 Xiao-Tong Li†
Xiao-Tong Li† Xing-Yu Feng
Xing-Yu Feng Zhu-Qing Shao
Zhu-Qing Shao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet., 23 November 2021
Sec. Genomics of Plants and the Phytoecosystem
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.801218
This article is part of the Research TopicInheritance and Improvement of Disease Resistance or Stress Tolerance for Triticeae CropsView all 11 articles
Plants have evolved sophisticated systems to cope with the environmental stresses, with the heat shock factor (HSF) family proteins composing an integral part of the transcriptional regulation system. Understanding the evolutionary history and functional diversity of HSFs will facilitate improving tolerance of crops to adverse environmental conditions. In this study, genome-wide analysis of Secale cereale identified 31 HSF genes. The total number of HSF genes in S. cereale is larger than that in barley and the three subgenomes of wheat, suggesting it is a valuable resource for mining functional HSFs. Chromosome analysis revealed an uneven distribution of HSF genes among the 7 S. cereale chromosomes, with no HSF gene was detected on chromosome 4. Further interspecies synteny analysis revealed that chromosome reorganization during species-speciation may lead to the escape of HSF genes from the S. cereale chromosome 4. Phylogenetic analysis revealed that S. cereale experienced more HSF gene duplications than barley and the three wheat subgenomes. Expression analysis demonstrated that S. cereale HSF genes showed diverse expression patterns across plant developmental stages and upon drought and freezing treatment, suggesting functional diversity of the gene family. Notably, we detected distinct expression patterns for a recently duplicated HSF gene pair, indicating functional divergence may have occurred between the two genes. The study presents the genome organization, evolutionary features and expression patterns of the S. cereale HSF genes. These results provide new insights into the evolution of HSF genes in Triticeae and may serve as a resource for Triticeae molecular breeding.
Plants are consistently affected by biotic and abiotic stresses in the environment during their whole lifespan, including drought, salt, heat, cold and pathogens’ infection. In the long-term evolution, plants have evolved sophisticated systems and regulatory networks to avoid or attenuate the deleterious effects of these stresses (Peck and Mittler, 2020). Many gene families have been reported for their distinct roles in responding to external environmental stresses, such as the nucleotide-binding leucine-rich-repeat (NLR) disease resistance gene family (Saur et al., 2021); and the cold-induced C-repeat binding factor (CBF) gene family (Zhou et al., 2011). There are also some gene families that can respond to multiple external stresses by their different family members (Javed et al., 2020; Wani et al., 2021). Among them, plant heat shock factor (HSF) family proteins compose an integral part of the transcriptional regulation system for plants against external environmental stresses, by modulating the expression of different sets of plant genes in responding to heat, cold, salt stresses and the infection of pathogens (Andrasi et al., 2021).
HSF is a conserved gene family that widely spreads in eukaryotes and prokaryotes. Proteins encoded by this family were initially identified as transcription factors that regulate the expression of HSPs, whose functions as molecular chaperones to maintain protein homeostasis in cells (Boston et al., 1996). However, increasing studies in plants have revealed that HSFs are important components of the complex signaling systems that control responses not only to high temperatures but also to a number of abiotic stresses such as cold, drought, hypoxic conditions, soil salinity, and to pathogen threats (Andrasi et al., 2021). Compared to the single copy HSF gene in yeast and four HSF genes in human genome, plant genomes have expanded the HSF genes to several dozens (Wang et al., 2018). For example, 22 and 25 HSF genes have been identified from the dicot plant Arabidopsis thaliana and monocot species Oryza sativa, respectively (Guo et al., 2008).
The plant HSF family proteins have several characteristic domains that are essential for their functions, including a N-terminal DNA-binding domain (DBD) that recognizes heat shock elements in the promoter region of target genes, a following oligomerization domain (OD or HR-A/B motif) that is responsible for protein–protein interactions and trimerization during transcriptional activation (Scharf et al., 2012). Based on the length of the linker between the DBD and OD domains and the number of amino acid residues inserted into the HR-A/B regions, plant HSFs are classified into three subgroups: HSFA, HSFB and HSFC (Scharf et al., 2012). The subgroup A HSFs have additional nuclear localization signal (NLS) and nuclear export signal (NES) sequences, and a C-terminal aromatic and hydrophobic amino acid motif (AHA), which is needed for its transcriptional activation activity (Andrasi et al., 2021). The subgroup B HSFs contain a C-terminus tetrapeptide (LFGV) that functions as a repressor domain (RD) (Scharf et al., 2012; Andrasi et al., 2021).
Due to the functional importance, genome-wide identification and functional exploration of HSF genes have been carried out in model plants and many crops (Wang et al., 2018; Andrasi et al., 2021). Some HSF members have been used to enhance plant tolerance to different stresses and molecular breeding. For example, overexpression of AtHSFA2 in A. thaliana increased the motolerance, salt/osmotic stress tolerance, and enhanced callus growth of the plant (Charng et al., 2007; Ogawa et al., 2007; Nishizawa et al., 2006). Overexpression of GmHSFA1 in soybean and SlHSFA1 in tomato enhanced thermotolerance of the transgenic plants (Mishra et al., 2002; Zhu et al., 2006).
Triticeae crops, including wheat (Triticum aestivum), barley (Hordeum vulgare) and rye (Secale cereale), are important grain crops, which are frequently challenged by various biotic and abiotic stresses. Recent studies revealed that overexpression of several HSF genes could enhance plants tolerance to multiple stresses (Bi et al., 2020; Poonia et al., 2020), suggesting HSF genes have significant potential for molecular breeding of Triticeae species. Genome-wide analysis of HSF genes has been conducted in wheat and barley, which provides primary resources for mining and utilizing functional HSF genes in the two species (Duan et al., 2019; Zhou et al., 2019; Mishra et al., 2020; Ye et al., 2020). However, the HSF genes composition in rye has not been investigated yet. In this study, we performed genome-wide identification and evolutionary analysis of HSF genes in a recently published rye genome (Li et al., 2021), and performed comparative analysis of HSF genes in Triticeae species.
The genome sequences of S. cereale, H. vulgare, T. aestivum (three subgenomes), O. sativa and A. thaliana were downloaded from public databases (Table S1). HSF genes were identified as described using a method by Shao et al. (2014) with some modifications. Briefly, the annotated proteins in each genome were screened for the HSF domain (Pfam accession: PF00447) by using the hmmsearch program implemented in the hmmer3.0 software (Johnson et al., 2010). The amino acid sequences of obtained HSF genes were then used to run a genome-wide BLASTp analysis for each genome. All hits were further analyzed using the hmmscan program in hmmer3.0 against the local Pfam-A database to confirm a detectable HSF domain in each sequence, with a e-value setting as 0.0001. All obtained HSF candidates were validated by subjecting to the Heatster database (Fan et al., 2021). Only genes simultaneously encoding DBD, HR-A and HR-B were recognized as true HSF genes, which were classified into the A, B, and C classes by the Heatster database.
The gene structure analysis for identified HSFs was constructed using the Gene Structure Display Server (GSDS) (http://gsds.cbi.pku.edu.cn/) (Suyama et al., 2006), while domain composition was presented by using the Tbtools (Chen et al., 2020).
Amino acid sequences of the HSF domain were aligned using ClustalW program (Edgar, 2004) that is integrated in MEGA 7.0 (Kumar et al., 2016) with default options, and then was manually corrected. ModelFinder was used to estimate the best-fit model of nucleotide substitution (Kalyaanamoorthy et al., 2017). Phylogenetic analyses were performed using the IQ-TREE with the maximum likelihood algorithm (Nguyen et al., 2015). Branch support values were calculated using the SH-aLRT (Anisimova et al., 2011) and the UFBoot2 (Minh et al., 2013) methods with 1,000 bootstrap replicates.
Inter-species synteny analysis of the HSF genes from S. cereale, T. aestivum and H. vulgare were performed by using the MCScanX program that is integrated in the TBtools (Wang et al., 2012; Chen et al., 2020). Syntenic relationships were then drawn using Tbtools (Chen et al., 2020).
RNA-seq raw reads of rye in different tissues and under different stresses were downloaded from the SRA database (Accesion: SRX9567472). The adaptors were removed using Trim_Galore (https://github.com/FelixKrueger/TrimGalore). The resulted clean reads were mapped to the rye reference genome using Hisat2 (Kim et al., 2019). Quantitation of gene expression was performed using feature Counts (Liao et al., 2014). The resulted read counts of each gene were normalized to FPKM.
A total of 31 HSF family members were identified from the S. cereale genome by searching the annotated proteome of Weining rye (Figure 1 and Supplementary Table S1). Gene structure analysis revealed that the annotated transcripts for 28 of the 31 HSF genes have both 5′ and 3′ untranslated regions (UTRs), whereas the transcript of one gene (ScWN3R01G480000) only has 3’ UTR and transcripts of two genes (ScWN5R01G655400 and ScWN5R01G653500) do not have annotated UTR region, suggesting an overall high quality of the annotation. The annotated transcripts showed a high diversity of exon-intron composition among different HSF members, with 1–4 introns were found from the transcripts of the 29 genes, and 2 genes not having annotated introns. Among them, 24 HSF genes have only 1 intron, and introns for 23 of them are located at the coding sequences (CDS) of the transcripts. In contrast, 5 transcripts have 2 to 4 annotated introns. The large diversity of exon-intron composition in S. cereale may serve as primary resources for generating potential mRNA alternative splicing, which has been reported for several HSF genes in other plants (Ling et al., 2021). The amino acid numbers of the translated proteins from annotated CDSs of HSF genes range from 678 to 1,560 (Supplementary Table S2), suggesting potential fusion of additional domains by some HSF proteins. However, domain structure analysis showed that only two HSF proteins have an additional domain, namely Golgin_A5, at the C-terminal (Figure 1A), indicating a functional innovation of the two HSFs.
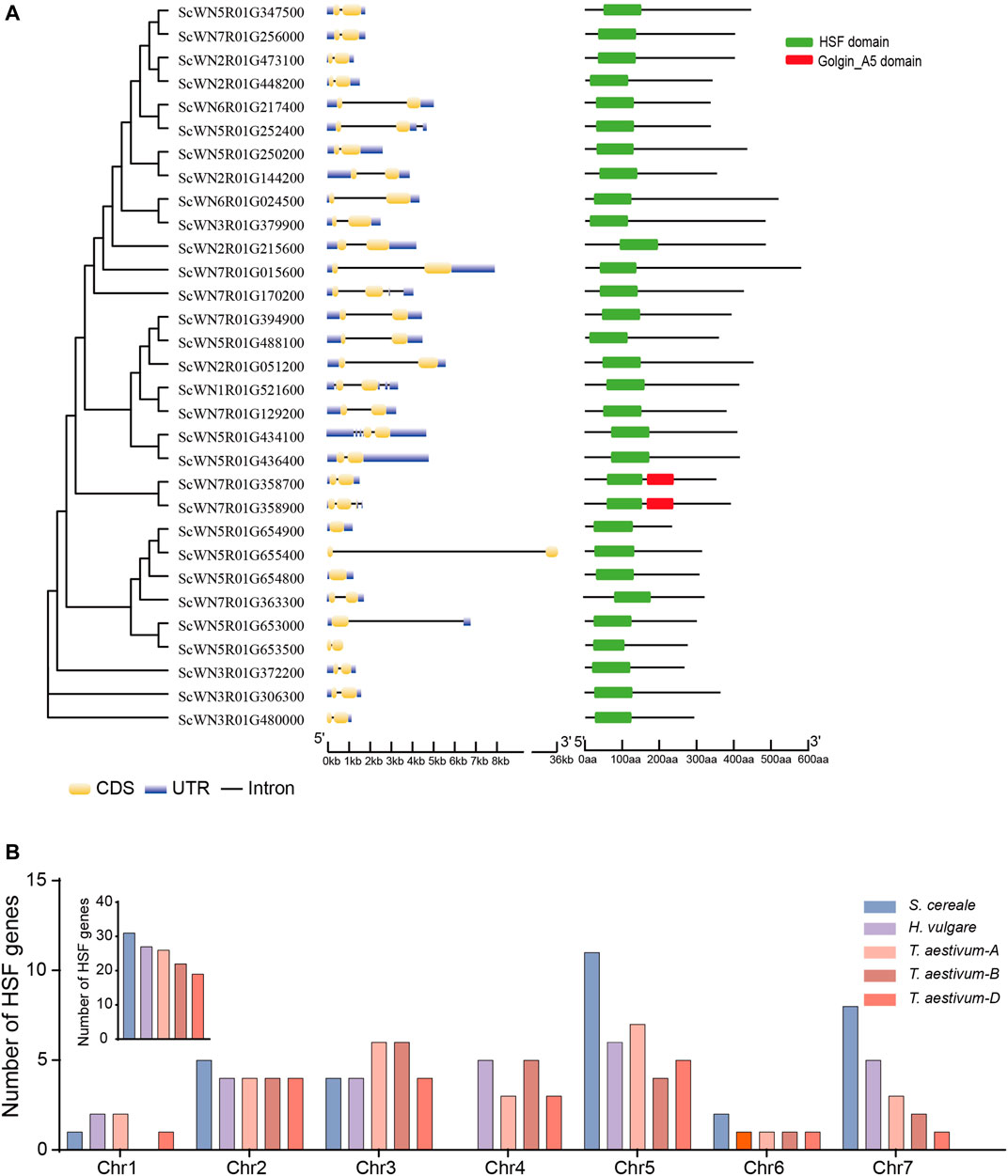
FIGURE 1. Gene structure, domain composition and chromosome distribution of S. cereale HSF genes. (A). HSF genes in the S. cereale genome were ordered by the phylogeny. Gene structure and domain composition for each gene were shown following the gene name. (B). The number of HSF genes on each chromosome in S. cereale were shown and compared to those of T. aestivum and H. vulgare.
Chromosomal distribution analysis revealed that the 31 HSF genes are unevenly distributed on seven chromosomes of S. cereale. Chromosome 5 contains the largest number of HSF genes (11 genes), whereas no HSF genes were identified on chromosome 4 (Figure S1). The chromosome 1, 2, 3, 6, 7 have 1, 5, 4, 2, and 8 HSF genes, respectively (Supplementary Figure S1). Since chromosome introgression has been frequently used for Triticeae crops (Li et al., 2021), we compared the chromosomal distribution pattern of HSF genes among S. cereale, H. vulgare, and T. aestivum. The results showed that the overall HSF gene number in S. cereale (R genome) is great than that in the H. vulgare (H genome) and the subgenome A, B and D of wheat (Figure 1B). In accordance with the high HSF gene number in S. cereale, the numbers of HSF genes on the chromosomes 2, 5, 6, and 7 of S. cereale each ranks the first among the five genomes, respectively. However, in contrast to the lack of HSF genes on chromosome 4 in S. cereale, the H. vulgare genome and the subgenome A, B and D of wheat each has 5, 3, 5, and 3 HSF genes, respectively (Figure 1B). Considering the three species were only diverged from the common ancestor within twenty million years, it is possible that the HSF genes on the chromosome 4 of S. cereale have translocated to other chromosomes or underwent gene loss.
Plant HSFs have been classified into three classes, HSFA, B, and C, based on the linker length of the DBD and HR-A/B regions and the inserted amino acid residues number into the HR-A/B regions (Scharf et al., 2012). According to this criterion, 14 S. cereale HSF genes were assigned to class A, while 8 and 9 genes were assigned to class B and C, respectively (Figure 2A; Supplementary Table S3). The characteristic feature of different insertion size between HR-A/B regions could be clearly observed in the alignment, with 21, and 7 amino acid residues detected in class A and C HSFs, respectively (Figure 2A). No amino acid was detected between the HR-A/B regions for the sequences in class B. The boundary separating HR-A/B regions at the end of HR-A is conserved within each class but differs among the three classes. For example, a conserved motif of ‘RQEQ’ is readily detected at the end of HR-A region in most class A HSFs, whereas nearly all class C HSFs have a ‘MWRR’ motif. In comparison, the boundary of HR-B is less conserved in each class.
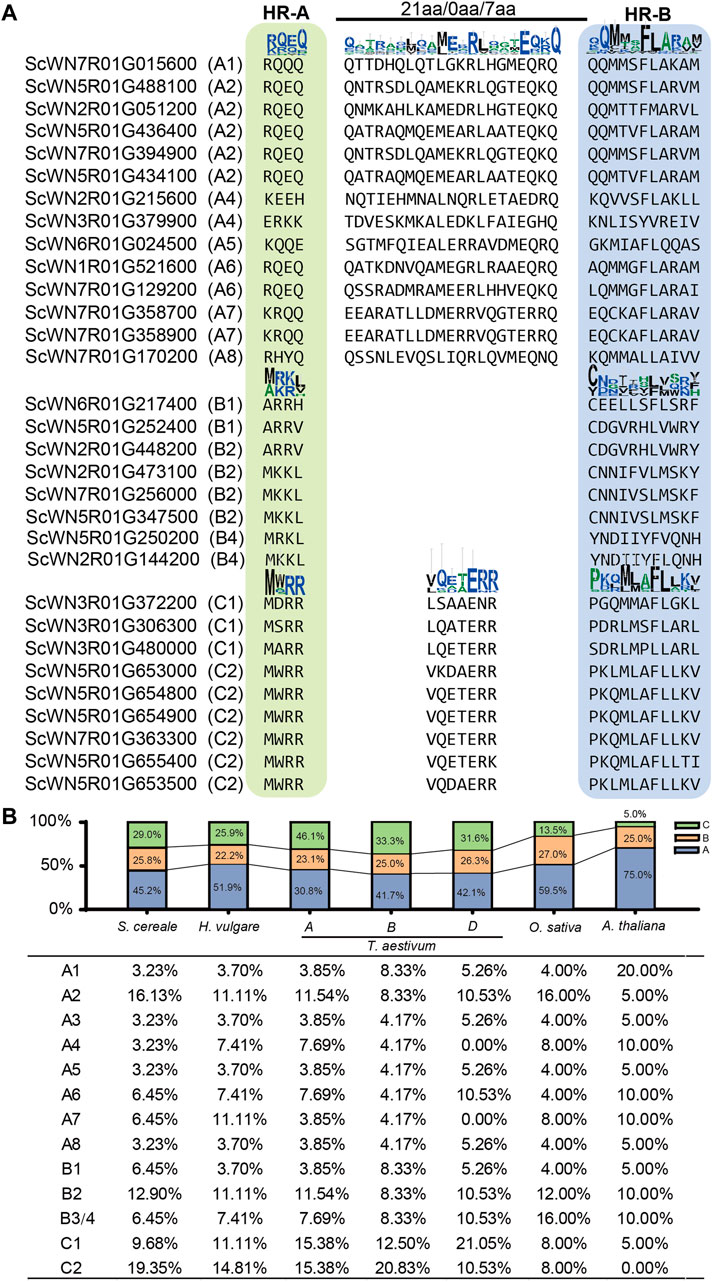
FIGURE 2. Class and subclass division of S. cereale HSF genes. (A). Classification of HSF genes in S. cereale based on protein sequence characteristics. (B). Proportion of different classes and subclasses of HSF genes in S. cereale, H. vulgare, T. aestivum (three subgenomes), O. sativa and A. thaliana respectively.
The proportion of HSF genes in each class only varied slightly among S. cereale (A: 29.0%; B: 25.8%; C: 45.2), H. vulgare (A: 25.9%; B: 22.2%; C: 51.9%) and the subgenome B (A: 33.3%; B: 25%; C: 41.7%) and D (A: 31.6%; B: 26.3%; C: 42.1%) of T. aestivum. In contrast, the wheat A subgenome showed an elevated proportion of class A (46.1%) HSFs and a decreased proportion of class C (30.8%) HSFs. The HSF genes from O. sativa and A. thaliana were also identified and their class compositions were compared with those in the three Triticeae species. The results showed that O. sativa has a more expanded class A (59.5%) and more contracted class C (13.5%) than that in the wheat subgenome A. The A. thaliana genome has the highest ratio of HSFA members (75.0%) and the lowest ratio of HSFC members (5%) among all investigated genomes. This result is consistent with results from previous studies that the class C HSFs have undergone expansion in monocot species (Guo et al., 2016). The proportion of HSFB members is stable among all investigated genomes, ranging from 22.2 to 27.0%. The subclass composition of HSF family also varies cross-species (Figure 2B). For example, subclass C2 occupied the highest proportion of HSFs in S. cereale, H. vulgare and the wheat subgenome A and B, whereas subclass C1, A2 and A1 occupied the highest proportion of HSFs in the wheat subgenome D, O. sativa and A. thaliana, respectively.
To clarify the evolutionary relationship of HSF genes among S. cereale, H. vulgare, and T. aestivum, and trace the evolutionary trajectory during species-speciation of Triticeae, a phylogenetic analysis was performed for HSF genes from the three Triticeae species with those from O. sativa and A. thaliana (Figure 3A). The result showed that members of HSFB and HSFC form two separate monophyletic clades, containing 43 and 47 genes respectively, whereas members of HSFA form a paraphyletic group containing 81 genes. The topology of the HSF phylogeny is also highly consistent with a previous study which showed that HSFC may be diverged from HSFA in a rooted plant HSF phylogeny (Wang et al., 2018). This HSF phylogeny also supports the classification result based on characteristic features of the protein sequences. Tracing the evolutionary history of different HSF classes revealed that the monocot and dicot HSFA genes are inherited from at least 13 ancestral lineages, while the HSFB genes are inherited from 7 ancestral lineages that are presented in the common ancestor of monocot and dicot species (Figure 3A). Interestingly, there is only 1 A. thaliana (At3G24520) gene presented in the HSFC clade which contains 46 monocot sequences (Figure 3A), including 9, 9, 24 and 4 genes from S. cereale, H. vulgare, T. aestivum and O. sativa, suggesting the ancestral HSFC lineage that presented in the common ancestor of monocot and dicot has experienced drastic expansion in monocot species.
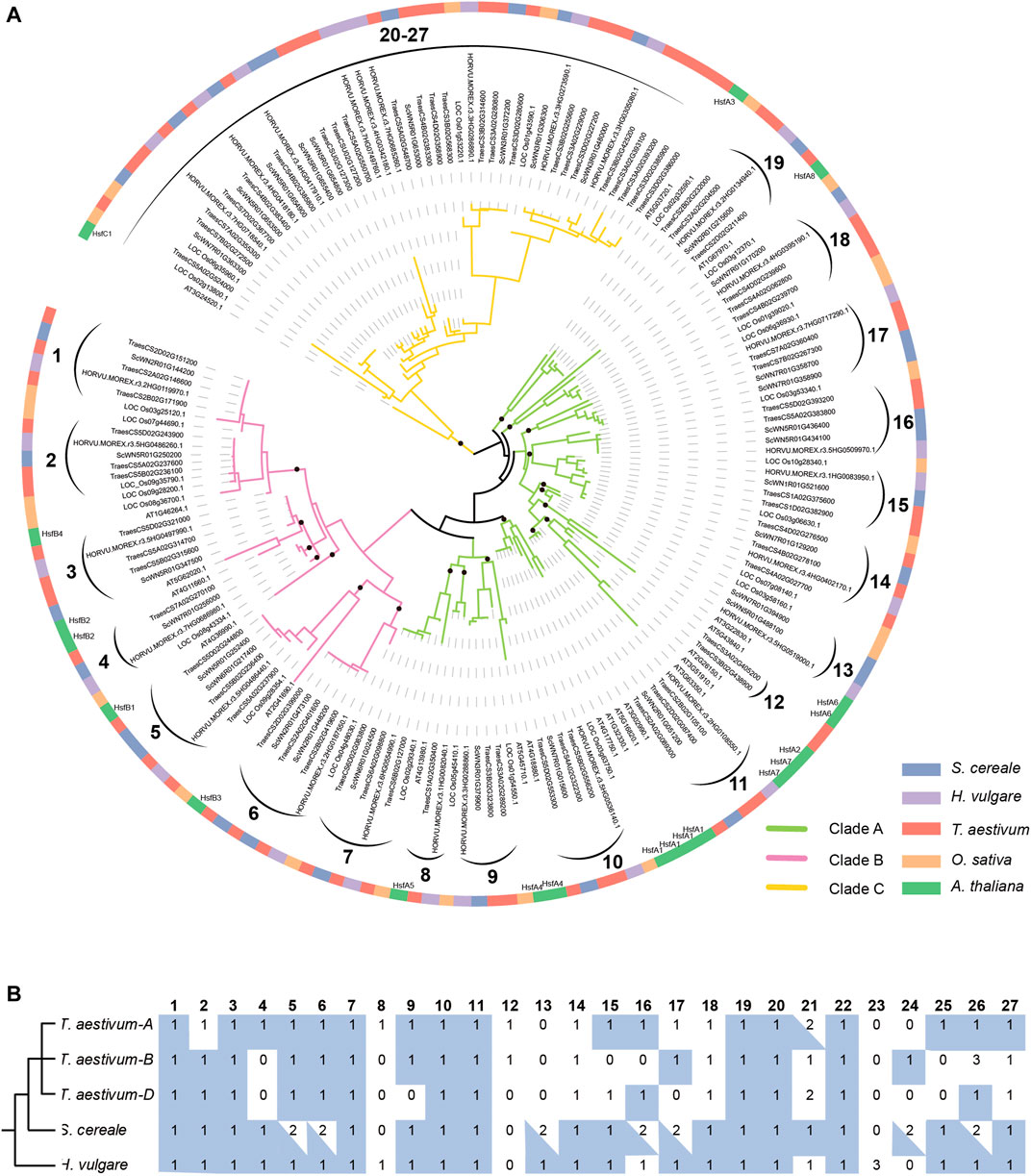
FIGURE 3. Phylogenetic analysis of HSF genes from S. cereale, H. vulgare, T. aestivum, O. sativa and A. thaliana. (A). A phylogeny of HSF genes from S. cereale, H. vulgare, T. aestivum (three subgenomes), O. sativa and A. thaliana based on the amino acid sequences of the HSF domain. Black brackets indicate a group of orthologous HSFs in Triticeae. The identification of class C Triticeae HSF orthologous group were based on an additional phylogeny provided in Supplementary Figure S2, which is constructed by using the full-length protein sequences encoded by HSFC genes. Black dots on the basal of several lineages indicate ancestral HSF lineages of monocots and dicots. (B). The number of HSF genes from different species in each Triticeae HSF orthologous group. A blue rectangle background indicates the HSF gene and HSF genes from at least one other species are in collinear blocks. A blue triangle indicates one of the HSFs in this orthologous group has a collinear relationship with HSF genes from at least one other species.
Further analysis of the phylogeny revealed that HSF genes from the S. cereale, H. vulgare and T. aestivum form 27 independent groups, suggesting that the 21 ancestral HSF genes in the common ancestor of monocot and dicot further diverged into at least 27 Triticeae HSF lineages before the separation of the three Triticeae species (Figure 3A). Interspecific synteny analysis showed that 24 genes of the 27 Triticeae HSF lineages could be detected at syntenic chromosomal blocks from at least two species, providing additional evidence to support the orthologous relationship of genes in each lineage. Interestingly, several interspecies syntenic blocks were detected among chromosome 7 of S. cereale, chromosome 4 or 5 of H. vulgare and/or the subgenome A, B of wheat (Supplementary Figure S3; Supplementary Table S4). The result suggests that chromosome rearrangement may have occurred in the S. cereale genome, causing HSF genes escaped from its chromosome 4.
Different extent of gene duplication and gene loss could be traced from S. cereale, H. vulgare, and the three subgenomes of T. aestivum. Among them, S. cereale has the most duplicated genes for the 27 Triticeae HSF lineages. Seven of the Triticeae HSF lineages have duplicated in the S. cereale genome, including 3, 2 and 2 lineages in the HSF A, B and C classes, respectively (Figures 3A,B; Supplementary Figure S2). The duplicated gene pairs are presented at adjacent, distant region of the same chromosome or at different chromosomes, suggesting tandem, dispersed, small-scale segmental duplications and ectopic duplications have been evolved in generating new HSF copies in the genome. In contrast, H. vulgare and the three subgenomes of T. aestivum each has one duplicated Triticeae HSF lineage. Loss of Triticeae HSF lineages was also detected in the three species. The H. vulgare genome lost 2 Triticeae HSF lineages (lineage 12 and 24), while the S. cereale and the subgenome A, B and D of wheat lost 3, 3, 7 and 9 Triticeae HSF lineages, respectively.
To explore the potential involvement of HSF members in development and resistance to environmental stresses, we analyzed their expression patterns using the public data (Li et al., 2021). An obvious tissue-specific or developmental stage-specific high expression was observed for nearly all HSF members (Figure 4). Furthermore, the expressions of most HSF genes are higher in root than in leaf and stem, except four genes, including 3 from the class A and 1 from the class B, which have the highest expression in stem among the three tissues (Figure 4). During S. cereal development after flowering, four genes show the highest expression in spike 1 week after flowering, whereas five genes have the highest expression in 40-days seed. The expression levels of most HSFs gradually increased during 10-days to 40-days after pollination.
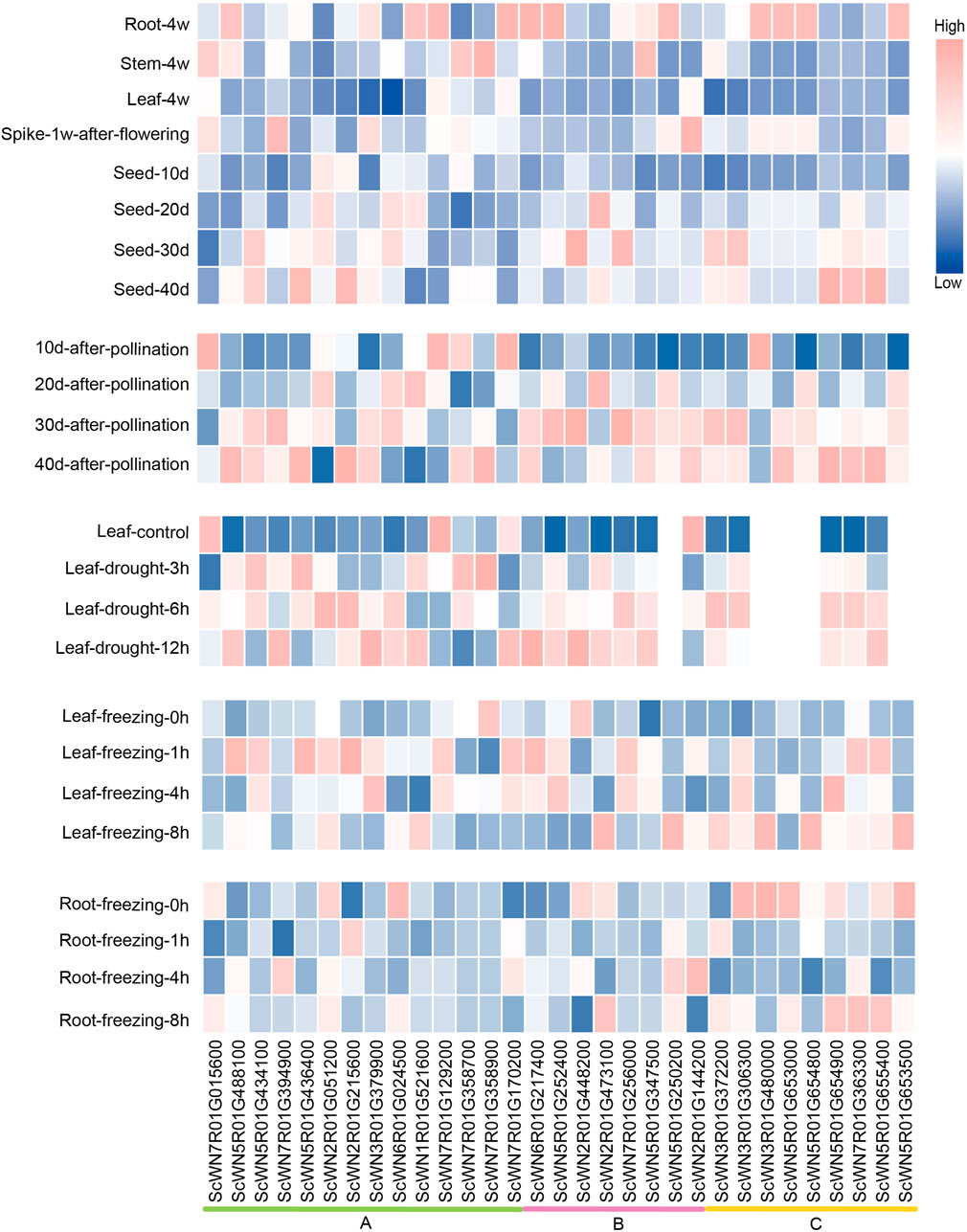
FIGURE 4. Expression analysis of HSF genes in S. cereale across different tissues, development stages and different stress treatments.
We also analyzed the expressions of HSFs in leaves and roots under drought and freezing conditions. The results show that most HSF genes were induced by drought in leaf at least at one time point compared to the control, except ScWN7R01G015600, ScWN7R01G129200 and ScWN2R01G144200, which were down-expressed at the drought condition. A similar pattern was also observed for a large number of HSF genes in freezing leaves, except ScWN7R01G358900 and ScWN2R01G448200, the expressions of which were repressed by freezing treatment. In contrast to the pattern observed in leaves, only a few HSFs showed induced expression upon freezing treatment in root, whereas a considerable number of HSFs genes were down-expressed (Figure 4). The result suggests that leaf and root may adapt different combinations of HSF genes to respond to the freezing stress. We also observed expression divergence of a newly duplicated gene with its parental gene. The ScWN7R01G394900, which is duplicated by ectopic duplication from the ScWN5R01G488100, shows the highest expression in spike, whereas ScWN5R01G488100 only highly expressed in root but not in other all detected tissues. Similar pattern of expression divergence could also be detected in stress treatment. ScWN7R01G394900 was induced in root upon freezing treatment, whereas ScWN5R01G488100 showed no obvious expression alteration. The results suggest functional divergence may have occurred in the gene pairs.
Understanding the molecular mechanisms of how plants respond to abiotic and biotic stresses is important for improving plant tolerance to stresses and crop productivity. HSF family proteins can modulate the expression of genes in responding to heat, cold, salt stresses and pathogens (Andrasi et al., 2021). The present study identified 31 HSF genes from the recently released S. cereale genome and traced the dynamic evolution of the HSF genes in three Triticeae species. S. cereale is an important cereal crop, which has high tolerance to many biotic and abiotic stresses (Li et al., 2021). The identification of HSF genes from S. cereale in this study provides a primary resource for mining functional genes that will facilitate the molecular breeding of S. cereale. Moreover, S. cereal plays an important role in the improvement of wheat breeding (Merker, 1984). It has great potential to expand the genetic variability of T. aestivum. Actually, genes with different functions have been transformed from S. cereal to T. aestivum to improve the growth or resistance to biotic and abiotic stress (Szakacs et al., 2020). The S. cereale HSF genes should also be served as a potential resource for wheat and other crops’ breeding.
Different numbers of HSF genes have been identified from model and crop plants (Wang et al., 2018), however the evolutionary history of HSF genes in a specific plant lineage has rarely been investigated. By incorporating HSF genes from O. sativa and A. thaliana for the phylogenetic analysis, we found that the HSF gene family had undergone extensive expansion prior to the divergence of monocot and dicot, with at least 21 ancestral lineages being recovered. This number exceeded the quantity of the currently defined HSF subclass (Berz et al., 2019), suggesting an updated subclass definition by including HSF genes from more plant genomes is needed to help functional distinguish anciently diverged lineages. The ancestral HSF lineages further expanded before the radiation of Triticeae, with at least 27 ancestral Triticeae HSF lineages could be traced to the common ancestor of the three Triticeae species. These genes were differentially inherited by S. cereale, H. vulgare, and T. aestivum. S. cereale lost three of the ancestral Triticeae HSF lineages that presented in the common ancestor the three Triticeae species, while H. vulgare and T. aestivum both lost two. However, the S. cereale genome has more HSF genes than the H. vulgare genome and the three T. aestivum subgenomes, because of more specie-specific gene duplications and fewer gene loss events have been occurred after it separated with the other two species. A recent study of wheat revealed that both A subgenome and D subgenome have more HSF genes than their wild ancestors (T. urartu, 15 genes and A. tauschii, 16 genes) and concluded that the number of HSF increased in transition from diploidy to hexaploidy (Zhou et al., 2019). However, our data does not support this notion, because only one gene has gained by the wheat A genome after its separation from rye and barley, whereas three gene lost occurred. Similarly, only one gene has gained by the wheat D genome after its separation from rye and barley, whereas 9 gene lost occurred. Therefore, the fewer HSF genes in the genome of T. urartu and A. tauschii than those in wheat A and D subgenomes is more likely due to the increased gene loss in the two genomes.
Gene duplication provides raw resource for gene function innovation (Guo et al., 2019). As shown by the expression data, HSF genes from different ancestral lineages show diverse expression patterns across different tissues, development stages and stress treatments. This is consistent with the previous studies in wheat, suggesting multiple functions of HSF genes (Ye et al., 2020). Recent gene duplications also contribute to plant adaptive evolution (Zhang and Long, 2014). While few HSF gene duplications were detected in the barley genome, the wheat genome obviously benefitted from harboring three subgenomes to have a neatly tripled HSF number. The S. cereale genome adopted a different strategy to amplify its HSF content, with six gene duplications generated by different mechanisms detected. The newly birthed genes provide opportunities for functional innovation of HSF genes in S. cereale. To support our speculation, we found that a pair of genes from one duplication showed inconsistent expression patterns, suggesting functional innovation may have occurred in the recently duplicated S. cereale HSF genes.
In summary, this study presents a complete profile of HSF genes in S. cereale, which is composed by 31 genes from three classes. Chromosomal reorganization may have contributed to the HSF escape from chromosome 4 in S. cereale. Phylogenetic and syntenic analysis supported that at least 27 ancestral HSF lineages were presented in the common ancestor of S. cereale, H. vulgare, and T. aestivum. S. cereale experienced the most HSF gene duplications among the Triticeae A, B, D, R and H genomes. Expression analysis revealed the potential involvement of HSF genes in growth, development and response to abiotic stress of S. cereale, and indicated the functional innovation of recently duplicated HSF genes. The results provide new insights into the evolution of HSF genes in Triticeae and may serve as a resource for Triticeae molecular breeding.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Z-QS and YL conceived and designed the study. X-TL, X-YF, ZZ, and YL obtained and analyzed the data. X-TL and X-YF wrote the manuscript. Z-QS and YL revised the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We greatly appreciate the Frontiers editors and reviewers for handling our manuscript and providing critical suggestions.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.801218/full#supplementary-material
Andrási, N., Pettkó-Szandtner, A., and Szabados, L. (2021). Diversity of Plant Heat Shock Factors: Regulation, Interactions, and Functions. J. Exp. Bot. 72, 1558–1575. doi:10.1093/jxb/eraa576
Anisimova, M., Gil, M., Dufayard, J.-F., Dessimoz, C., and Gascuel, O. (2011). Survey of Branch Support Methods Demonstrates Accuracy, Power, and Robustness of Fast Likelihood-Based Approximation Schemes. Syst. Biol. 60, 685–699. doi:10.1093/sysbio/syr041
Berz, J., Simm, S., Schuster, S., Scharf, K. D., Schleiff, E., and Ebersberger, I. (2019). Heatster: A Database and Web Server for Identification and Classification of Heat Stress Transcription Factors in Plants. Bioinform Biol. Insights 13, 1177932218821365. doi:10.1177/1177932218821365
Bi, H., Zhao, Y., Li, H., and Liu, W. (2020). Wheat Heat Shock Factor TaHsfA6f Increases ABA Levels and Enhances Tolerance to Multiple Abiotic Stresses in Transgenic Plants. Int. J. Mol. Sci. 21, 3121. doi:10.3390/ijms21093121
Boston, R. S., Viitanen, P. V., and Vierling, E. (1996). Molecular Chaperones and Protein Folding in Plants. Plant Mol. Biol. 32, 191–222. doi:10.1007/bf00039383
Charng, Y.-Y., Liu, H.-C., Liu, N.-Y., Chi, W.-T., Wang, C.-N., Chang, S.-H., et al. (2007). A Heat-Inducible Transcription Factor, HsfA2, Is Required for Extension of Acquired Thermotolerance in Arabidopsis. Plant Physiol. 143, 251–262. doi:10.1104/pp.106.091322
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 13, 1194–1202. doi:10.1016/j.molp.2020.06.009
Duan, S., Liu, B., Zhang, Y., Li, G., and Guo, X. (2019). Genome-Wide Identification and Abiotic Stress-Responsive Pattern of Heat Shock Transcription Factor Family in Triticum A L. Bmc Genomics 20, 257. doi:10.1186/s12864-019-5617-1
Edgar, R. C. (2004). MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 32, 1792–1797. doi:10.1093/nar/gkh340
Fan, K., Mao, Z., Ye, F., Pan, X., Li, Z., Lin, W., et al. (2021). Genome-Wide Identification and Molecular Evolution Analysis of the Heat Shock Transcription Factor (HSF) Gene Family in Four Diploid and Two Allopolyploid Gossypium Species. Genomics 113, 3112–3127. doi:10.1016/j.ygeno.2021.07.008
Guo, H., Jiao, Y., Tan, X., Wang, X., Huang, X., Jin, H., et al. (2019). Gene Duplication and Genetic Innovation in Cereal Genomes. Genome Res. 29, 261–269. doi:10.1101/gr.237511.118
Guo, J., Wu, J., Ji, Q., Wang, C., Luo, L., Yuan, Y., et al. (2008). Genome-Wide Analysis of Heat Shock Transcription Factor Families in rice and Arabidopsis. J. Genet. Genomics 35, 105–118. doi:10.1016/s1673-8527(08)60016-8
Guo, M., Liu, J.-H., Ma, X., Luo, D.-X., Gong, Z.-H., and Lu, M.-H. (2016). The Plant Heat Stress Transcription Factors (HSFs): Structure, Regulation, and Function in Response to Abiotic Stresses. Front. Plant Sci. 7, 114. doi:10.3389/fpls.2016.00114
Javed, T., Shabbir, R., Ali, A., Afzal, I., Zaheer, U., and Gao, S. J. (2020). Transcription Factors in Plant Stress Responses: Challenges and Potential for Sugarcane Improvement. Plants (Basel) 9, 491. doi:10.3390/plants9040491
Johnson, L. S., Eddy, S. R., and Portugaly, E. (2010). Hidden Markov Model Speed Heuristic and Iterative HMM Search Procedure. BMC Bioinformatics 11, 431. doi:10.1186/1471-2105-11-431
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 14, 587–589. doi:10.1038/nmeth.4285
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 37, 907–915. doi:10.1038/s41587-019-0201-4
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. doi:10.1093/molbev/msw054
Li, G., Wang, L., Yang, J., He, H., Jin, H., Li, X., et al. (2021). A High-Quality Genome Assembly Highlights rye Genomic Characteristics and Agronomically Important Genes. Nat. Genet. 53, 574–584. doi:10.1038/s41588-021-00808-z
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 30, 923–930. doi:10.1093/bioinformatics/btt656
Ling, Y., Mahfouz, M. M., and Zhou, M. (2021). Pre-mRNA Alternative Splicing as a Modulator for Heat Stress Response in Plants. Trends Plant Sci. 28, 18. doi:10.1016/j.tplants.2021.07.008
Minh, B. Q., Nguyen, M. A. T., and Von Haeseler, A. (2013). Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi:10.1093/molbev/mst024
Mishra, S. K., Poonia, A. K., Chaudhary, R., Baranwal, V. K., Arora, D., Kumar, R., et al. (2020). Genome-Wide Identification, Phylogeny and Expression Analysis of HSF Gene Family in Barley during Abiotic Stress Response and Reproductive Development. Plant Gene 23, 100231. doi:10.1016/j.plgene.2020.100231
Mishra, S. K., Tripp, J., Winkelhaus, S., Tschiersch, B., Theres, K., Nover, L., et al. (2002). In the Complex Family of Heat Stress Transcription Factors, HsfA1 Has a Unique Role as Master Regulator of Thermotolerance in Tomato. Genes Dev. 16, 1555–1567. doi:10.1101/gad.228802
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 32, 268–274. doi:10.1093/molbev/msu300
Nishizawa, A., Yabuta, Y., Yoshida, E., Maruta, T., Yoshimura, K., and Shigeoka, S. (2006). Arabidopsis Heat Shock Transcription Factor A2 as a Key Regulator in Response to Several Types of Environmental Stress. Plant J. 48, 535–547. doi:10.1111/j.1365-313x.2006.02889.x
Ogawa, D., Yamaguchi, K., and Nishiuchi, T. (2007). High-Level Overexpression of the Arabidopsis HsfA2 Gene Confers Not Only Increased Themotolerance but Also Salt/osmotic Stress Tolerance and Enhanced Callus Growth. J. Exp. Bot. 58, 3373–3383. doi:10.1093/jxb/erm184
Peck, S., and Mittler, R. (2020). Plant Signaling in Biotic and Abiotic Stress. J. Exp. Bot. 71, 1649–1651. doi:10.1093/jxb/eraa051
Poonia, A. K., Mishra, S. K., Sirohi, P., Chaudhary, R., Kanwar, M., Germain, H., et al. (2020). Overexpression of Wheat Transcription Factor (TaHsfA6b) Provides Thermotolerance in Barley. Planta 252, 53. doi:10.1007/s00425-020-03457-4
Saur, I. M. L., Panstruga, R., and Schulze-Lefert, P. (2021). NOD-Like Receptor-Mediated Plant Immunity: from Structure to Cell Death. Nat. Rev. Immunol. 21, 305–318. doi:10.1038/s41577-020-00473-z
Scharf, K.-D., Berberich, T., Ebersberger, I., and Nover, L. (2012). The Plant Heat Stress Transcription Factor (Hsf) Family: Structure, Function and Evolution. Biochim. Biophys. Acta (Bba) - Gene Regul. Mech. 1819, 104–119. doi:10.1016/j.bbagrm.2011.10.002
Shao, Z.-Q., Zhang, Y.-M., Hang, Y.-Y., Xue, J.-Y., Zhou, G.-C., Wu, P., et al. (2014). Long-Term Evolution of Nucleotide-Binding Site-Leucine-Rich Repeat Genes: Understanding Gained from and Beyond the Legume Family. Plant Physiol. 166, 217–234. doi:10.1104/pp.114.243626
Suyama, S., Abe, S., Inoue, Y., Toukairin, A., Ohtake, Y., and Ohkubo, Y. (2006). The Involvement of Transferrin in the Uptake of Iron-59 by Hepatocytes of Carbon Tetrachloride-Damaged Rats. Biol. Pharm. Bull. 29, 1387–1390. doi:10.1248/bpb.29.1387
Szakács, É., Szőke-Pázsi, K., Kalapos, B., Schneider, A., Ivanizs, L., Rakszegi, M., et al. (2020). 1RS Arm of Secale Cereanum 'Kriszta' Confers Resistance to Stripe Rust, Improved Yield Components and High Arabinoxylan Content in Wheat. Sci. Rep. 10, 1792. doi:10.1038/s41598-020-58419-3
Wang, X., Shi, X., Chen, S., Ma, C., and Xu, S. (2018). Evolutionary Origin, Gradual Accumulation and Functional Divergence of Heat Shock Factor Gene Family with Plant Evolution. Front. Plant Sci. 9, 71. doi:10.3389/fpls.2018.00071
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: A Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 40, e49. doi:10.1093/nar/gkr1293
Wani, S. H., Anand, S., Singh, B., Bohra, A., and Joshi, R. (2021). WRKY Transcription Factors and Plant Defense Responses: Latest Discoveries and Future Prospects. Plant Cel Rep 40, 1071–1085. doi:10.1007/s00299-021-02691-8
Ye, J., Yang, X., Hu, G., Liu, Q., Li, W., Zhang, L., et al. (2020). Genome-Wide Investigation of Heat Shock Transcription Factor Family in Wheat (Triticum A L.) and Possible Roles in Anther Development. Int. J. Mol. Sci. 21, 608. doi:10.3390/ijms21020608
Zhang, Y. E., and Long, M. (2014). New Genes Contribute to Genetic and Phenotypic Novelties in Human Evolution. Curr. Opin. Genet. Develop. 29, 90–96. doi:10.1016/j.gde.2014.08.013
Zhou, M. Q., Shen, C., Wu, L. H., Tang, K. X., and Lin, J. (2011). CBF-dependent Signaling Pathway: A Key Responder to Low Temperature Stress in Plants. Crit. Rev. Biotechnol. 31, 186–192. doi:10.3109/07388551.2010.505910
Zhou, M., Zheng, S., Liu, R., Lu, J., Lu, L., Zhang, C., et al. (2019). Genome-Wide Identification, Phylogenetic and Expression Analysis of the Heat Shock Transcription Factor Family in Bread Wheat (Triticum A L.). BMC Genomics 20, 505. doi:10.1186/s12864-019-5876-x
Keywords: S. cereale, HSF gene, stress tolerance, evolution, functional diversity
Citation: Li X-T, Feng X-Y, Zeng Z, Liu Y and Shao Z-Q (2021) Comparative Analysis of HSF Genes From Secale cereale and its Triticeae Relatives Reveal Ancient and Recent Gene Expansions. Front. Genet. 12:801218. doi: 10.3389/fgene.2021.801218
Received: 25 October 2021; Accepted: 08 November 2021;
Published: 23 November 2021.
Edited by:
Pengtao Ma, Yantai University, ChinaReviewed by:
Jiancai Li, Shanghai Institutes for Biological Sciences (CAS), ChinaCopyright © 2021 Li, Feng, Zeng, Liu and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhu-Qing Shao, emh1cWluZ3NoYW9Abmp1LmVkdS5jbg==; Yang Liu, bTE4ODQ1MDQzMTg3QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.