 Yuxiang Zhang1,2†
Yuxiang Zhang1,2† Peng Li1,2†Nachuan Liu1,2†Tao Jing1,2Zhiyong Ji3
Peng Li1,2†Nachuan Liu1,2†Tao Jing1,2Zhiyong Ji3 Chao Yang1,2
Chao Yang1,2 Liangyu Zhao1,2Ruhui Tian1,2Huixing Chen1,2Yuhua Huang1,2Erlei Zhi1,2
Liangyu Zhao1,2Ruhui Tian1,2Huixing Chen1,2Yuhua Huang1,2Erlei Zhi1,2 Ningjing Ou3Haowei Bai1,2
Ningjing Ou3Haowei Bai1,2 Yuchuan Zhou4*
Yuchuan Zhou4* Zheng Li1,2*Chencheng Yao1,2*
Zheng Li1,2*Chencheng Yao1,2*- 1Depart. of Andrology, Center for Men’s Health, Urologic Medical Center, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Shanghai Key Lab of Reproductive Medicine, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 3State Key Lab of Reproductive Medicine, Nanjing Medical University, Nanjing, China
- 4The International Peace Maternity and Child Health Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
Non-obstructive azoospermia (NOA) is the most severe disease in male infertility, but the genetic causes for the majority of NOA remain unknown. FANCM is a member of Fanconi Anemia (FA) core complex, whose defects are associated with cell hypersensitivity to DNA interstrand crosslink (ICL)-inducing agents. It was reported that variants in FANCM (MIM: 609644) might cause azoospermia or oligospermia. However, there is still a lack of evidence to explain the association between different FANCM variants and male infertility phenotypes. Herein, we identified compound heterozygous variants in FANCM in two NOA-affected brothers (c. 1778delG:p. R593Qfs*76 and c. 1663G > T:p. V555F), and a homozygous variant in FANCM (c. 1972C > T:p. R658X) in a sporadic case with NOA, respectively. H&E staining and immunohistochemistry showed Sertoli cell-only Syndrome (SCOS) in the three patients with NOA. Collectively, our study expands the knowledge of variants in FANCM, and provides a new insight to understand the genetic etiology of NOA.
Introduction
Infertility is a common reproductive disorder affecting about 8–12% of couples worldwide. Male-infertility-associated factors are found in approximately half of these cases (Agarwal et al., 2021). Azoospermia, which is defined as the complete absence of spermatozoa in the ejaculate, and accounts for 10–15% of male infertility cases. In terms of azoospermia, 70% of cases represent non-obstructive azoospermia (NOA) with the absence or reduction of germ cells owing to testicular atrophy (Stephen and Chandra, 2006). Sertoli cell-only Syndrome (SCOS) is the most severe type of NOA with impairment of spermatogenesis, which is characterized as complete loss of male germ cells in the seminiferous tubules with only Sertoli cells retained (Ezeh et al., 1998).
NOA is heterogeneous in etiology, including Y chromosome microdeletions, chromosomal abnormalities, hypogonadism, testicular tumor, cryptorchidism, varicocele, improper drug administration, and single gene mutations (Minhas et al., 2021). Recently, several causative genes have been identified by whole-exome sequencing (WES) in NOA pedigrees, such as DNA Meiotic Recombinase 1 (DMC1 MIM: 602721), Stromal Antigen 3 (STAG3 MIM:608489), Testis Expressed 11 (TEX11 MIM: 300311), Shortage In Chiasmata 1 (SHOC1, MIM: 618038), Synaptonemal Complex Central Element Protein 1 (SYCE1 MIM: 611486), Meiosis Specific With OB-Fold (MEIOB MIM: 617670), Coiled-Coil Domain Containing 155 (CCDC155 MIM: 618125), Testis Expressed 14 (TEX14 MIM: 605792), Testis Expressed 15 (TEX15 MIM: 605795), and X-Ray Repair Cross Complementing 2 (XRCC2 MIM: 600375) (Maor-Sagie et al., 2015; Okutman et al., 2015; Yang et al., 2015; Gershoni et al., 2017; Fakhro et al., 2018; He et al., 2018; Yang et al., 2018; Riera-Escamilla et al., 2019; Minhas et al., 2021; Yao et al., 2021). Recently, it was demonstrated that compound heterozygous FANCM variants (c. 1491dupA: p. Gln498Thrfs*7 and c. 4387–10A > G: p. Arg1436_ Ser1437insLeuLeu*) were identified in two NOA-affected brothers in an Estonian family. Histopathological analysis showed SCOS in the proband. Another two homozygous variants (c. 5101C > T: p. Gln1701* and c. 5791C > T: p. Arg 1931*) were identified in two sporadic NOA-affected cases (Kasak et al., 2018). Furthermore, bi-allelic FANCM pathogenic variants (PVs) were also reported in oligoasthenospermic or azoospermic patients. It was reported that a homozygous FANCM frameshift variant (c. 1946_1958del: p. P648Lfs*16) was identified in three brothers with idiopathic infertility in a family. Two affected cases were diagnosed with oligoasthenospermia, whereas another was diagnosed with azoospermia (Yin et al., 2019). However, there is still no evidence to explain the association between different types of FANCM variants and male infertility phenotypes.
The present study reports on three patients with SCOS and NOA due to novel bi-allelic variants in FANCM. Compound heterozygous FANCM variants (c. 1778delG: p. R593Qfs*76 and c. 1663G > T: p. V555F) were identified in two NOA-affected brothers. Another homozygous FANCM variant (c. 1972C > T: p. R658X) in a sporadic NOA-affected patient. Hematoxylin and eosin (H&E) and immunofluorescence (IF) staining showed SCOS in all three NOA-affected patients. Together, our study ascertained FANCM as the candidate gene for SCOS, and provided novel foci for NOA genetic counseling.
Materials and Methods
Subjects
All three patients with male infertility were diagnosed with idiopathic NOA at the Department of Andrology, Center for Men’s Health, Urologic Medical Center, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine. Family histories were collected. Furthermore, 479 patients with sporadic NOA-affected patients were enrolled in this study. Reproductive congenital diseases such as Klinefelter syndrome and genomic AZF deletions, or other azoospermia-associated factors including varicocele, radiation, chemotherapy, orchitis, cryptorchidism, and testicular cancer were excluded for these patients with NOA.
This study was approved by the Institutional Ethical Review Committee of Shanghai General Hospital, Shanghai Jiao Tong University (Permit Number: 2020SQ199). Written informed consent was obtained from the individual(s) and/or minor(s) legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
WES and Variant Filtration
Genomic DNA was extracted from blood samples using the DNeasy Blood and Tissue Kit (Qiagen). The quality and size of libraries were measured by 2,100 Bioanalyzer High Sensitivity DNA Assay (Agilent Technologies).
For next-generation sequencing, the qualified libraries were applied to 2 × 150-bp paired-end sequencing on the Illumina NovaSeq platform (Illumina, San Diego, and United States). Raw data files were obtained from Novaseq 6,000 and then were demultiplexed and converted to fastq format using bcl2fastq software for downstream analysis. Adapters and reads with low quality were trimmed using fastp software. The BAM files were obtained by aligning the sequence reads to the reference (hg19/GRCH37, fasta format) with the use of the SpeedSeq. Additionally, duplicate reads were flagged in the BAM files to prevent downstream variant call errors, sample contamination, and swaps using VerifyBamID. Then the UnifiedGenotyper tool of GAT was used to call SNVs. The variants were annotated using Annovar software. During the annotation, several public databases such as Clinvar, gnomAD, and dbNSFP, etc. were used. Variants with allele frequencies higher than 1% in any public databases (ExAC Browser and gnomAD) were excluded. Because autosomal recessive or X-linked inheritance was assumed for spermatogenic defect, genes with two alleles of potentially deleterious missense mutations (SIFT, PolyPhen-2, and MutationTaster), or loss-of-function (LoF) variants were kept for further analysis. Moreover, we compared candidate genes with human germline-enriched genes in the database and known pathogenic genes for azoospermia in mice. The aforementioned sequencing and bioinformatic analyses were conducted together with the Nuprobe company (Shanghai, China). The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Sanger Sequencing
Validation of FANCM variants in the patients with NOA was performed with Sanger sequencing. Genomic DNA was extracted from peripheral blood using the TIANamp Genomic DNA Blood Kit (TIANGEN, China, and Beijing) according to the manufacturer’s instructions. The exon regions of FANCM were amplified with PCR primers (Supplementary Table S1). The PCR products were bidirectionally sequenced through a 3730xl DNA Analyzer (Applied Biosystems, California, and United States).
H&E Staining
The testicular biopsies were obtained from the NOA-affected cases and the obstructive azoospermia (OA)-affected patient as the control. The testicular tissues were fixed in 4% paraformaldehyde solution overnight, embedded in paraffin, and sectioned at 5 μm thickness. Paraffin sections were dewaxed with xylene for 30 min, and then rehydrated sequentially in 95, 90, 85, and 70% ethanol each for 10 min, submerged in Phosphate-Buffered Saline (PBS) solution for 10 min, and stained with hematoxylin and eosin according to standard protocols (catalogue number: ab245880, Abcam, Cambridge, and United Kingdom). All the staining sections were captured by phase-contrast microscope to observe the structural changes of testicular tissues (Leica).
Immunofluorescence (IF) Staining
The testis tissues were dewaxed in xylene and rehydrated in descending alcohol. Antigen retrieval was performed by incubation with 10 mM sodium citrate pH 6.0, 0.05% Tween-20 at 90–98°C for 30 min. The slides were cooled to room temperature and washed with PBS-T (PBS + 0.5% Tween-20). Non-specific antibody binding was blocked in 5% donkey serum for 1 h at room temperature and then incubated overnight with anti-DEAD-Box Helicase 4 (DDX4; dilution: 1:500; catalogue number: ab13840, Abcam), anti-Ubiquitin C-Terminal Hydrolase L1 (UCHL1; dilution: 1:500; catalogue number: ab8189, Abcam), anti-Androgen Receptor (AR; dilution: 1:100; catalogue number: sc-7305, Santa Cruz), and anti-SRY-Box Transcription Factor 9 (SOX9; dilution: 1:400; catalogue number: AB5535, Millipore) at 4°C in a humidified chamber. After three washes in PBS-T, secondary antibodies were applied for 1 h at room temperature. Subsequently, the slides were washed three times in PBS-T and mounted with antifade medium with 4,6-diamidino-2-phenylindole (DAPI; Vector). The images were captured by a fluorescence microscope (Leica).
Results
Clinical Characteristics
The two brothers and the sporadic patient were diagnosed with idiopathic NOA at our center. There was no family history of consanguinity or fertility problems and no chronic diseases in all three NOA-affected cases (Figure 1). None of the patients had a history of cryptorchidism, hypogonadism, cancer, and drinking, or smoking. Physical examination revealed normal development of penis, epididymis, prostate, scrotum, and vas deferens. There was no varicocele in the three patients.
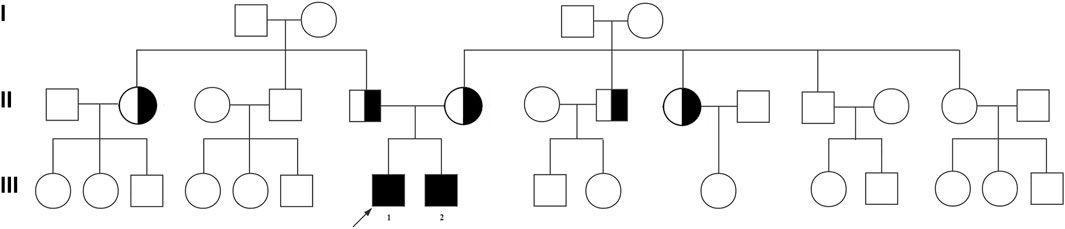
FIGURE 1. Pedigree of the affected family with FANCM variants. Family pedigree with black fill denoting NOA-affected patient P6649 (Ⅲ-1) and his elder brother (Ⅲ-2). The arrowheads indicate the probands.
The two brothers (P6612, Ⅲ-1 and P6612-B, Ⅲ-2) had a history of male infertility for 4 and 5 years respectively. Physical examination showed the small size of the testis (10 ml in the proband, and 8 ml in the elder brother, both sides). Routine semen analyses (three times) revealed normal volume but no sperm in the ejaculate, indicating complete azoospermia. Laboratory examination revealed elevated FSH levels (14.29 IU/L and 12.85 IU/L versus 1.5–12.5 IU/L in normal). Similarly, for another sporadic NOA-affected case (P6612) with a history of male infertility for 2 years (23 years old), the results of physical examination and semen analyses (three times) suggested normal testis volume (12 ml, both sides), however, complete azoospermia. Laboratory examination also revealed elevated FSH levels (24.95 IU/L versus 1.5–12.5 IU/L in normal). All patients have 46,XY karyotypes without microdeletions in the Y chromosome. The clinical characteristics mentioned above are summarized in Table 1.
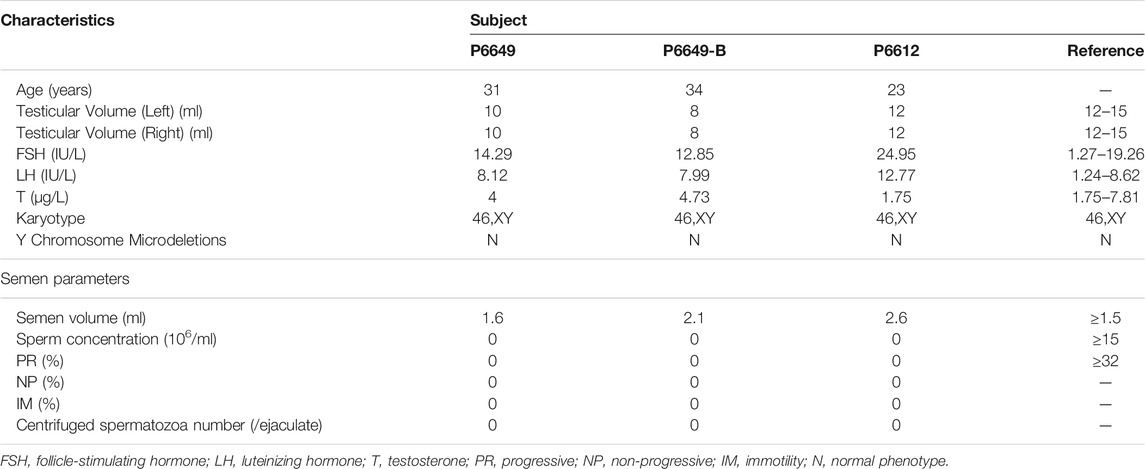
TABLE 1. Clinical and semen characteristics in patients with bi-allelic FANCM variants.
The two brothers with NOA and the NOA-affected sporadic cases were subjected to microsurgical testicular sperm extraction (micro-TESE) operation at the Center for Men’s Health, Urologic Medical Center, Shanghai General Hospital, and Shanghai Jiao Tong University School of Medicine. Histopathological analysis revealed SCOS phenotype in all three NOA-affected cases.
WES Revealed Bi-Allelic FANCM Variants in the NOA-Affected Patients
WES was performed in the two NOA-affected brothers (P6649 and P6649-B) to identify the potential PV for their male infertility. After the genetic analyses pipeline, compound heterozygous variants in FANCM (c. 1778delG: p. R593Qfs*76 and c. 1663G > T: p. V555F) were assumed as the most likely primary cause of spermatogenic defects in the two brothers (Figures 2A–C). Furthermore, we conducted WES in the 479 patients with NOA recruited from our center. Intriguingly, a sporadic individual (P6612) with a homozygous variant (c. 1972C > T: p. R658X) in FANCM was identified, and it was verified by Sanger sequencing (Figure 2D). Although this could be a homozygous LoF variant in FANCM, it is also possible that the individual carries a heterozygous R658X variant on the one allele and a heterozygous deletion in FANCM on the other allele. Thus, we employed the WES data for Copy Number Variant (CNV) analysis in this case according to the protocol as described previously (Yao et al., 2017; Marchuk et al., 2018; Tsuchida et al., 2018), and no evidence of FANCM deletion has been obtained in this NOA-affected case.
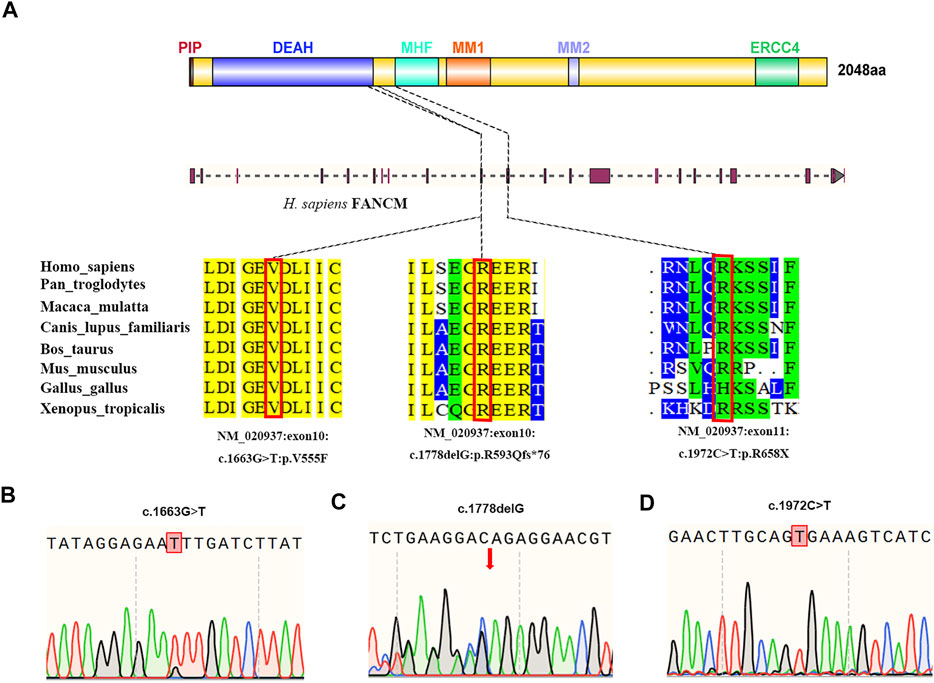
FIGURE 2. Structure of the FANCM protein and the genetic context of the FANCM variants detected in three cases diagnosed with NOA. (A) The positions of FANCM variants are shown and the conservation of the FANCM variants was analyzed. (B–D) Validation of FANCM variants identified by WES using Sanger sequencing in two brothers from the same family [(B,C), P6649, and P6649-B] and a sporadic patient [(D), P6612].
The Impact of FANCM Variants
All three variants are extremely rare and are absent in the ExAC database and 1,000 Genomes Project database, respectively. It has been identified that FANCM contains six independent domains with separable functions so far, including PIP-box (aa 5–12), DEAH domain (aa 77–590), MHF binding domain (aa 661–800), MM1domain (aa 826–967), MM2 domain (aa 1,218–1,251), and ERCC4 domain (aa 1818–1956) (Domingues-Silva et al., 2019; O'Rourke et al., 2019). Correspondingly, the FANCM variants (c. 1778delG: p. R593Qfs*76 and c. 1972C > T: p. R658X) resulted in truncated FANCM proteins without expression of MHF binding domain, MM1 domain, MM2 domain, and ERCC4 domain. Furthermore, the variant (c. 1663G > T: p. V555F) was predicted to be deleterious by in silico analysis, including SIFT, PoyPhen-2, and MutationTaster analysis. Altogether, all three FANCM variants were assumed to be deleterious in the study, and the results are summarized in Table 2.
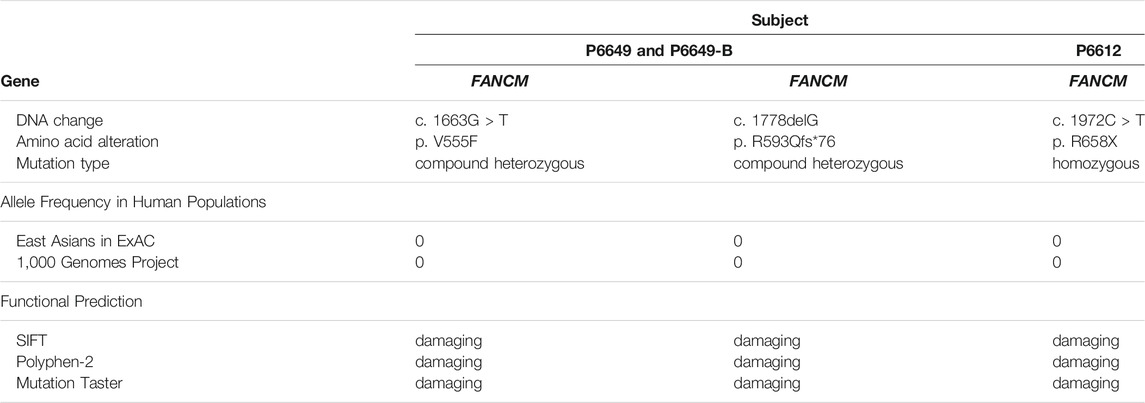
TABLE 2. Bi-allelic FANCM variants identified in the three NOA-effected cases.
SCOS Phenotype in the Subjects With Bi-Allelic Variants in FANCM
SCOS phenotype was verified in NOA-affected patients with bi-allelic FANCM variants using H&E and IF staining. H&E staining revealed that all germ cells were lost except for a single layer of Sertoli cells at the basement membrane within the seminiferous tubules of the two brothers (P6649 and P6649-B) with compound heterozygous FANCM variants (NM_020,937: c. 1778delG: p. R593Qfs*76) (Figures 3A,B). In contrast, there was normal spermatogenesis in the patients with OA as the positive control (Figure 3D). IF staining results showed that there were no expressions of UCHL1 (a marker of spermatogonia) and DDX4 (a hallmark of germ cells) in the testis of the two brothers, whereas all of the cells in seminiferous tubules were positive for AR or SOX9 (markers of Sertoli cells) (Figure 4). However, there was normal spermatogenesis in the OA-affected patients as shown by the positive signals of germ cells as well as somatic cells. Consistent with the two brothers in the pedigree, H&E staining also showed SCOS phenotype in the sporadic patient (P6612) with homozygous FANCM variant (NM_020937: c. 1972C > T: p. R658X) (Figure 3C). Altogether, these results suggested that bi-allelic variants in FANCM were associated with the SCOS phenotype.
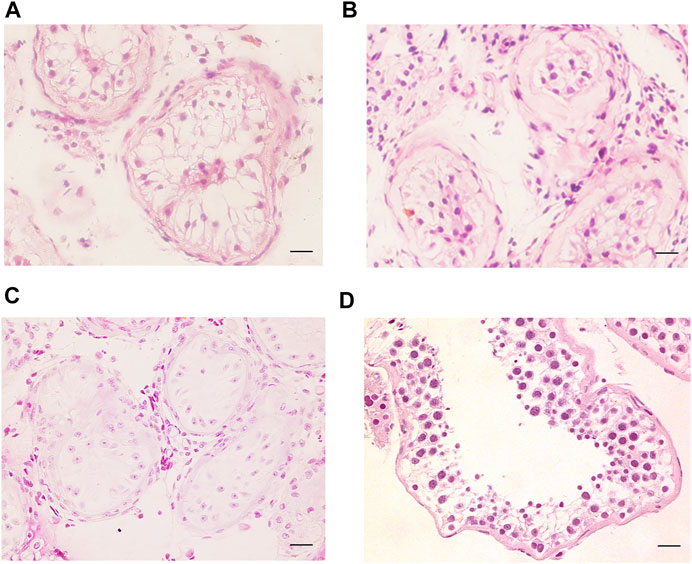
FIGURE 3. H&E staining of cross-sections of testis in NOA-affected patients. (A–C) H&E staining of cross-sections of testicular biopsy in the patient with NOA (A), P6649; (B), P6649-B; (C), P6612). (D) H&E staining of cross-sections of seminiferous tubule in the OA-affected patient as the positive control. Scale bars = 20 μm in (A–D).
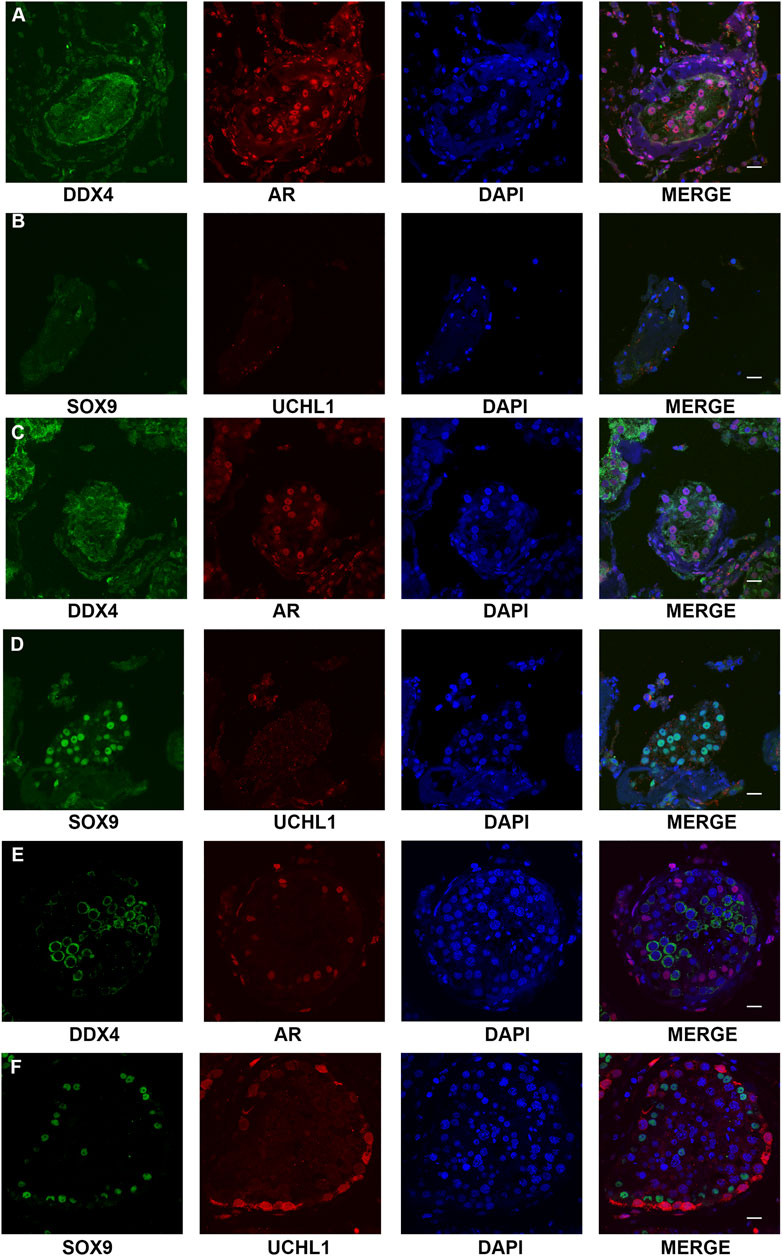
FIGURE 4. Expression of the markers of germ cells and somatic cells respectively in the testis of NOA-affected patients and patients with OA as the positive control. (A–D) IF staining results showed that there were no expressions of UCHL1 (a marker of spermatogonia) and DDX4 (hallmark of germ cells) in the testis of the proband (A,B) and his elder brother (C,D), whereas all of the cells in seminiferous tubules were positive for AR or SOX9 (markers of Sertoli cells-affected). (E,F) IF staining results showed that there was normal spermatogenesis in the OA patients. Scale bars = 20 μm in (A–E).
Discussion
In the present study, it is demonstrated that bi-allelic variants in FANCM could result in SCOS, and NOA. In the Chinese pedigree, compound heterozygous FANCM variants (c. 1778delG: p. R593Qfs*76 and c. 1663G > T: p. V555F) were identified in two NOA-affected brothers. Both variants were absent in the ExAC and 1,000 Genomes Project database. SIFT, PoyPhen-2, and MutationTaster analysis revealed that both variants were potentially deleterious. Furthermore, we identified another homozygous FANCM variant (c. 1972C > T: p. R658X) in a sporadic NOA-affected patient. SCOS phenotype was ascertained using H&E and IF staining in all three NOA-affected patients.
Fanconi Anemia (FA) is a rare genetic disease with bone marrow failure, organ defects, physical abnormalities, an increased risk of certain cancers, and cell hypersensitivity to DNA ICL-inducing agents (Deans and West, 2009; Neidhardt et al., 2017). FA core complex is composed of FANC-A, -B, -C, -E, -F, -G, -L, -M, FAAP24, and FAAP100 (Ciccia et al., 2007; Collis et al., 2008; Yang et al., 2013), which has been determined to play diverse and complex biological roles including involvement in DNA replication, repair, and in anti-crossover function to maintain genomic stability (Yan et al., 2010; Xue et al., 2015). Notably, FA mouse models show sub-fertile phenotypes in both genders, from reduced fertility to complete sterility (Tsui and Crismani, 2019), and suggesting that the FA pathway is associated with reproductive disorders.
FANCM is the most conserved member of the FA core complex, and it is reported that FANCM could affect different stages in male gametogenesis, including primordial germ cells (PGCs) proliferation, maintenance of spermatogonial stem cells (SSCs), and spermiogenesis. Fancm-deficient mice displayed a drastically decreased rate of proliferation of PGCs, a progressive reduction of SSCs, and partial maturation arrest at the round spermatid stage (Bakker et al., 2009; Yin et al., 2019). More seriously, some subsets of seminiferous tubules showed SCOS phenotype, and which is supposed to result from defective repair of ICL occurring in DNA replication of germ cells.
Consistent with the mice model, bi-allelic FANCM PV was also associated with both male and female infertility in humans. It was reported that females with bi-allelic LoF variants in FANCM presented with premature ovarian failure (POI) (Yang et al., 2020). Correspondingly, Kasak, et al. revealed that bi-allelic FANCM PV could result in SCOS (Kasak, et al., 2018). Recently, another study identified a homozygous FANCM PV in three brothers with idiopathic male infertility. Two suffered from oligoasthenospermia and another sporadic case was diagnosed with NOA (Yin et al., 2019). Similarly, in the current study, and novel variants in FANCM were identified in three patients with SCOS. However, none of our three patients has developed any systemic diseases or cancers like previous patients (Bogliolo et al., 2018; Yin et al., 2019). This discrepancy might be attributed to the difference of PV that is independent of the canonical FA pathway.
It is noteworthy that the age of the three NOA-affected patients in our study (P6649, 31 years; P6649-B, 34 years; P6612, 23 years) is younger than those in Bogliolo et al. Thus, future cancer incidence of the patients should be monitored.
FANCM protein has six critical functional domains including PIP-box, DEAH domain, MHF binding domain, MM1 domain, MM2 domain, and ERCC4 domain as mentioned above. Therein, the DEAH is the primary component that allows FANCM to transfer the core complex along with DNA during repair (Xue et al., 2015). It should be noted that the missense variant FANCM (c. 1663G > T) in this study is located in the DEAH domain, thus probably affecting the recruitment of the FA core complex. The MHF-FANCM complex could maintain genomic stability during cell division by inducing FACNM to bind to double-strand DNA and interact with chromatin (Yang et al., 2013). Furthermore, it has been reported that the MM1 domain could interact with FANCF within the FA core complex (Deans and West, 2009), and the MM2 domain could interact with RecQ-Mediated genome Instability protein 1 (RMI1) (Deans and West, 2009). In addition, the ERCC4 domain is necessary for the FAAP24 protein-binding function, and forming a heterodimer that is critical in protecting cells from ICL (Tao et al., 2012). Therefore, the LoF variants in FANCM (P6649 and P6649-B: c. 1778delG: p. R593Qfs*76; P6612: c. 1972C > T: p. R658X) may result in the instability of genome and a plethora of ICLs especially in PGCs and SSCs stages, which are supposed to be linked with the SCOS phenotypes of the three NOA-affected patients in our study. In short, the specific domain of FANCM accounts for different functions, and thus different PVs may cause the discrepancy of phenotypes. Moreover, the ICL repair pathway is indispensable in the DNA replication of germ cells, and can be affected by a variety of environmental agents, which have also been proposed as explanations for the clinical variability in male patients suffering from infertility.
However, there are also limitations in the current study. The missense variant FANCM (c. 1663G > T) in the two NOA-affected brothers, which is assumed to be deleterious by bioinformatics analysis, is located at the DEAH domain, and probably affecting the recruitment of the FA core complex. Thus, further studies should be performed to reveal the pathogenic mechanism of the variant (c. 1663G > T) through the FANCMV555F mutant mice model.
Conclusion
In conclusion, we identified compound heterozygous variants in FANCM in two NOA-affected brothers and a homozygous LoF variant in FANCM in a sporadic case with NOA, respectively. H&E and immunohistochemistry showed SCOS in the three patients with NOA. Our study expands the knowledge of variants in FANCM and provides a new insight to understand the genetic etiology of NOA.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors
Ethics Statement
This study was approved by the Institutional Ethical Review Committee of Shanghai General Hospital, Shanghai Jiao Tong University (Permit Number 2020SQ199). Written informed consent was obtained from the individual(s) and/or minor(s) legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
Conceptualization: YZ, PL, CY, ZL, and YZ; Data curation: NL, TJ, CY, and RT; Formal Analysis: LZ, ZJ, HC, and YH; Funding acquisition: CY, ZL, and YZ; Investigation: EZ, NO, and HB; Project administration: YZ and PL; Supervision: NL and TJ; Visualization: PL; Writing—original draft: YZ and PL; Writing—review and editing: CY, ZL, and YZ.
Funding
This work was supported by National Natural Science Foundation of China (82001530, 81903417, 81871215, and 81971368), Clinical Research Innovation Plan of Shanghai General Hospital (KD007-ly01 and CTCCR-C04), Clinical Research Plan of SHDC (SHDC2020CR3077B), and the Key Project of Research and Development of Ningxia Hui Autonomous Region of China (2020BFH02002), and Shanghai Sailing Program (20YF1439500 and 20YF1453700).
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Yan Hong, Wei Chen, Xiaobo Wang, Cunzhong Deng, and Jianxiong Zhang for the coordinated study recruitment and sample collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.799886/full#supplementary-material
References
Agarwal, A., Baskaran, S., Parekh, N., Cho, C.-L., Henkel, R., Vij, S., et al. (2021). Male Infertility. The Lancet 397, 319–333. doi:10.1016/s0140-6736(20)32667-2
Bakker, S. T., Van De Vrugt, H. J., Rooimans, M. A., Oostra, A. B., Steltenpool, J., Delzenne-Goette, E., et al. (2009). Fancm-deficient Mice Reveal Unique Features of Fanconi Anemia Complementation Group M. Hum. Mol. Genet. 18, 3484–3495. doi:10.1093/hmg/ddp297
Bogliolo, M., Bluteau, D., Lespinasse, J., Pujol, R., Vasquez, N., D'enghien, C. D., et al. (2018). Biallelic Truncating FANCM Mutations Cause Early-Onset Cancer but Not Fanconi Anemia. Genet. Med. 20, 458–463. doi:10.1038/gim.2017.124
Ciccia, A., Ling, C., Coulthard, R., Yan, Z., Xue, Y., Meetei, A. R., et al. (2007). Identification of FAAP24, a Fanconi Anemia Core Complex Protein that Interacts with FANCM. Mol. Cel 25, 331–343. doi:10.1016/j.molcel.2007.01.003
Collis, S. J., Ciccia, A., Deans, A. J., Hořejší, Z., Martin, J. S., Maslen, S. L., et al. (2008). FANCM and FAAP24 Function in ATR-Mediated Checkpoint Signaling Independently of the Fanconi Anemia Core Complex. Mol. Cel 32, 313–324. doi:10.1016/j.molcel.2008.10.014
Deans, A. J., and West, S. C. (2009). FANCM Connects the Genome Instability Disorders Bloom's Syndrome and Fanconi Anemia. Mol. Cel 36, 943–953. doi:10.1016/j.molcel.2009.12.006
Domingues-Silva, B., Silva, B., and Azzalin, C. M. (2019). ALTernative Functions for Human FANCM at Telomeres. Front. Mol. Biosci. 6, 84. doi:10.3389/fmolb.2019.00084
Ezeh, U. I., Moore, H. D., and Cooke, I. D. (1998). A Prospective Study of Multiple Needle Biopsies versus a Single Open Biopsy for Testicular Sperm Extraction in Men with Non-obstructive Azoospermia. Hum. Reprod. 13, 3075–3080. doi:10.1093/humrep/13.11.3075
Fakhro, K. A., Elbardisi, H., Arafa, M., Robay, A., Rodriguez-Flores, J. L., Al-Shakaki, A., et al. (2018). Point-of-care Whole-Exome Sequencing of Idiopathic Male Infertility. Genet. Med. 20, 1365–1373. doi:10.1038/gim.2018.10
Gershoni, M., Hauser, R., Yogev, L., Lehavi, O., Azem, F., Yavetz, H., et al. (2017). A Familial Study of Azoospermic Men Identifies Three Novel Causative Mutations in Three New Human Azoospermia Genes. Genet. Med. 19, 998–1006. doi:10.1038/gim.2016.225
He, W.-B., Tu, C.-F., Liu, Q., Meng, L.-L., Yuan, S.-M., Luo, A.-X., et al. (2018). DMC1 Mutation that Causes Human Non-obstructive Azoospermia and Premature Ovarian Insufficiency Identified by Whole-Exome Sequencing. J. Med. Genet. 55, 198–204. doi:10.1136/jmedgenet-2017-104992
Kasak, L., Punab, M., Nagirnaja, L., Grigorova, M., Minajeva, A., Lopes, A. M., et al. (2018). Bi-allelic Recessive Loss-Of-Function Variants in FANCM Cause Non-obstructive Azoospermia. Am. J. Hum. Genet. 103, 200–212. doi:10.1016/j.ajhg.2018.07.005
Maor-Sagie, E., Cinnamon, Y., Yaacov, B., Shaag, A., Goldsmidt, H., Zenvirt, S., et al. (2015). Deleterious Mutation in SYCE1 Is Associated with Non-obstructive Azoospermia. J. Assist. Reprod. Genet. 32, 887–891. doi:10.1007/s10815-015-0445-y
Marchuk, D. S., Crooks, K., Strande, N., Kaiser-Rogers, K., Milko, L. V., Brandt, A., et al. (2018). Increasing the Diagnostic Yield of Exome Sequencing by Copy Number Variant Analysis. PLoS One 13, e0209185. doi:10.1371/journal.pone.0209185
Minhas, S., Bettocchi, C., Boeri, L., Capogrosso, P., Carvalho, J., Cilesiz, N. C., et al. (2021). European Association of Urology Guidelines on Male Sexual and Reproductive Health: 2021 Update on Male Infertility. Eur. Urol. doi:10.1016/j.eururo.2021.08.014
Neidhardt, G., Hauke, J., Ramser, J., Gross, E., Gehrig, A., Müller, C. R., et al. (2017). Association between Loss-Of-Function Mutations within the FANCM Gene and Early-Onset Familial Breast Cancer. JAMA Oncol. 3, 1245–1248. doi:10.1001/jamaoncol.2016.5592
O'rourke, J. J., Bythell-Douglas, R., Dunn, E. A., and Deans, A. J. (2019). ALT Control, Delete: FANCM as an Anti-cancer Target in Alternative Lengthening of Telomeres. Nucleus 10, 221–230. doi:10.1080/19491034.2019.1685246
Okutman, O., Muller, J., Baert, Y., Serdarogullari, M., Gultomruk, M., Piton, A., et al. (2015). Exome Sequencing Reveals a Nonsense Mutation inTEX15causing Spermatogenic Failure in a Turkish Family. Hum. Mol. Genet. 24, 5581–5588. doi:10.1093/hmg/ddv290
Riera-Escamilla, A., Enguita-Marruedo, A., Moreno-Mendoza, D., Chianese, C., Sleddens-Linkels, E., Contini, E., et al. (2019). Sequencing of a 'mouse Azoospermia' Gene Panel in Azoospermic Men: Identification of RNF212 and STAG3 Mutations as Novel Genetic Causes of Meiotic Arrest. Hum. Reprod. 34, 978–988. doi:10.1093/humrep/dez042
Stephen, E. H., and Chandra, A. (2006). Declining Estimates of Infertility in the United States: 1982-2002. Fertil. Sterility 86, 516–523. doi:10.1016/j.fertnstert.2006.02.129
Tao, Y., Jin, C., Li, X., Qi, S., Chu, L., Niu, L., et al. (2012). The Structure of the FANCM-MHF Complex Reveals Physical Features for Functional Assembly. Nat. Commun. 3, 782. doi:10.1038/ncomms1779
Tsuchida, N., Nakashima, M., Kato, M., Heyman, E., Inui, T., Haginoya, K., et al. (2018). Detection of Copy Number Variations in Epilepsy Using Exome Data. Clin. Genet. 93, 577–587. doi:10.1111/cge.13144
Tsui, V., and Crismani, W. (2019). The Fanconi Anemia Pathway and Fertility. Trends Genet. 35, 199–214. doi:10.1016/j.tig.2018.12.007
Xue, X., Sung, P., and Zhao, X. (2015). Functions and Regulation of the Multitasking FANCM Family of DNA Motor Proteins. Genes Dev. 29, 1777–1788. doi:10.1101/gad.266593.115
Yan, Z., Delannoy, M., Ling, C., Daee, D., Osman, F., Muniandy, P. A., et al. (2010). A Histone-fold Complex and FANCM Form a Conserved DNA-Remodeling Complex to Maintain Genome Stability. Mol. Cel 37, 865–878. doi:10.1016/j.molcel.2010.01.039
Yang, F., Silber, S., Leu, N. A., Oates, R. D., Marszalek, J. D., Skaletsky, H., et al. (2015). TEX 11 Is Mutated in Infertile Men with Azoospermia and Regulates Genome‐wide Recombination Rates in Mouse. EMBO Mol. Med. 7, 1198–1210. doi:10.15252/emmm.201404967
Yang, H., Zhang, T., Tao, Y., Wang, F., Tong, L., and Ding, J. (2013). Structural Insights into the Functions of the FANCM-FAAP24 Complex in DNA Repair. Nucleic Acids Res. 41, 10573–10583. doi:10.1093/nar/gkt788
Yang, Y., Guo, J., Dai, L., Zhu, Y., Hu, H., Tan, L., et al. (2018). XRCC2 Mutation Causes Meiotic Arrest, Azoospermia and Infertility. J. Med. Genet. 55, 628–636. doi:10.1136/jmedgenet-2017-105145
Yang, Y., Guo, T., Liu, R., Ke, H., Xu, W., Zhao, S., et al. (2020). FANCL Gene Mutations in Premature Ovarian Insufficiency. Hum. Mutat. 41, 1033–1041. doi:10.1002/humu.23997
Yao, C., Yang, C., Zhao, L., Li, P., Tian, R., Chen, H., et al. (2021). Bi-allelic SHOC1 Loss-Of-Function Mutations Cause Meiotic Arrest and Non-obstructive Azoospermia. J. Med. Genet. 58, 679–686. doi:10.1136/jmedgenet-2020-107042
Yao, R., Zhang, C., Yu, T., Li, N., Hu, X., Wang, X., et al. (2017). Evaluation of Three Read-Depth Based CNV Detection Tools Using Whole-Exome Sequencing Data. Mol. Cytogenet. 10, 30. doi:10.1186/s13039-017-0333-5
Keywords: FANCM, gene mutation, male infertility, Sertoli cell-only syndrome, non-obstructive azoospermia
Citation: Zhang Y, Li P, Liu N, Jing T, Ji Z, Yang C, Zhao L, Tian R, Chen H, Huang Y, Zhi E, Ou N, Bai H, Zhou Y, Li Z and Yao C (2021) Novel Bi-Allelic Variants of FANCM Cause Sertoli Cell-Only Syndrome and Non-Obstructive Azoospermia. Front. Genet. 12:799886. doi: 10.3389/fgene.2021.799886
Received: 22 October 2021; Accepted: 16 November 2021;
Published: 15 December 2021.
Edited by:
Maria Grazia Giansanti, Italian National Research Council, ItalyReviewed by:
Andrew J. Deans, University of Melbourne, AustraliaSusana S.bmF2YXJyb0BjaWVtYXQuZXM=, Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas, Spain
Copyright © 2021 Zhang, Li, Liu, Jing, Ji, Yang, Zhao, Tian, Chen, Huang, Zhi, Ou, Bai, Zhou, Li and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuchuan Zhou, emhvdXljaEBzaWJjYi5hYy5jbg==; Zheng Li, bGl6aGVuZ2Jvc2hpQHNqdHUuZWR1LmNu; Chencheng Yao, eWFvY2hlbmNoZW5nQDEyNi5jb20=
†These authors have contributed equally to this work