 Zhaomin Yao
Zhaomin Yao Gancheng Zhu
Gancheng Zhu Jingwei Too
Jingwei Too Meiyu Duan
Meiyu Duan Zhiguo Wang
Zhiguo Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 08 March 2022
Sec. Computational Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.793629
This article is part of the Research Topic Biomarker Detection Algorithms and Tools for Medical Imaging or Omic Data View all 23 articles
OMIC datasets have high dimensions, and the connection among OMIC features is very complicated. It is difficult to establish linkages among these features and certain biological traits of significance. The proposed ensemble swarm intelligence-based approaches can identify key biomarkers and reduce feature dimension efficiently. It is an end-to-end method that only relies on the rules of the algorithm itself, without presets such as the number of filtering features. Additionally, this method achieves good classification accuracy without excessive consumption of computing resources.
The OMIC data includes genomes, transcriptomes, metabolomes, and proteomes. (Karczewski and Snyder 2018). Its quantity and quality have been improved significantly during the rapid development and continuous innovation of high-throughput sequencing and mass spectrum technologies (Margolis et al., 2014). Generally, biomedical data has the characteristics of “large p and small n,” that is, the species of features is far larger than the species of samples (Liao and Chin 2007). Thus, it is necessary for biomedical dataset dimension reduction to protect against potential dimension disaster.
Feature selection has been proven with excellent performance in data preprocessing, especially for high dimensional data (Dash and Liu 1997; Bolón-Canedo, Sánchez-Maroño, and Alonso-Betanzos 2015). Its goals consist of cleaning out understandable and analyzable data, constructing simple and efficient models, and improving the efficiency of data mining (Li et al., 2017). It has achieved prominent results in the bioinformation field (Fu et al., 2018; Qiu, Ching, and Zou 2021). Swarm intelligence (SI) is the decentralized self-organizing collective behavior at the collective level (Hu et al., 2021b). It usually consists of a group of simple agents that interact with each other locally and with their environment. The agents follow very simple rules, and there is no centralized control structure to specify the behavior of a single agent. However, the interaction among these agents will lead to the emergence of “intelligent” global behavior (Hu et al., 2021a). Therefore, the whole problem-solving process will not be affected by the failure of one or several agents, so this method has good robustness and potential global search ability. Additionally, SI can transmit and coordinate information through indirect communication. With the increase in the number of individuals, the increase in communication overhead is small. Thus, it also has good scalability. Because of these advantages, SI is widely used in feature selection; its combination with machine learning has especially proven to be able to obtain outstanding results. Through the research and development of the genetic algorithm (Malakar et al., 2019) and the firefly algorithm (Bacanin et al., 2021), the features extracted from each handwritten word image have been significantly optimized so that the performance of the handwritten word recognition technique has been increased visibly.
Various computational feature selection models have been proposed to reduce the dimension of OMIC datasets (Ge et al., 2016; Liu et al., 2017; Yuanyuan, Lan, and Fengfeng 2021). However, these algorithms need to design the number of features in advance as an intervention. Meanwhile, the heuristic rules applied are almost mathematical principles. Thus, this study was intended to investigate the performance of the features screened based on biological or natural rules, instead of traditional mathematical principles, and manually specify the number.
This article is organized as follows: details of the datasets and overview of the methods are described in Section 2. Experimental results and a corresponding analysis of these results are presented in Section 3. Finally, a brief conclusion is drawn in Section 4.
As shown in Figure 1, this study involved six major stages: Dataset curation, data preprocessing, feature selection, model training and validation, feature intersection and union combination, and prediction. First, a large number of OMIC datasets are collected, including transcriptome datasets (Dataset 1) and methylation datasets (Dataset 2). Then, all the features with missing values in the collected datasets will be deleted. Next, all the transcriptome datasets will have features extracted by twelve advanced swarm intelligent algorithms, and then these features will be input into five different representative classifiers and finally classification performance will be obtained. According to these results, the best classifier and the top three algorithms that use this classifier to get the best results will be selected to apply to methylation datasets. Later, these subsets will generate different combinations through union and intersection. Finally, the classification performance of these combinations will be evaluated by the best classifiers. The details of each process are described in the following sections.
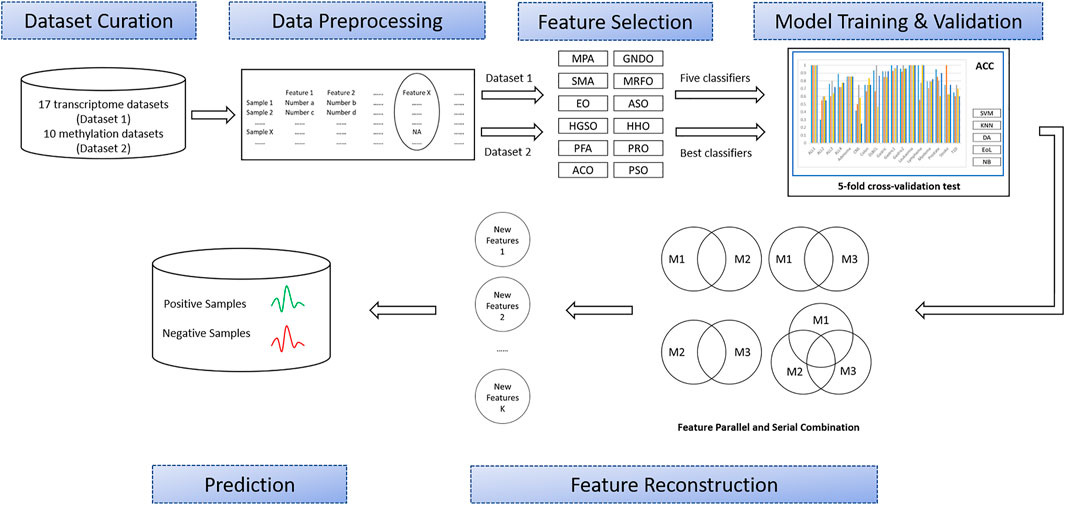
FIGURE 1. Overview of the proposed methodology.
This study concentrated on binary classification and analyzed the relevant publicly available OMIC databases. As shown in Supplementary Table S1, these data include 17 transcriptome datasets and 10 methylation datasets. Methylation is an important modification of proteins and nucleic acids; it reveals the influence of genetic and environmental factors on the occurrence and development of complex diseases (Barros and Offenbacher 2009). Compared with transcriptome data, methylation data usually have more feature dimension and are more challenging in classification.
First, all transcriptome datasets (Dataset 1) were used to test the performance of the algorithm. As shown in Supplementary Table S1, they were DLBCL (Shipp et al., 2002), Pros (Aalinkeel et al., 2004), Colon (Alon et al., 1999), Leuk (Golub et al., 1999), Mye (Tian et al., 2003), All (All1/All2/All3/All4) (Chiaretti et al., 2004), CNS (Pomeroy et al., 2002), Lym (Alizadeh et al., 2000), Adeno (Notterman et al., 2001), Gas (Wu et al., 2013), Gas1/Gas2 (Wang et al., 2013) , T1D (Levy et al., 2012), and Stroke (Krug et al., 2012). These datasets were obtained and preprocessed as similar in Mctwo (Ge et al., 2016).
Additionally, ten methylation datasets (Dataset 2) were used to demonstrate the binary classification performances, as shown in Supplementary Table S1. The dataset GSE74845 profiled 110 Fimbria and 106 proximal tubal DNA samples of fallopian tube fimbriae in BRCA mutation carriers (Bartlett et al., 2016). The dataset GSE80970 provided the methylomes of 148 Alzheimer’s disease samples and 138 controls (Smith et al., 2018). The dataset GSE103186 illustrated 130 gastric light or mild intestinal metaplasia and 61 gastric normal samples (Huang et al., 2017). The dataset GSE139032 investigated 77 lung adenocarcinomas and 77 matched non-malignant lung samples (Enfield et al., 2019). The dataset GSE139404 compared 40 low-grade adenoma and high-grade adenoma in colorectal and 20 normal tissues (Fan et al., 2020). The dataset GSE144910 collected a total of 88 genomic DNA samples taken from the postmortem superior temporal gyrus of the human brain with 44 schizophrenia and paired non-psychiatric controls (Mckinney et al., 2020). The dataset GSE164269 generated 33 discovery and 46 independent validation cohorts of malignant pleural mesothelioma samples (Bertero et al., 2021). The dataset GSE166787 contrasted DNA methylation data throughout human muscle cell differentiation in 28 individuals with type 2 diabetes and 28 controls (Davegårdh et al., 2021). The dataset GSE173330 supplied DNA methylation data from several tissues in toothed whales (N = 254) and dolphin (N = 291) (Robeck et al., 2021). The last dataset GSE174613 analyzed samples of non-malignancy obtained from prostatectomy specimens (n = 12) and of bone metastasis tissue samples obtained from separate prostate cancer patients (n = 70) (Ylitalo et al., 2021).
Due to various experimental reasons, gene expression data universally suffer from the missing value problem. The features with missing values can adversely affect the classifiers (Varsha et al., 2016). Considering the number of features with missing values in the datasets accounts for less than 0.1% of the total number of features, direct removal also has little impact on the overall datasets. Thus, these features affected by missing values are removed directly. For example, for a feature X, the value of X is missing in only one sample, but there is a definite value in all other samples. The X must be removed from all samples.
Twelve swarm intelligence methods are used in the study, including ten state-of-the-art methods from the last 2 years and two classic methods. The methods are briefly described below.
Marine predator algorithm (MPA) is a natural heuristic optimization algorithm. It follows the rule of natural dominance in the optimal foraging strategy and encounters the rate strategy between predator and prey in the marine ecosystem. This algorithm is inspired by the predator–prey strategy in nature and considers that the top predator has the greatest search ability, that is, the decision of a top predator is a solution of the problem (Faramarzi et al., 2020a).
Generalized normal distribution optimization (GNDO) is a novel metaheuristic algorithm inspired by normal distribution theory. It can solve optimization problems by natural phenomenon distribution and fitting minimum standard variance of the positions of all individuals. Generally speaking, GNDO consists of two main strategies: local exploitation and global exploration. The former focuses on building the generalized distribution model while the latter explores the search region based on three randomly selected individuals (Zhang et al., 2020).
Slime mould algorithm (SMA) is based on the diffusion and foraging behavior of slime mould in nature. It calculates the optimal path by simulating the relationship between morphological changes and contraction patterns of slime mould during foraging. SMA performs the search relying on three stages: Find approach, wrap food, and oscillation (Li et al., 2020).
Manta ray foraging optimization (MRFO) mathematically models and mimics three unique foraging strategies of manta rays, including chain foraging, cyclone foraging, and somersault foraging, for solving global optimization problems. In chain foraging, the manta rays update their solutions by following the best solution and the solution in front of it. For cyclone foraging, the manta rays move toward the global optima along a spiral path. Last, in somersault foraging, the manta rays tend to update their position around the best solution in the population (Zhao et al., 2020).
Equilibrium optimizer (EO) is inspired by a physical phenomenon of controlling volume mass balance. It simulates the physical process of mass entering, leaving, and generating in the control volume to finally reach the equilibrium state as optimal results. In EO, there is an equilibrium pool that used to store the current four best-so-far solutions. Iteratively, these stored solutions will be applied to enhance the quality of solutions in the population. Additionally, EO integrates the particle memory saving to benefit the exploitation capability (Faramarzi et al., 2020b).
Atom search optimization (ASO) is a novel algorithm based on a basic molecular dynamics model. In a molecular system, there are interaction forces between neighboring atoms, and the globally optimal atoms constrain other atoms. Gravitation makes atoms explore the whole search space extensively, and repulsion makes them develop the potential region effectively. It simulates this phenomenon to find the global optimal solution (Zhao et al., 2019).
Henry gas solubility optimization (HGSO) is a novel metaheuristic algorithm; it imitates the huddling behavior of gas described in Henry’s law to balance the exploitation ability and the exploration ability of the algorithm for searching the global optimum and avoid trapping into local optima (Hashim et al., 2019).
Harris hawks optimization (HHO) is a novel population-based, natural heuristic optimization. Its main inspiration comes from Harris’s eagle’s cooperative behavior and pursuit in nature. It is unique because it has a unique cooperative foraging activity with other family members in the group. Because of this, it is very suitable to simulate the unique predatory behavior of Harris’s hawk as a swarm intelligence optimization process (Heidari et al., 2019).
Path finder algorithm (PFA) is inspired by the hunting behavior of group animals. The algorithm realizes the optimization process through the communication between pathfinder and follower from the population in the process of the population searching for food. Naturally, PFA stores the best-so-far solution (pathfinder), in which the pathfinder is used to enhance the exploitation and exploration capability (Yapici and Cetinkaya 2019).
Poor and rich optimization (PRO) is developed based on the real social phenomenon, that is, the attempt of the rich and the poor to improve their economic conditions. This social behavior can be regarded as a solution for complex optimization problems. In PRO, a mutation operator is designed to improve the compound population. Even though PRO is a promising algorithm, it suffers from the high computational complexity (Moosavi and Bardsiri 2019).
Ant colony algorithm is inspired by the foraging behavior of ants in nature. In the process of ant foraging, an ant colony can always find an optimal path between the ant nest and food source. This is because the ants in the ant colony can transmit information through some information mechanism. After further research, it is found that ants will release a substance called “pheromone” on their path. Ants in the ant colony have the ability to perceive the “pheromone.” They will walk along the path with high concentration of “pheromone,” and each passing ant will leave “pheromone” on the road, which forms a mechanism similar to positive feedback; in this way, after a period of time, the whole ant colony will reach the food source along the shortest path (Dorigo et al., 2006).
Particle swarm optimization is inspired by the study of bird predation behavior. Specifically, birds find the optimal destination through collective information sharing. In PSO, the potential solution of each optimization problem is a bird in the search space, which is called a particle. All particles have a fitness value determined by the optimized function, and each particle also has a speed to determine their flying direction and distance. Then the particles follow the current optimal particle to search in the solution space (Kennedy and Eberhart 1995).
K-fold cross-validation is one of the most commonly used evaluation strategies. This experimental procedure is performed by the 5-fold cross-validation, that is, the baseline dataset is randomly divided into five equal parts (the number and distribution of samples are the same) and the test processes are repeated five times; for each cross-validation test, one subset is used for testing while the remains are used for training the model. The final performance is represented by the average of five experimental results.
Leave one method cross-validation is to treat each data sample as an independent dataset, use one sample each time as the test set, and use all the remaining samples as the training set. The result obtained using this method is closest to the expected value of the whole test set, but the computing cost is excessively expensive.
Higher classification accuracy and fewer features are the objectives of generating models; however, it is difficult to achieve both at the same time. Here, the first consideration in this study is the classification accuracy. For achieving a more comprehensive and stable performance, five widely used classifiers are applied to the models, that is, support vector machine (SVM), K-Nearest Neighbor (KNN), discriminant analysis (DA), ensemble of learners (EoL), and naive Bayes (NB). This study evaluates a feature subset through the best classification performance of multiple classifiers. Generally, prediction accuracy is defined as follows:
where TP, FP, TN, and FN represent the value of true positives, false positives, true negatives, and false negatives, respectively.
Intersection and union combination approaches were employed to ensemble the selected features. As shown in Figure 2, two or three different feature selection results were combined into eight subsets for performance comparison.
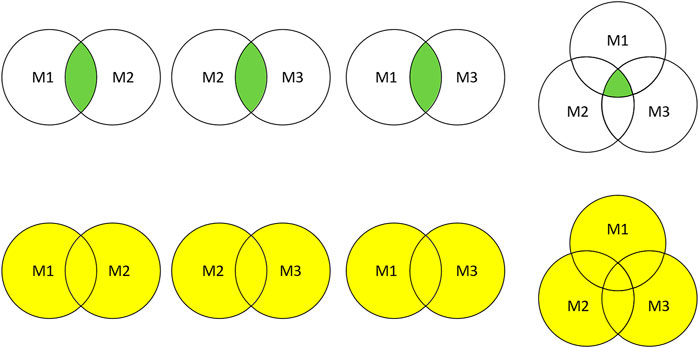
FIGURE 2. Feature subsets combination. M1, M2, and M3 represent the feature subsets extracted by three different methods, respectively. The green part and yellow part represent the combination results obtained by intersection and union.
This study used these transcriptome datasets for testing the performance of baseline swarm intelligence algorithms and classifiers. Enough iterations are used to satisfy the fitness value. Here, the random 5-fold cross-validation and leave-one-out cross-validation are used to evaluate the performance, respectively. The results are shown in Supplementary Tables S2, S3. Both of the tables show that KNN can make most datasets achieve the best classification effect in most algorithms. Additionally, in the other three algorithms, where KNN cannot achieve the best results, the gap between KNN and the best classifier in the number of datasets for best performance is small, only one to three datasets.
Through the information combination of two tables, when using KNN, the number of best results obtained by PFA and SMA is 12 and 8, respectively, ranking first and second. ASO, GNDO, PSO, and HGSO all get 7 best results, and the number is equal. As shown in Supplementary Table S4, considering the average number of features used on each dataset, HGSO is chosen as the last algorithm to be applied to the next stage.
Because there is little performance difference between 5-fold cross-validation and leave-one-out cross-validation in these transcriptome datasets and the computing cost of leave-one-out cross-validation is relatively high, the subsequent evaluation is only based on the random 5-fold cross-validation.
In the FS phase, a fitness function is adopted to evaluate the quality of the initial and newly generated solutions. This study evaluates the solutions by considering the minimum classification error and minimum size of features (Emary et al., 2016a). Mathematically, the fitness function is defined as follows:
where ER is the classification error rate computed by the k-nearest neighbor classifier (KNN, k-value = 5), |SF| is the number of the selected features, |AF| is the total number of features, and β is the weight factor between 0 and 1. This study adopts β = 0.99 since the classification performance is the most importance measurement (Emary, Zawbaa, and Hassanien 2016b; Mafarja et al., 2019). In the fitness evaluation stage, the dataset is partitioned into training and validation sets using the k-fold cross-validation method. Consequently, the dataset is divided into 5 folds, in which k-1 folds are used to build the training set while the rest is kept for accessing the selected features.
The T1D dataset is used as an example to show the convergence of the top three algorithms. As shown in Figure 3, PFA and HGSO converge in about 22 iterations, while SMA converges faster, and the convergence can be completed in about 10 iterations.

FIGURE 3. The convergence speed of top three swarm intelligence algorithms on T1D.
This section evaluated the performance of SMA, PFA, and HGSO on the methylation datasets, and the classifier is KNN.
Although methylome datasets may be a challenge for many feature selection algorithms, the swarm intelligence algorithm has achieved good results on many datasets. As shown in Figure 4, PFA achieves more than 90% accuracy on four datasets. Meanwhile, SMA obtains about 90% accuracy on the GSE139032 and GSE139404, where PFA does not get good results. In addition, the consumption of computing resources and time is also within an acceptable range; the average time consumption (CPU: i9-11900H) of SMA, PFA, and HGSO are 101.83, 415.21, and 312.31 s, respectively.

FIGURE 4. Performance of three swarm intelligence algorithms on methylation datasets.
Besides accuracy, other evaluation indicators are also very important. They can reveal the characteristics of the algorithm in other aspects. Therefore, another four commonly used indicators for classification evaluation (precision, recall, F1-score, and AUC ROC) have also been tested, and the results are shown in the Supplementary Table S5. It can be seen from the results that there is little difference between precision and recall of most models. However, the precision of PFA reaches 100% but the corresponding recall just obtains about 12% on GSE164269. It may be caused by the insensitivity of the dataset to the algorithm, that is, the algorithm cannot filter the core features of the dataset. Thus, many positive samples are identified as negative samples.
Statistical tests on the results obtained using the three methods were performed. The statistics are described in Table 1. The result of Wilcoxon signed ranks test are shown in Table 2. Through the nonparametric test of paired samples, the p-values are greater than the significance level, indicating that there is no difference in the measurement accuracy of these 10 samples after three methods. Additionally, the Friedman test was also applied, and the chi-squared, df, and p-value are 0.2, 2, and 0.906, respectively. It also proved that there was no significant difference in accuracy.

TABLE 1. Descriptive statistics of the results on methylation datasets.
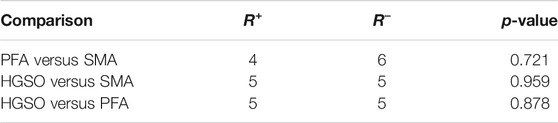
TABLE 2. Wilcoxon signed ranks test.
Generally, for a given dataset, the feature subsets for different feature selection are individually somewhat different due to the different theories. So, their different combinations will be more diverse. These subsets are evaluated in this section. What is more, there is no duplicate selection of the same features by different methods.
Figure 5 shows the classification performance obtained by intersection and union combination-based feature subset ensemble methods. In some feature subset combinations, no classification accuracy is available because there is no repeat selection of the same features by the applied methods. As we can see, the performance of the union combination method with PFA is not obvious. The reason may be that PFA selects too many features, which is over 2000 times that of SMA and about 200 times that of HGSO. Additionally, the performance of union combination between SMA and HGSO is always better than just using HGSO but not always better than just using SMA. The reason may be that the number of features used by HGSO is ten times than that of SMA. Therefore, the characteristics of SMA can only be used for auxiliary adjustment. What is more, the performance of some intersection methods does not decrease so much. This may be because the features selected by all these algorithms are the core features of the datasets.
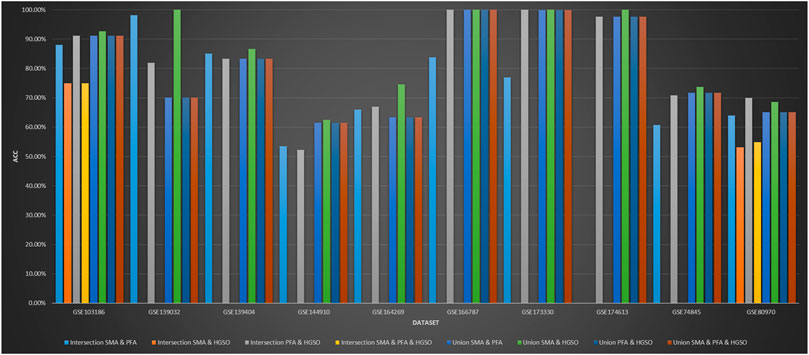
FIGURE 5. Performance of feature intersection and union combination on methylation datasets.
Table 3 shows the feature selection rates of single and different combination swarm intelligence methods on methylation datasets. Note that the feature selection rate is the percentage of the features that are extracted from the original features.
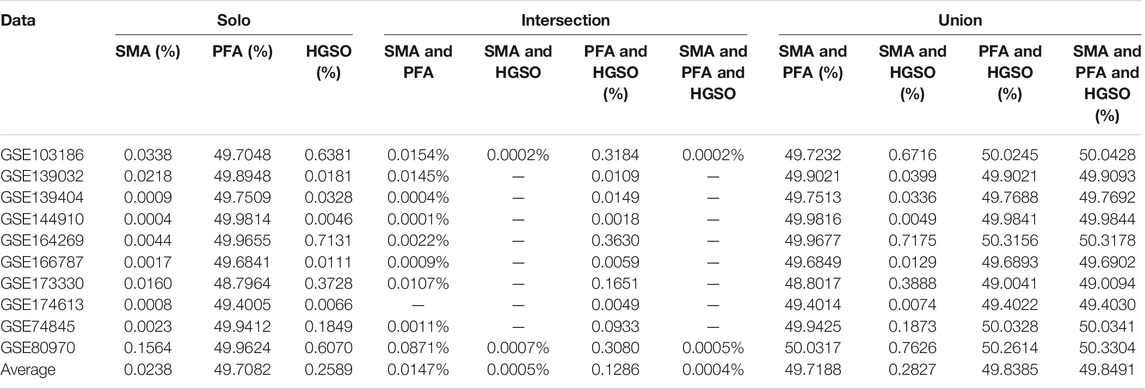
TABLE 3. Feature selection rates of all used feature subsets on methylation datasets.
As we can see, SMA produces the lowest feature reduction rate in a single model, that is, the average is 0.0238%. This means that applying SMA as the embedded feature selection method may cause “over selection,” with too many informative features filtered out. On the other hand, PFA not only allows selection of the most informative features but also avoids the risk of over selection. However, using the intersection combination with HGSO and PFA not only can reduce the number of features further but also not reduce the accuracy in many datasets. The results indicate that intersection combination method-based ensemble feature selection is likely to play a positive role in filtering out information redundancy among the feature selection methods that retain too much information after use.
In addition, using the combination among feature subsets with widely different feature numbers will not lead to excessive changes in classification performance, and most of the classification results will be the result of the feature subset with the highest number of features, because its feature distribution has not changed.
The internal metaplasia samples contained in GSE103186 can also be more finely divided into classic and mild. Therefore, GSE103186 is regarded as a three-category dataset for testing the multi-classification performance. The performance of SMA, PFA, and HGSO is 81.69, 80.63, and 83.78%, respectively. Although the proposed method mainly focuses on binary classification problems, the results show that it still has the potential to be used in multi-classification problems.
The dataset GSE144910 collected DNA samples from the superior temporal gyrus of the human brain for researching schizophrenia. The features detected by the union combination of SMA and HGSO as the classification biomarkers and these methylation features are related to 18 genes, which are C1orf168, CAMLG, SMOX, KCNIP4, MIR658, CENPA, ASRGL1, PISD, HNRNPL, EEF2K, GMDS, MPPED1, ANKRD54, PLEK2, ADA, RNF121, KRT6A, and EPHA2. In order to explore the biological functions of the selected genes, pathway analysis was conducted. Figure 6 showed the mainly obtained four biological process pathways (GO: 0033627, 072657, 00488872, and 0044089). We found that schizophrenia may be related to the function of cell adhesion.
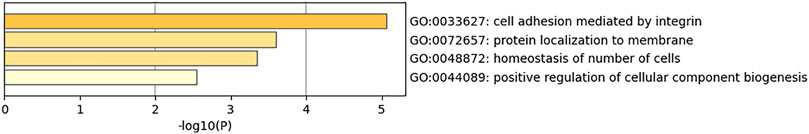
FIGURE 6. Performance of feature intersection and union combination on methylation datasets.
This study focuses on examining the binary classification performance of swarm intelligence algorithms on OMIC datasets. The experimental results suggest that swarm intelligence algorithms can achieve high accuracy on the collected OMIC datasets, significantly reduce feature dimensions, and identify key features. Meanwhile, this study finds some rules to improve ensemble feature subset performance through intersection and union combination methods. However, there are still some limitations in the proposed study. For example, the methodology framework has not been improved, and there is no methodological fusion of different swarm intelligence algorithms. Our future research will focus on combining machine learning and swarm intelligence approaches for reducing the feature dimension and improve the accuracy further in OMIC data and other biological data.
Publicly available datasets were analyzed in this study. The data can be found at: http://www.broadinstitute.org/cgi-bin/cancer/datasets.cgi (Broad Institute Genome Data Analysis Center) and https://www.ncbi.nlm.nih.gov/geo/ [NCBI Gene Expression Omnibus (GEO) database].
ZW and ZY designed the project, GZ collected the datasets, ZY and JT carried out the coding of the computational analysis, ZY, JT, and MD drafted the manuscript, and ZW revised and polished the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Key Research and Development Program of Liaoning Province (2019JH2/10300010).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.793629/full#supplementary-material
Aalinkeel, R., Nair, M. P. N., Sufrin, G., Mahajan, S. D., Chadha, K. C., Chawda, R. P., et al. (2004). Gene Expression of Angiogenic Factors Correlates with Metastatic Potential of Prostate Cancer Cells. Cancer Res. 64, 5311–5321. doi:10.1158/0008-5472.can-2506-2
Alizadeh, A. A., Eisen, M. B., Davis, R. E., Ma, C., Lossos, I. S., Rosenwald, A., et al. (2000). Distinct Types of Diffuse Large B-Cell Lymphoma Identified by Gene Expressionprofiling. Nature 403, 503. doi:10.1038/35000501
Alon, U., Barkai, N., Notterman, D. A., Gish, K., Ybarra, S., Mack, D., et al. (1999). Broad Patterns of Gene Expression Revealed by Clustering Analysis of Tumor and normal colon Tissues Probed by Oligonucleotide Arrays. Proc. Natl. Acad. Sci. 96, 6745–6750. doi:10.1073/pnas.96.12.6745
Bacanin, N., Stoean, R., Zivkovic, M., Petrovic, A., Rashid, T. A., and Bezdan, T. (2021). Performance of a Novel Chaotic Firefly Algorithm with Enhanced Exploration for Tackling Global Optimization Problems: Application for Dropout Regularization. Mathematics 9, 2705. doi:10.3390/math9212705
Barros, S. P., and Offenbacher, S. (2009). Epigenetics: Connecting Environment and Genotype to Phenotype and Disease. J. Dental Res. 88, 400–408. doi:10.1177/0022034509335868
Bartlett, T. E., Chindera, K., Mcdermott, J., Breeze, C. E., Cooke, W. R., Jones, A., et al. (2016). Epigenetic Reprogramming of Fallopian Tube Fimbriae in BRCA Mutation Carriers Defines Early Ovarian Cancer Evolution. Nat. Commun. 7, 11620. doi:10.1038/ncomms11620
Bertero, L., Righi, L., Collemi, G., Koelsche, C., and Deimling, A. V. (2021). DNA Methylation Profiling Discriminates between Malignant Pleural Mesothelioma and Neoplastic or Reactive Histological Mimics. J. Mol. Diagn. 23, 834–846. doi:10.1016/j.jmoldx.2021.04.002
Bolón-Canedo, V., Sánchez-Maroño, N., and Alonso-Betanzos, A. (2015). Feature Selection for High-Dimensional Data. Berlin/Heidelberg, Germany: Springer.
Chiaretti, S., Li, X., Gentleman, R., Vitale, A., Vignetti, M., Mandelli, F., et al. (2004). Gene Expression Profile of Adult T-Cell Acute Lymphocytic Leukemia Identifies Distinct Subsets of Patients with Different Response to Therapy and Survival. Blood 103, 2771–2778. doi:10.1182/blood-2003-09-3243
Dash, M., and Liu, H. (1997). Feature Selection for Classification. Intell. Data Anal. 1, 131–156. doi:10.1016/s1088-467x(97)00008-5
Davegårdh, C., Säll, J., Anna, B., Broholm, C., Volkov, P., Alexander, P., et al. (2021). VPS39-deficiency Observed in Type 2 Diabetes Impairs Muscle Stem Cell Differentiation via Altered Autophagy and Epigenetics. Nat. Commun. 12, 2431. doi:10.1038/s41467-021-22068-5
Dorigo, M., Birattari, M., and Stutzle, T. (2006). Ant colony Optimization. IEEE Comput. intelligence Mag. 1, 28–39. doi:10.1109/ci-m.2006.248054
Emary, E., Zawbaa, H. M., and Hassanien, A. E. (2016a). Binary Ant Lion Approaches for Feature Selection. Neurocomputing 213, 54–65. doi:10.1016/j.neucom.2016.03.101
Emary, E., Zawbaa, H. M., and Hassanien, A. E. (2016b). Binary Grey Wolf Optimization Approaches for Feature Selection. Neurocomputing 172, 371–381. doi:10.1016/j.neucom.2015.06.083
Enfield, K. S. S., Marshall, E. A., AndersonNg, C. K. W., and Wan, L. L. (2019). Epithelial Tumor Suppressor ELF3 Is a Lineage-specific Amplified Oncogene in Lung Adenocarcinoma. Nat. Commun. 10, 5438. doi:10.1038/s41467-019-13295-y
Fan, J., Li, J., Guo, S., Tao, C., and Zeng, C. (2020). Genome-wide DNA Methylation Profiles of Low- and High-Grade Adenoma Reveals Potential Biomarkers for Early Detection of Colorectal Carcinoma. Clin. Epigenetics 12, 56. doi:10.1186/s13148-020-00851-3
Faramarzi, A., Heidarinejad, M., Mirjalili, S., and Gandomi, A. H. (2020a). Marine Predators Algorithm: A Nature-Inspired Metaheuristic. Expert Syst. Appl. 152, 113377. doi:10.1016/j.eswa.2020.113377
Faramarzi, A., Heidarinejad, M., Stephens, B., and Mirjalili, S. (2020b). Equilibrium Optimizer: A Novel Optimization Algorithm. Knowledge-Based Syst. 191, 105190. doi:10.1016/j.knosys.2019.105190
Fu, G., Wang, J., Domeniconi, C., and Yu, G. (2018). Matrix Factorization-Based Data Fusion for the Prediction of lncRNA–Disease Associations. Bioinformatics 34, 1529–1537. doi:10.1093/bioinformatics/btx794
Ge, R., Zhou, M., Luo, Y., Meng, Q., and Zhou, F. (2016). McTwo: A Two-step Feature Selection Algorithm Based on Maximal Information Coefficient. BMC Bioinformatics 17, 142. doi:10.1186/s12859-016-0990-0
Golub, T. R., Slonim, D. K., Tamayo, P., Huard, C., and Lander, E. S. (1999). Molecular Classification of Cancer: Class Discovery and Class Prediction by Gene Monitoring. Science 286, 531–537. doi:10.1126/science.286.5439.531
Hashim, F. A., Houssein, E. H., Mai, S. M., Al-Atabany, W., and Mirjalili, S. (2019). Henry Gas Solubility Optimization: A Novel Physics-Based Algorithm. Future Generation Comput. Syst. 101, 646–667. doi:10.1016/j.future.2019.07.015
Heidari, A. A., Mirjalili, S., Faris, H., Aljarah, I., Mafarja, M., and Chen, H. (2019). Harris Hawks Optimization: Algorithm and Applications. Future Generation Comput. Syst. 97, 849–872. doi:10.1016/j.future.2019.02.028
Hu, J., Bhowmick, P., Jang, I., Arvin, F., and Lanzon, A. (2021a). A Decentralized Cluster Formation Containment Framework for Multirobot Systems. IEEE Trans. Robotics 37, 1. doi:10.1109/tro.2021.3071615
Hu, J., Turgut, A. E., Lennox, B., and Arvin, F. (2021b). Robust Formation Coordination of Robot Swarms with Nonlinear Dynamics and Unknown Disturbances: Design and Experiments. IEEE Trans. Circuits Syst. Express Briefs 69, 114–118. doi:10.1109/TCSII.2021.3074705
Huang, K. K., Ramnarayanan, K., Zhu, F., Srivastava, S., Xu, C., Tan, A. L. K., et al. (2017). Genomic and Epigenomic Profiling of High-Risk Intestinal Metaplasia Reveals Molecular Determinants of Progression to Gastric Cancer. Cancer Cell 33, 137–150. doi:10.1016/j.ccell.2017.11.018
Karczewski, K. J., and Snyder, M. P. (2018). Integrative Omics for Health and Disease. Nat. Rev. Genet. 19, 299–310. doi:10.1038/nrg.2018.4
Kennedy, J., and Eberhart, R. (1995). “Particle Swarm Optimization,” in Proceedings of ICNN'95 - International Conference on Neural Networks, Perth, WA, Australia, 27 Nov.-1 Dec. 1995.
Krug, T., Gabriel, J. P., Taipa, R., Fonseca, B. V., Domingues-Montanari, S., Fernandez-Cadenas, I., et al. (2012). TTC7B Emerges as a Novel Risk Factor for Ischemic Stroke through the Convergence of Several Genome-wide Approaches. J. Cereb. Blood Flow Metab. 32, 1061–1072. doi:10.1038/jcbfm.2012.24
Levy, H., Wang, X., Kaldunski, M., Shuang, J., and Hessner, M. J. (2012). Transcriptional Signatures as a Disease-specific and Predictive Inflammatory Biomarker for Type 1 Diabetes. Genes Immun. 13, 593–604. doi:10.1038/gene.2012.41
Li, J., Cheng, K., Wang, S., Morstatter, F., Trevino, R. P., Tang, J., et al. (2017). Feature Selection: A Data Perspective. ACM Comput. Surv. (Csur) 50, 1–45. doi:10.1145/3136625
Li, S., Chen, H., Wang, M., Heidari, A. A., and Mirjalili, S. (2020). Slime Mould Algorithm: A New Method for Stochastic Optimization. Future Generation Comput. Syst. 111, 300–323. aliasgharheidari.com. doi:10.1016/j.future.2020.03.055
Liao, J. G., and Chin, K.-V. (2007). Logistic Regression for Disease Classification Using Microarray Data: Model Selection in a Large P and Small N Case. Bioinformatics 23, 1945–1951. doi:10.1093/bioinformatics/btm287
Liu, J., Cheng, X., Yang, W., Shu, Y., and Zhou., F. (2017). Multiple Similarly-Well Solutions Exist for Biomedical Feature Selection and Classification Problems. Scientific Rep. 7, 838. doi:10.1038/s41598-017-13184-8
Mafarja, M., Aljarah, I., Faris, H., Hammouri, A. I., Al-Zoubi, A.’M., and Mirjalili, S. (2019). Binary Grasshopper Optimisation Algorithm Approaches for Feature Selection Problems. Expert Syst. Appl. 117, 267–286. doi:10.1016/j.eswa.2018.09.015
Malakar, S., Ghosh, M., Bhowmik, S., Sarkar, R., and Nasipuri, M. (2019). A GA Based Hierarchical Feature Selection Approach for Handwritten Word Recognition. Neural Comput. Appl. 32 (7), 2533–2552. doi:10.1007/s00521-018-3937-8
Margolis, R., Derr, L., Dunn, M., Huerta, M., Larkin, J., Sheehan, J., et al. (2014). The National Institutes of Health's Big Data to Knowledge (BD2K) Initiative: Capitalizing on Biomedical Big Data. J. Am. Med. Inform. Assoc. 21, 957–958. doi:10.1136/amiajnl-2014-002974
Mckinney, B. C., Hensler, C. M., Wei, Y., Lewis, D. A., and Sweet, R. A. (2020). Schizophrenia-associated Differential DNA Methylation in the superior Temporal Gyrus Is Distributed to many Sites across the Genome and Annotated by the Risk Gene MAD1L1. medRxiv. doi:10.1101/2020.08.02.20166777
Moosavi, Shs., and Bardsiri, V. K. (2019). Poor and Rich Optimization Algorithm: A New Human-Based and Multi Populations Algorithm. Eng. Appl. Artif. Intelligence 86, 165–181. doi:10.1016/j.engappai.2019.08.025
Notterman, D. A., Alon, U. A., Sierk, A. J., and Levine, A. J. (2001). Transcriptional Gene Expression Profiles of Colorectal Adenoma, Adenocarcinoma, and Normal Tissue Examined by Oligonucleotide Arrays. Cancer Res. 61, 3124–3130. https://cancerres.aacrjournals.org/content/61/7/3124.long
Pomeroy, S. L., Tamayo, P., Gaasenbeek, M., and Sturia, L. M. (2002). Prediction of central Nervous System Embryonal Tumour Outcome Based on Gene Expression. Nature 415, 436–36. doi:10.1038/415436a
Qiu, Y., Ching, W.-K., and Zou, Q. (2021). Prediction of RNA-Binding Protein and Alternative Splicing Event Associations during Epithelial–Mesenchymal Transition Based on Inductive Matrix Completion. Brief. Bioinform. 22, bbaa440. doi:10.1093/bib/bbaa440
Robeck, T. R., Fei, Z., Lu, A. T., Haghani, A., Jourdain, E., Zoller, J. A., et al. (2021). Multi-species and Multi-Tissue Methylation Clocks for Age Estimation in Toothed Whales and Dolphins. Commun. Biol. 4, 1–11. doi:10.1038/s42003-021-02179-x
Shipp, M. A., Ross, K. N., Tamayo, P., Weng, A. P., Kutok, J. L., Aguiar, R. C. T., et al. (2002). Diffuse Large B-Cell Lymphoma Outcome Prediction by Gene-Expression Profiling and Supervised Machine Learning. Nat. Med. 8, 69–74. doi:10.1038/nm0102-68
Smith, R. G., Hannon, E., De Jager, P. L., Chibnik, L., Lott, S. J., Condliffe, D., et al. (2018). Elevated DNA Methylation across a 48-kb Region Spanning the HOXA Gene Cluster Is Associated with Alzheimer's Disease Neuropathology. Alzheimer Demen. 14, 1580–1588. doi:10.1016/j.jalz.2018.01.017
Tian, E., Zhan, F., Walker, R., Rasmussen, E., Ma, Y., Barlogie, B., et al. (2003). The Role of the Wnt-Signaling Antagonist DKK1 in the Development of Osteolytic Lesions in Multiple Myeloma. N. Engl. J. Med. 349, 2483–2494. doi:10.1056/nejmoa030847
Varsha, D., Gibbs, D. L., Theo, K., Roger, K., Joseph, V., John, N., et al. (2016). Using Incomplete Trios to Boost Confidence in Family Based Association Studies. Front. Genet. 7, 34. doi:10.3389/fgene.2016.00034
Wang, G., Hu, N., Yang, H. H., Wang, L., Hua, S., Wang, C., et al. (2013). Comparison of Global Gene Expression of Gastric Cardia and Noncardia Cancers from a High-Risk Population in China. Plos One 8, e63826. doi:10.1371/journal.pone.0063826
Wu, Y., Grabsch, H., Ivanova, T., Tan, I. B., Murray, J., Ooi, C. H., et al. (2013). Comprehensive Genomic Meta-Analysis Identifies Intra-tumoural Stroma as a Predictor of Survival in Patients with Gastric Cancer. Gut 62, 1100–1111. doi:10.1136/gutjnl-2011-301373
Yapici, H., and Cetinkaya, N. (2019). A New Meta-Heuristic Optimizer: Pathfinder Algorithm. Appl. Soft Comput. 74, 545–568. doi:10.1016/j.asoc.2019.03.012
Ylitalo, E. B., Thysell, E., Landfors, M., Brattsand, M., Jernberg, E., Crnalic, S., et al. (2021). A Novel DNA Methylation Signature Is Associated with Androgen Receptor Activity and Patient Prognosis in Bone Metastatic Prostate Cancer. Clin. Epigenetics 13, 1–15. doi:10.1186/s13148-021-01119-0
Yuanyuan, H., Huang, L., and Zhou, F. (2021). A Dynamic Recursive Feature Elimination Framework (dRFE) to Further Refine a Set of OMIC Biomarkers. Bioinformatics, 37 (15), 2183–2189. doi:10.1093/bioinformatics/btab055
Zhang, Y., Jin, Z., and Mirjalili, S. (2020). Generalized normal Distribution Optimization and its Applications in Parameter Extraction of Photovoltaic Models. Energ. Convers. Manage. 224, 113301. doi:10.1016/j.enconman.2020.113301
Zhao, W., Wang, L., and Zhang, Z. (2019). Atom Search Optimization and its Application to Solve a Hydrogeologic Parameter Estimation Problem. Knowledge-Based Syst. 163, 283–304. doi:10.1016/j.knosys.2018.08.030
Keywords: swarm intelligence (SI), feature selection (FS), transcriptome data, methylation data, intersection and union combination
Citation: Yao Z, Zhu G, Too J, Duan M and Wang Z (2022) Feature Selection of OMIC Data by Ensemble Swarm Intelligence Based Approaches. Front. Genet. 12:793629. doi: 10.3389/fgene.2021.793629
Received: 12 October 2021; Accepted: 22 December 2021;
Published: 08 March 2022.
Edited by:
Lin Hua, Capital Medical University, ChinaReviewed by:
Yushan Qiu, Shenzhen University, ChinaCopyright © 2022 Yao, Zhu, Too, Duan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiguo Wang, d2FuZ3poaWd1bzU3NzhAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.