a challenging obstacle for replication forks. Furthermore, the displaced G-rich single strand DNA during TERRA R-loop formation promotes the generation of another noncanonical secondary DNA structure: G-quadruplex (G4). G4 is also known to obstruct DNA replication resulting in RS and genome instability (Wang et al., 2019). Therefore, TERRA R-loop may be a major source for telomere fragility. Interestingly, the level of TERRA transcription is negatively correlated with DNA replication process, with the highest being at the G1-S transition and the lowest at the S-G2 transition (Flynn et al., 2015), which can reduce the chance of replication machineries colliding with TERRA R-loops. Besides, several R-loops processing factors have been found to be essential for telomere stability. Specifically, RNaseH1 can regulate TERRA-telomeric DNA hybrids to maintain telomere stability in cells employing non-telomerase approach to maintain the length of their telomers (Arora et al., 2014). FEN1 and UPF1 (Up-frameshift 1) can promote telomeric leading stand DNA synthesis by processing TERRA R-loops through the flap endonuclease activity and 5′-3′ helicase activity, respectively, (Azzalin et al., 2007; Teasley et al., 2015).
Pathways in Dealing With Transcription Associated RS at CFSs
Despite the high incidence of transcription and replication collisions and the prevalence of R-loops at chromosome fragile loci, cells seem to proliferate with a fairly stable genome. Therefore, it is conceivable that cells have evolved surveillance pathways to avoid or minimize the deleterious effects cause by these transcription and replication collision events. Indeed, multiple strategies or pathways have been identified that can prevent or resolve transcription and replication collisions and R-loops, such as spatial and temporal separation of transcription and replication (Wei et al., 1998), the coupling of transcription with mRNA processing (Aguilera and García-Muse, 2012; Huertas and Aguilera, 2003; Li and Manley, 2005), RNAP anti-backtracking and clearance (Castel et al., 2014; Saponaro et al., 2014; Urban et al., 2016; Cheung and Cramer, 2011; Thomas et al., 1998; Roghanian et al., 2015; Ganesan et al., 2012; Epshtein et al., 2014; Zaratiegui et al., 2011; Zatreanu et al., 2019), Fanconi anemia pathway (Madireddy et al., 2016; Schwab et al., 2015; García-Rubio, 2015a), ATR-mediated DNA damage response and the expression of specific R-loop processing factors including RNase H1 (Lockhart et al., 2019) and SETX (García-Muse et al., 2019). Some of the key proteins involved in these pathways are summarized in Table 1. Interestingly, deficiency in some of these proteins such as FANCD2, ATR, RNase H1, and Dicer, have been linked to CFSs fragility (Casper et al., 2002; Madireddy et al., 2016; Di Marco et al., 2017; Fragkos et al., 2019), indicating cells employ these pathways to counteract transcription induced obstacles or RS to maintain CFS stability. Of note, most of these pathways seem to largely operate in S phase of the cell cycle.
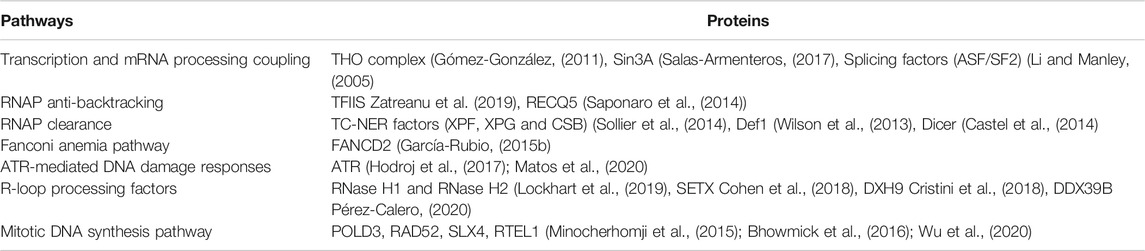
TABLE 1. Key proteins involved in preventing or resolving transcription associated RS.
Recently, an unscheduled mitotic DNA synthesis (MiDAS) pathway functioning in early mitosis has been found to be essential for CFS stability and is recognized as a “salvage pathway” to alleviate RS accumulated at CFSs (Minocherhomji et al., 2015). In response to RS, CFSs can remain under-replicated from interphase till mitosis without necessarily activating G2/M checkpoint. In early mitosis, under-replicated DNA (URD) can activate MiDAS to complete the under-replicated regions. Failure in filling up URD can cause DNA bridges (chromatin bridges and ultra-fine DNA bridges), which can affect sister chromatids separation and lead to genome instability or mitotic catastrophe (Ying et al., 2013). MiDAS is thought to be a break-induced replication (BIR) like pathway. Consistently, more than half of MiDAS foci, defined by visualizing nascent DNA synthesis in mitosis, were found to be on only one of the two sister chromatids, which is consistent with the conserved DNA synthesis feature in BIR (Özer and Hickson, 2018). Until now, many proteins have been identified to be essential for MiDAS. TRAIP is an E3 ubiquitin ligase required for replisome unloading at G2/M boundary, which is a prerequisite for SLX4-dependent endonucleases to access and cleave incomplete DNA structures. After that, RAD52 and POLD3 dependent DNA synthesis can take place. Our recent data has linked R-loop to MiDAS (Wu et al., 2020). In the presence of RS, RNase H1 depletion can stimulate robust MiDAS, while overexpression of RNase H1 can reduce MiDAS. The simplest and more understandable interpretation to these results could be envisaged as R-loop induced RS cannot be completely resolved in interphase, making the cells enter mitosis with incompletely replicated DNA, and consequently triggering the MiDAS pathway. However, there is another possibility that R-loop might directly take part in MiDAS pathway and regulate MiDAS. Indeed, R-loop can stimulate RAD52-POLD3-mediated BIR at ROS-induced telomeric DNA breaks (Tan et al., 2020). Which of the two possibilities is closer to what truly happens in cells warrants further investigation.
Conclusion
Tumorigenesis is a process involving mutations in tumor suppressor genes and the activation of oncogenes. In the early stage of tumor, oncogene activation induces RS and drives genomic instability. Many oncogenes act as growth factors to support proliferation by upregulating transcription factors which in turn can stimulate RNA synthesis (Kotsantis et al., 2016). The increased level of transcription might enhance the conflicts between transcription and replication machineries and its associated R-loops, giving rise to transcription associated RS in S phase. Indeed, accumulating evidence suggests transcription mediated RS is one of the major sources of genome instability, especially at difficult-to-replicate loci mentioned above. Of note, these loci are preferably targeted by RS generated during cancer development (Tsantoulis et al., 2008; Barlow et al., 2013), indicating these loci might be constantly being challenged and mutated during tumorigenesis. Understanding the mechanisms underlying their fragility and identifying molecular pathways in preventing or resolving transcription associated RS will help us understand the molecular basis of cancer development and identify more cancer specific druggable targets.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by National Natural Science Foundation of China (Project 82103232, to WW).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aguilera, A., and García-Muse, T. (2012). R Loops: from Transcription Byproducts to Threats to Genome Stability. Mol. Cel 46 (2), 115–124. doi:10.1016/j.molcel.2012.04.009
Arora, R., Lee, Y., Wischnewski, H., Brun, C. M., Schwarz, T., and Azzalin, C. M. (2014). RNaseH1 Regulates TERRA-Telomeric DNA Hybrids and Telomere Maintenance in ALT Tumour Cells. Nat. Commun. 5, 5220. doi:10.1038/ncomms6220
Azvolinsky, A., Giresi, P. G., Lieb, J. D., and Zakian, V. A. (2009). Highly Transcribed RNA Polymerase II Genes Are Impediments to Replication Fork Progression in Saccharomyces cerevisiae. Mol. Cel 34 (6), 722–734. doi:10.1016/j.molcel.2009.05.022
Azzalin, C. M., Reichenbach, P., Khoriauli, L., Giulotto, E., and Lingner, J. (2007). Telomeric Repeat-Containing RNA and RNA Surveillance Factors at Mammalian Chromosome Ends. Science 318 (5851), 798–801. doi:10.1126/science.1147182
Barlow, J. H., Faryabi, R. B., Callén, E., Wong, N., Malhowski, A., Chen, H. T., et al. (2013). Identification of Early Replicating Fragile Sites that Contribute to Genome Instability. Cell 152 (3), 620–632. doi:10.1016/j.cell.2013.01.006
Berger, C., Horlebein, A., Gögel, E., and Grummt, F. (1997). Temporal Order of Replication of Mouse Ribosomal RNA Genes during the Cell Cycle. Chromosoma 106 (8), 479–484. doi:10.1007/s004120050269
Bermejo, R., Lai, M. S., and Foiani, M. (2012). Preventing Replication Stress to Maintain Genome Stability: Resolving Conflicts between Replication and Transcription. Mol. Cel 45 (6), 710–718. doi:10.1016/j.molcel.2012.03.001
Bhowmick, R., Minocherhomji, S., and Hickson, I. D. (2016). RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cel 64 (6), 1117–1126. doi:10.1016/j.molcel.2016.10.037
Casper, A. M., Nghiem, P., Arlt, M. F., and Glover, T. W. (2002). ATR Regulates Fragile Site Stability. Cell 111 (6), 779–789. doi:10.1016/s0092-8674(02)01113-3
Castel, S. E., Ren, J., Bhattacharjee, S., Chang, A.-Y., Sánchez, M., Valbuena, A., et al. (2014). Dicer Promotes Transcription Termination at Sites of Replication Stress to Maintain Genome Stability. Cell 159 (3), 572–583. doi:10.1016/j.cell.2014.09.031
Chávez, S., and Aguilera, A. (1997). The Yeast HPR1 Gene Has a Functional Role in Transcriptional Elongation that Uncovers a Novel Source of Genome Instability. Genes Dev. 11 (24), 3459–3470.
Chen, L., Chen, J.-Y., Zhang, X., Gu, Y., Xiao, R., Shao, C., et al. (2017). R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-Loops with Transcriptional Pausing at Gene Promoters. Mol. Cel. 68 (4), 745–757. doi:10.1016/j.molcel.2017.10.008
Cheung, A. C. M., and Cramer, P. (2011). Structural Basis of RNA Polymerase II Backtracking, Arrest and Reactivation. Nature 471 (7337), 249–253. doi:10.1038/nature09785
Coffman, F. D., He, M., Diaz, M.-L., and Cohen, S. (2005). DNA Replication Initiates at Different Sites in Early and Late S Phase within Human Ribosomal RNA Genes. Cell Cycle 4 (9), 1223–1226. doi:10.4161/cc.4.9.1961
Cohen, S., Puget, N., Lin, Y.-L., Clouaire, T., Aguirrebengoa, M., Rocher, V., et al. (2018). Senataxin Resolves RNA:DNA Hybrids Forming at DNA Double-Strand Breaks to Prevent Translocations. Nat. Commun. 9 (1), 533. doi:10.1038/s41467-018-02894-w
Cristini, A., Groh, M., Kristiansen, M. S., and Gromak, N. (2018). RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cel Rep. 23 (6), 1891–1905. doi:10.1016/j.celrep.2018.04.025
de Lange, T. (2009). How Telomeres Solve the End-protection Problem. Science 326 (5955), 948–952. doi:10.1126/science.1170633
Deshpande, A. M., and Newlon, C. S. (1996). DNA Replication fork Pause Sites Dependent on Transcription. Science 272 (5264), 1030–1033. doi:10.1126/science.272.5264.1030
Di Marco, S., Hasanova, Z., Kanagaraj, R., Chappidi, N., Altmannova, V., Menon, S., et al. (2017). RECQ5 Helicase Cooperates with MUS81 Endonuclease in Processing Stalled Replication Forks at Common Fragile Sites during Mitosis. Mol. Cel 66 (5), 658–671. doi:10.1016/j.molcel.2017.05.006
El Hage, A., French, S. L., Beyer, A. L., and Tollervey, D. (2010). Loss of Topoisomerase I Leads to R-Loop-Mediated Transcriptional Blocks during Ribosomal RNA Synthesis. Genes Dev. 24 (14), 1546–1558. doi:10.1101/gad.573310
El Hage, A., Webb, S., Kerr, A., and Tollervey, D. (2014). Genome-wide Distribution of RNA-DNA Hybrids Identifies RNase H Targets in tRNA Genes, Retrotransposons and Mitochondria. Plos Genet. 10 (10), e1004716. doi:10.1371/journal.pgen.1004716
Epshtein, V., Kamarthapu, V., McGary, K., Svetlov, V., Ueberheide, B., Proshkin, S., et al. (2014). UvrD Facilitates DNA Repair by Pulling RNA Polymerase Backwards. Nature 505 (7483), 372–377. doi:10.1038/nature12928
et al. (2013). Common Fragile Site Profiling in Epithelial and Erythroid Cells Reveals that Most Recurrent Cancer Deletions Lie in Fragile Sites Hosting Large Genes. Cell Rep 4 (3), 420–428. doi:10.1016/j.celrep.2013.07.003
Flynn, R. L., Cox, K. E., Jeitany, M., Wakimoto, H., Bryll, A. R., Ganem, N. J., et al. (2015). Alternative Lengthening of Telomeres Renders Cancer Cells Hypersensitive to ATR Inhibitors. Science 347 (6219), 273–277. doi:10.1126/science.1257216
Fragkos, M., Barra, V., Egger, T., Bordignon, B., Lemacon, D., Naim, V., et al. (2019). Dicer Prevents Genome Instability in Response to Replication Stress. Oncotarget 10 (43), 4407–4423. doi:10.18632/oncotarget.27034
French, S. L., Osheim, Y. N., Cioci, F., Nomura, M., and Beyer, A. L. (2003). In Exponentially Growing Saccharomyces cerevisiae Cells, rRNA Synthesis Is Determined by the Summed RNA Polymerase I Loading Rate rather Than by the Number of Active Genes. Mol. Cel Biol 23 (5), 1558–1568. doi:10.1128/mcb.23.5.1558-1568.2003
Gómez‐González, B. (2012). Genome‐wide Function of THO/TREX in Active Genes Prevents R‐loop‐dependent Replication Obstacles. 30(15). 3106–3119.
Ganesan, A., Spivak, G., and Hanawalt, P. C. (2012). “Transcription-Coupled DNA Repair in Prokaryotes,” in Progress in Molecular Biology and Translational Science. Editor P. W. Doetsch (Academic Press), 25–40. doi:10.1016/b978-0-12-387665-2.00002-x
Gao, G., Johnson, S. H., Vasmatzis, G., Pauley, C. E., Tombers, N. M., Kasperbauer, J. L., et al. (2017). Common Fragile Sites (CFS) and Extremely Large CFS Genes Are Targets for Human Papillomavirus Integrations and Chromosome Rearrangements in Oropharyngeal Squamous Cell Carcinoma. Genes Chromosomes Cancer 56 (1), 59–74. doi:10.1002/gcc.22415
García-Rubio, M. L. (2015b). The Fanconi Anemia Pathway Protects Genome Integrity from R-Loops. 11(11): e1005674.
García-Muse, T., Aguilera, A., and Loops, R. (2019). From Physiological to Pathological Roles. Cell 179 (3), 604–618.
García-Rubio, M. L., Pérez-Calero, C., Barroso, S. I., Tumini, E., Herrera-Moyano, E., Rosado, I. V., et al. (2015). The Fanconi Anemia Pathway Protects Genome Integrity from R-Loops. Plos Genet. 11 (11), e1005674. doi:10.1371/journal.pgen.1005674
García-Rubio, M. L. (2015). The Fanconi Anemia Pathway Protects Genome Integrity from R-Loops. PLOS Genet. 11 (11), e1005674.
Guy, L., and Roten, C.-A. H. (2004). Genometric Analyses of the Organization of Circular Chromosomes: a Universal Pressure Determines the Direction of Ribosomal RNA Genes Transcription Relative to Chromosome Replication. Gene 340 (1), 45–52. doi:10.1016/j.gene.2004.06.056
Halazonetis, T. D., Gorgoulis, V. G., and Bartek, J. (2008). An Oncogene-Induced DNA Damage Model for Cancer Development. Science 319 (5868), 1352–1355. doi:10.1126/science.1140735
Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T., and Cimprich, K. A. (2017). Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 170 (4), 774–786. doi:10.1016/j.cell.2017.07.043
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of Cancer: the Next Generation. Cell 144 (5), 646–674. doi:10.1016/j.cell.2011.02.013
Hellman, A., Rahat, A., Scherer, S. W., Darvasi, A., Tsui, L.-C., and Kerem, B. (2000). Replication Delay along FRA7H, a Common Fragile Site on Human Chromosome 7, Leads to Chromosomal Instability. Mol. Cel Biol 20 (12), 4420–4427. doi:10.1128/mcb.20.12.4420-4427.2000
Helmrich, A., Ballarino, M., and Tora, L. (2011). Collisions between Replication and Transcription Complexes Cause Common Fragile Site Instability at the Longest Human Genes. Mol. Cel 44 (6), 966–977. doi:10.1016/j.molcel.2011.10.013
Hodroj, D., Recolin, B., Serhal, K., Martinez, S., Tsanov, N., Abou Merhi, R., et al. (2017). An ATR ‐dependent Function for the Ddx19 RNA Helicase in Nuclear R‐loop Metabolism. Embo J. 36 (9), 1182–1198. doi:10.15252/embj.201695131
Huertas, P., and Aguilera, A. (2003). Cotranscriptionally Formed DNA:RNA Hybrids Mediate Transcription Elongation Impairment and Transcription-Associated Recombination. Mol. Cel. 12 (3), 711–721. doi:10.1016/j.molcel.2003.08.010
Keil, R. L., and Roeder, G. S. (1984). Cis-acting, Recombination-Stimulating Activity in a Fragment of the Ribosomal DNA of S. cerevisiae. Cell 39 (2 Pt 1), 377–386. doi:10.1016/0092-8674(84)90016-3
Kim, N., and Jinks-Robertson, S. (2012). Transcription as a Source of Genome Instability. Nat. Rev. Genet. 13 (3), 204–214. doi:10.1038/nrg3152
Kobayashi, T. (2014). Ribosomal RNA Gene Repeats, Their Stability and Cellular Senescence. Proc. Jpn. Acad. Ser. B: Phys. Biol. Sci. 90 (4), 119–129. doi:10.2183/pjab.90.119
Kotsantis, P., Silva, L. M., Irmscher, S., Jones, R. M., Folkes, L., Gromak, N., et al. (2016). Increased Global Transcription Activity as a Mechanism of Replication Stress in Cancer. Nat. Commun. 7 (1), 13087. doi:10.1038/ncomms13087
Kumari, D., Hayward, B., Nakamura, A. J., Bonner, W. M., and Usdin, K. (2015). Evidence for Chromosome Fragility at the Frataxin Locus in Friedreich Ataxia. Mutat. Research/Fundamental Mol. Mech. Mutagenesis 781, 14–21. doi:10.1016/j.mrfmmm.2015.08.007
Kwapisz, M., and Morillon, A. (2020). Subtelomeric Transcription and its Regulation. J. Mol. Biol. 432 (15), 4199–4219. doi:10.1016/j.jmb.2020.01.026
Le Beau, M. (1998). Replication of a Common Fragile Site, FRA3B, Occurs Late in S Phase and Is Delayed Further upon Induction: Implications for the Mechanism of Fragile Site Induction. 7(4). 755–761.doi:10.1093/hmg/7.4.755
Letessier, A., Millot, G. A., Koundrioukoff, S., Lachagès, A.-M., Vogt, N., Hansen, R. S., et al. (2011). Cell-type-specific Replication Initiation Programs Set Fragility of the FRA3B Fragile Site. Nature 470 (7332), 120–123. doi:10.1038/nature09745
Li, X., and Manley, J. L. (2005). Inactivation of the SR Protein Splicing Factor ASF/SF2 Results in Genomic Instability. Cell 122 (3), 365–378. doi:10.1016/j.cell.2005.06.008
Lindström, M. S. (2018). Nucleolus as an Emerging Hub in Maintenance of Genome Stability and Cancer Pathogenesis. Oncogene 37 (18), 2351–2366.
Linskens, M. H., and Huberman, J. A. (1988). Organization of Replication of Ribosomal DNA in Saccharomyces cerevisiae. Mol. Cel Biol 8 (11), 4927–4935. doi:10.1128/mcb.8.11.4927-4935.1988
Liu, B., Lie Wong, M., Tinker, R. L., Geiduschek, E. P., and Alberts, B. M. (1993). The DNA Replication fork Can Pass RNA Polymerase without Displacing the Nascent Transcript. Nature 366 (6450), 33–39. doi:10.1038/366033a0
Lockhart, A., Pires, V. B., Bento, F., Kellner, V., Luke-Glaser, S., Yakoub, G., et al. (2019). RNase H1 and H2 Are Differentially Regulated to Process RNA-DNA Hybrids. Cel Rep. 29 (9), 2890–2900. doi:10.1016/j.celrep.2019.10.108
Macheret, M., and Halazonetis, T. D. (2018). Intragenic Origins Due to Short G1 Phases Underlie Oncogene-Induced DNA Replication Stress. Nature 555 (7694), 112–116. doi:10.1038/nature25507
Madireddy, A., Kosiyatrakul, S. T., Boisvert, R. A., Herrera-Moyano, E., García-Rubio, M. L., Gerhardt, J., et al. (2016). FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cel 64 (2), 388–404. doi:10.1016/j.molcel.2016.09.017
Magenis, R. E., Hecht, F., and Lovrien, E. W. (1970). Heritable Fragile Site on Chromosome 16: Probable Localization of Haptoglobin Locus in Man. Science 170 (3953), 85–87. doi:10.1126/science.170.3953.85
Matos, D. A., Zhang, J.-M., Ouyang, J., Nguyen, H. D., Genois, M.-M., and Zou, L. (2020). ATR Protects the Genome against R Loops through a MUS81-triggered Feedback Loop. Mol. Cel 77 (3), 514–527. doi:10.1016/j.molcel.2019.10.010
Merrikh, H., Machón, C., Grainger, W. H., Grossman, A. D., and Soultanas, P. (2011). Co-directional Replication-Transcription Conflicts lead to Replication Restart. Nature 470 (7335), 554–557. doi:10.1038/nature09758
Merrikh, H., Zhang, Y., Grossman, A. D., and Wang, J. D. (2012). Replication-transcription Conflicts in Bacteria. Nat. Rev. Microbiol. 10 (7), 449–458. doi:10.1038/nrmicro2800
Minocherhomji, S., Ying, S., Bjerregaard, V. A., Bursomanno, S., Aleliunaite, A., Wu, W., et al. (2015). Replication Stress Activates DNA Repair Synthesis in Mitosis. Nature 528 (7581), 286–290. doi:10.1038/nature16139
Mortusewicz, O., Herr, P., and Helleday, T. (2013). Early Replication Fragile Sites: where Replication-Transcription Collisions Cause Genetic Instability. Embo J. 32 (4), 493–495. doi:10.1038/emboj.2013.20
Özer, Ö., and Hickson, I. D. (2018). Pathways for Maintenance of Telomeres and Common Fragile Sites during DNA Replication Stress. Open Biol. 8 (4), 180018.
Pérez-Calero, C. (2020). UAP56/DDX39B Is a Major Cotranscriptional RNA–DNA Helicase that Unwinds Harmful R Loops Genome-wide. 34(13-14). 898.
Palm, W., and de Lange, T. (2008). How Shelterin Protects Mammalian Telomeres. Annu. Rev. Genet. 42, 301–334. doi:10.1146/annurev.genet.41.110306.130350
Pentzold, C., Shah, S. A., Hansen, N. R., Le Tallec, B., Seguin-Orlando, A., Debatisse, M., et al. (2018). FANCD2 Binding Identifies Conserved Fragile Sites at Large Transcribed Genes in Avian Cells. Nucleic Acids Res. 46 (3), 1280–1294. doi:10.1093/nar/gkx1260
Petryk, N., Kahli, M., d'Aubenton-Carafa, Y., Jaszczyszyn, Y., Shen, Y., Silvain, M., et al. (2016). Replication Landscape of the Human Genome. Nat. Commun. 7, 10208. doi:10.1038/ncomms10208
Pomerantz, R. T., and O’Donnell, M. (2008). The Replisome Uses mRNA as a Primer after Colliding with RNA Polymerase. Nature 456 (7223), 762–766. doi:10.1038/nature07527
Prado, F., and Aguilera, A. (2005). Impairment of Replication fork Progression Mediates RNA polII Transcription-Associated Recombination. Embo j 24 (6), 1267–1276. doi:10.1038/sj.emboj.7600602
Pruitt, S. C., Qin, M., Wang, J., Kunnev, D., and Freeland, A. (2017). A Signature of Genomic Instability Resulting from Deficient Replication Licensing. Plos Genet. 13 (1), e1006547. doi:10.1371/journal.pgen.1006547
Rippe, K., and Luke, B. (2015). TERRA and the State of the Telomere. Nat. Struct. Mol. Biol. 22 (11), 853–858. doi:10.1038/nsmb.3078
Rocha, E. P. C., and Danchin, A. (2003). Essentiality, Not Expressiveness, Drives Gene-Strand Bias in Bacteria. Nat. Genet. 34 (4), 377–378. doi:10.1038/ng1209
Roghanian, M., Zenkin, N., and Yuzenkova, Y. (2015). Bacterial Global Regulators DksA/ppGpp Increase Fidelity of Transcription. Nucleic Acids Res. 43 (3), 1529–1536. doi:10.1093/nar/gkv003
Salas‐Armenteros, I. (2017). Human THO–Sin3A Interaction Reveals New Mechanisms to Prevent R‐loops that Cause Genome Instability. 36(23). 3532–3547.
Santos-Pereira, J. M., and Aguilera, A. (2015). R Loops: New Modulators of Genome Dynamics and Function. Nat. Rev. Genet. 16 (10), 583–597. doi:10.1038/nrg3961
Saponaro, M., Kantidakis, T., Mitter, R., Kelly, G. P., Heron, M., Williams, H., et al. (2014). RECQL5 Controls Transcript Elongation and Suppresses Genome Instability Associated with Transcription Stress. Cell 157 (5), 1037–1049. doi:10.1016/j.cell.2014.03.048
Schwab, R. A., Nieminuszczy, J., Shah, F., Langton, J., Lopez Martinez, D., Liang, C.-C., et al. (2015). The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cel 60 (3), 351–361. doi:10.1016/j.molcel.2015.09.012
Shen, W., Sun, H., De Hoyos, C. L., Bailey, J. K., Liang, X.-h., and Crooke, S. T. (2017). Dynamic Nucleoplasmic and Nucleolar Localization of Mammalian RNase H1 in Response to RNAP I Transcriptional R-Loops. Nucleic Acids Res. 45 (18), 10672–10692. doi:10.1093/nar/gkx710
Sollier, J., Stork, C. T., García-Rubio, M. L., Paulsen, R. D., Aguilera, A., and Cimprich, K. A. (2014). Transcription-coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cel. 56 (6), 777–785. doi:10.1016/j.molcel.2014.10.020
Srivatsan, A., Tehranchi, A., MacAlpine, D. M., and Wang, J. D. (2010). Co-Orientation of Replication and Transcription Preserves Genome Integrity. Plos Genet. 6 (1), e1000810. doi:10.1371/journal.pgen.1000810
Sugimoto, N., Maehara, K., Yoshida, K., Ohkawa, Y., and Fujita, M. (2018). Genome-wide Analysis of the Spatiotemporal Regulation of Firing and Dormant Replication Origins in Human Cells. 46(13). 6683–6696.doi:10.1093/nar/gky476
Sutherland, G. R. (1979). Heritable Fragile Sites on Human Chromosomes I. Factors Affecting Expression in Lymphocyte Culture. Am. J. Hum. Genet. 31 (2), 125–135.
Takeuchi, Y., Horiuchi, T., and Kobayashi, T. (2003). Transcription-dependent Recombination and the Role of fork Collision in Yeast rDNA. Genes Dev. 17 (12), 1497–1506. doi:10.1101/gad.1085403
Tan, J., Duan, M., Yadav, T., Phoon, L., Wang, X., Zhang, J.-M., et al. (2020). An R-Loop-Initiated CSB-RAD52-POLD3 Pathway Suppresses ROS-Induced Telomeric DNA Breaks. Nucleic Acids Res. 48 (3), 1285–1300. doi:10.1093/nar/gkz1114
Teasley, D. C., Parajuli, S., Nguyen, M., Moore, H. R., Alspach, E., Lock, Y. J., et al. (2015). Flap Endonuclease 1 Limits Telomere Fragility on the Leading Strand. J. Biol. Chem. 290 (24), 15133–15145. doi:10.1074/jbc.m115.647388
Thomas, M. J., Platas, A. A., and Hawley, D. K. (1998). Transcriptional Fidelity and Proofreading by RNA Polymerase II. Cell 93 (4), 627–637. doi:10.1016/s0092-8674(00)81191-5
Thorland, E. C., Myers, S. L., Gostout, B. S., and Smith, D. I. (2003). Common Fragile Sites Are Preferential Targets for HPV16 Integrations in Cervical Tumors. Oncogene 22 (8), 1225–1237. doi:10.1038/sj.onc.1206170
Tran, P. L. T., Pohl, T. J., Chen, C.-F., Chan, A., Pott, S., and Zakian, V. A. (2017). PIF1 Family DNA Helicases Suppress R-Loop Mediated Genome Instability at tRNA Genes. Nat. Commun. 8, 15025. doi:10.1038/ncomms15025
Tsantoulis, P. K., Kotsinas, A., Sfikakis, P. P., Evangelou, K., Sideridou, M., Levy, B., et al. (2008). Oncogene-induced Replication Stress Preferentially Targets Common Fragile Sites in Preneoplastic Lesions. A Genome-wide Study. Oncogene 27 (23), 3256–3264. doi:10.1038/sj.onc.1210989
Tuduri, S., Crabbé, L., Conti, C., Tourrière, H., Holtgreve-Grez, H., Jauch, A., et al. (2009). Topoisomerase I Suppresses Genomic Instability by Preventing Interference between Replication and Transcription. Nat. Cel Biol 11 (11), 1315–1324. doi:10.1038/ncb1984
Urban, V., Dobrovolna, J., Hühn, D., Fryzelkova, J., Bartek, J., and Janscak, P. (2016). RECQ5 Helicase Promotes Resolution of Conflicts between Replication and Transcription in Human Cells. J. Cel Biol 214 (4), 401–415. doi:10.1083/jcb.201507099
Voelkel-Meiman, K., Keil, R. L., and Roeder, G. S. (1987). Recombination-stimulating Sequences in Yeast Ribosomal DNA Correspond to Sequences Regulating Transcription by RNA Polymerase I. Cell 48 (6), 1071–1079. doi:10.1016/0092-8674(87)90714-8
Wang, Y., Yang, J., Wild, A. T., Wu, W. H., Shah, R., Danussi, C., et al. (2019). G-quadruplex DNA Drives Genomic Instability and Represents a Targetable Molecular Abnormality in ATRX-Deficient Malignant Glioma. Nat. Commun. 10 (1), 943. doi:10.1038/s41467-019-08905-8
Wei, X., Samarabandu, J., Devdhar, R. S., Siegel, A. J., Acharya, R., and Berezney, R. (1998). Segregation of Transcription and Replication Sites into Higher Order Domains. Science 281 (5382), 1502–1505. doi:10.1126/science.281.5382.1502
Wilson, M. D., Harreman, M., Taschner, M., Reid, J., Walker, J., Erdjument-Bromage, H., et al. (2013). Proteasome-mediated Processing of Def1, a Critical Step in the Cellular Response to Transcription Stress. Cell 154 (5), 983–995. doi:10.1016/j.cell.2013.07.028
Wilson, T. E., Arlt, M. F., Park, S. H., Rajendran, S., Paulsen, M., Ljungman, M., et al. (2015). Large Transcription Units Unify Copy Number Variants and Common Fragile Sites Arising under Replication Stress. Genome Res. 25 (2), 189–200. doi:10.1101/gr.177121.114
Wu, W., Bhowmick, R., Vogel, I., Özer, Ö., Ghisays, F., Thakur, R. S., et al. (2020). RTEL1 Suppresses G-Quadruplex-Associated R-Loops at Difficult-To-Replicate Loci in the Human Genome. Nat. Struct. Mol. Biol. 27 (5), 424–437. doi:10.1038/s41594-020-0408-6
Ying, S., Minocherhomji, S., Chan, K. L., Palmai-Pallag, T., Chu, W. K., Wass, T., et al. (2013). MUS81 Promotes Common Fragile Site Expression. Nat. Cel Biol 15 (8), 1001–1007. doi:10.1038/ncb2773
Zaratiegui, M., Castel, S. E., Irvine, D. V., Kloc, A., Ren, J., Li, F., et al. (2011). RNAi Promotes Heterochromatic Silencing through Replication-Coupled Release of RNA Pol II. Nature 479 (7371), 135–138. doi:10.1038/nature10501
Keywords: transcription, replication, mitotic DNA synthesis (MiDAS), replication stress, fragile sites
Citation: Wu W, He JN, Lan M, Zhang P and Chu WK (2021) Transcription-Replication Collisions and Chromosome Fragility. Front. Genet. 12:804547. doi: 10.3389/fgene.2021.804547
Received: 29 October 2021; Accepted: 29 November 2021;
Published: 10 December 2021.
Edited by:
Advaitha Madireddy, Rutgers, The State University of New Jersey, United StatesReviewed by:
Kirill Lobachev, Georgia Institute of Technology, United StatesCopyright © 2021 Wu, He, Lan, Zhang and Chu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wai Kit Chu, d2Fpa2l0QGN1aGsuZWR1Lmhr