 Fengyu Che
Fengyu Che Jiangang Zhao
Jiangang Zhao Yujuan Zhao
Yujuan Zhao Zhi Wang
Zhi Wang Liyu Zhang
Liyu Zhang Ying Yang
Ying Yang- 1Shaanxi Institute for Pediatric Diseases, Xi’an Children’s Hospital, Xi’an, China
- 2Department of Neonatology, Xi’an Children’s Hospital, Xi’an, China
Aim: To determine the etiology of a Chinese family with thrombocytopenia by analyzing the clinical features and genetic variation.
Methods: Clinical profiles and genomic DNA extracts of the family members were collected for the study. Whole exome sequencing and Sanger sequencing was used to detect the associated genetic variation and verify the family co-segregation respectively. Bioinformatics analysis assessed the pathogenicity of missense mutations.
Results: The study reported a 3-generation pedigree including eight family members with thrombocytopenia. The platelet counts of the patients were varied, ranging from 38 to 110 × 109/L (reference range: 150–450 x 109/L). The mean volumes and morphology of the sampled platelet were both normal. The bleeding abnormality and mitochondriopathy were not observed in all the patients. Clinical signs of thrombocytopenia were mild. A novel heterozygous missense variant c.79C > T (p.His27Tyr) was identified in CYCS gene associated with autosomal dominant thrombocytopenia.
Conclusion: We report the first large family with autosomal dominant non-syndromic thrombocytopenia 4 in a Chinese family, a novel heterozygous missense variant c.79C > T (p.His27Tyr) was identified. The whole exome sequencing is an efficient tool for screening the variants specifically associated with the disease. The finding enriches the mutation spectrum of CYCS gene and laid a foundation for future studies on the correlation between genotype and phenotype.
1 Introduction
Thrombocytopenia is defined as a haematological condition with blood platelet count below 100–150 × 109/L (Greenberg and Kaled, 2013). It is correlated with inherited hematologic diseases, immune diseases (e.g.rheumatoid), radiation/chemotherapy injuries, infection, and drug-related thrombocytopenia (Lee and Lee, 2016; Franchini et al., 2017). At present, more than 20 genes have been reported to be associated with the incidence of inherited thrombocytopenia, including syndromic, and non-syndromic (Nurden and Nurden, 2020). Hence, understanding the disease from a genetic perspective is a key of providing effective diagnosis and prognostic risk assessment.
The CYCS gene encodes the Cytochrome C (Cyt-c), which is a small and stable heme protein with heme C as its auxiliary group (Stevens, 2011). The Cyt-c protein is an essential component anchoring in the inner membrane of the mitochondrion for multiple bio-functions (Santucci et al., 2019). It is mainly responsible of transferring electrons from cytochrome b to the cytochrome oxidase complex (Kagan et al., 2005; Li et al., 2006; Ow et al., 2008), and also could initiate cell apoptosis as an antioxidant agent (Wang et al., 2003; Zhao et al., 2003; Lee and Xu, 2007). It is reported that the heterozygous mutation of CYCS gene on chromosome 7p15 can caused autosomal dominant non-syndromic thrombocytopenia-4 (THC4, OMIM: 612004) (Morison et al., 2008). The clinical features of THC4 were characterized with mild thrombocytopenia, normal platelet size and morphology, and no increased bleeding tendency. So far, there are only 4 pedigrees with nonsyndrome thrombocytopenia and 1 pedigree with hemophilia A, associated with variants in CYCS gene (Morison et al., 2008; De Rocco et al., 2014; Johnson et al., 2016; Uchiyama et al., 2018; Turro et al., 2020). None of the patients in these reports showed symptoms resulting from cell apoptosis and abnormal mitochondrial oxidative respiratory chain, whereas thrombocytopenia was present.
We studied a first pedigree with autosomal dominant non-syndrome thrombocytopenia 4 in a Chinese family. All patients in the family were found to carry a heterozygotic missense variant in CYCS gene by whole exome sequencing (WES), while the pathogenicity of the genetic variation was also evaluated. According to the clinical and genetic features of the patients in the pedigree, thrombocytopenia 4 was diagnosed. This work could be a scientific evidence to support on thrombocytopenia diagnosis and its prognostic management.
2 Materials and Methods
2.1 Patient Clinical Information
The probands (IV-2) was the second son of a non-consanguineous Chinese couple (Figure 1A). He was born at gestational age of 39 + 5 weeks by spontaneous vaginal delivery with normal birth history (birth weight, 3500 g). Multiple hemorrhagic spots were found on the facial and back skin of the proband at birth. After 7 days, he was admitted for further treatment. Physical examination was normal. Brain magnetic resonance imaging (MRI) showed mild cerebral hemorrhage in the left ventricle and focal white matter injury near the posterior horn of the left ventricle. The full blood count test yielded a low platelet count at 54 × 109/L (reference range: 150–450 × 109/L) and the increased leucocyte count at 30.68 × 109/L (reference range: 10.4–12.21 × 109/L). After antibiotics and immunoglobulin treatment, the hemorrhagic spots disappeared with normal leucocyte counts and improved platelet counts (ranges from 56 to 96 × 109/L).
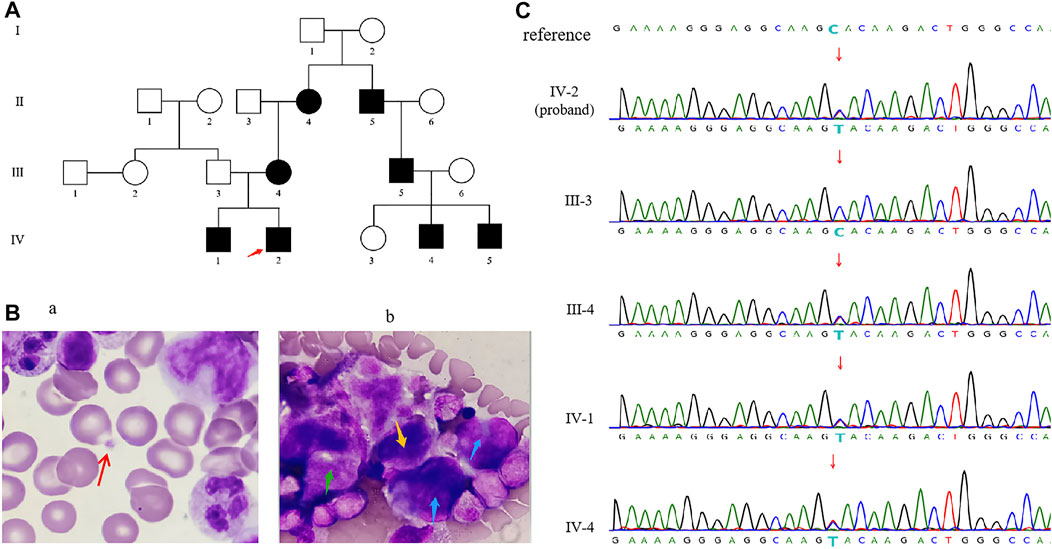
FIGURE 1. (A) Familial pedigree of CYCS-mutated thrombocytopenia. The red arrow indicates the proband. (B) Optical microscopy of bone marrow cells from the proband showed the number of platelets were reduced. The red arrow on the left (Ba) showed a single platelet was sporadically visible, and the blue arrow points to promegakaryocytes (Bb), the green arrow points to granular megakaryocytes (Bb), and the yellow arrow points to thromocytogenic megakaryocytes (Bb). (C) Sanger sequencing of CYCS gene c.79C > T (p.His27Tyr) variant in genomic DNA from the family.
At 16 days of age, the patient showed no signs of hemorrhage with normal abdominal ultrasound and urine organic acids. Blood tandem mass spectrometry analysis showed camitine deficiency. Optical microscope of bone marrow showed megaloblasts and toxic granules below the stage of myelocyte and metamyelocyte, indicating as bone marrow infection. In addition, the whole bone marrow smear showed 550 megakaryocytes, among which promegakaryocytes accounted for 30%, granular megakaryocytes accounted for 62%, thromocytogenic megakaryocytes accounted for 8%. These abnormal proportions of megakaryocytes indicate that megakaryocytes maturation problem leaded to a decrease in platelet count (Figure 1B). Subsequently, he was instructed to continue antibiotics therapy and oral administration of levocarnitine. At 34 days of age, the patient showed no signs of hemorrhage. The results of the secondary MRI and blood tandem mass spectrometry analysis were normal. The platelet counts maintained at decreased levels in multiple regular examinations, one of which showed normal level at 108 × 109/L (reference range: 150–450 × 109/L).
Clinical features of his family members were summarized in supplemental Table 1, which details a history of thrombocytopenia in the proband’s maternal family member. The patient’s father is healthy. Of the 11 subjects, 8 presented low platelet counts (IV-2, IV-1, III-4, II-4, II-5, III-5, IV-4, IV-5), and the remaining subjects showed normal platelet level (III-3, III-6, IV-3). The platelet counts of IV-2, IV-1, III-4, and II-4 were examined multiple times, while the rest examined once. The studied family members, including the proband, had platelet counts ranging from 38 to 110 × 109/L with a reference interval of 150–450 × 109/L. The mean volumes and morphology of the sampled platelet were normal. Most studied patients had mild bleeding of skin mucosa, except IV-5 with epistaxis, and the proband showed the spontaneous skin bleeding (Supplemental Table 1). All the patients had no symptom of mitochondrial disease such as neurodegeneration, diabetes, myopathy, eye, and kidney diseases.

TABLE 1. Clinical features of the family members from the current study.
2.2 Sample Collection
This study was approved by the Ethics Committee of Xi’an Children’s Hospital, and written informed consent was obtained from each participant or their guardian(s). The peripheral blood (3 ml) was individually sampled from the proband and his family members and collected in EDTA anticoagulant tube.
2.3 Whole Exome Sequencing
1 μg genomic DNA was extracted from 200 µL peripheral blood using a Qiagen DNA Blood Midi/Mini kit (Qiagen GmbH, Hilden, Germany) following the manufacturer’s protocol. Library preparation was performed using NanoWES (Berry Genomics, China) according to the manufacturer’s Protocol. Novaseq6000 platform (Illumina, San Diego, United States) was used for sequencing. The exome sequencing was performed with a minimum median coverage of 80X. The sequencing reads were aligned to the human reference genome (hg38/GRCh38) using Burrows–Wheeler Aligner tool. Verita Trekker® Variants Detection System by Berry Genomics and the third-party software GATK were employed for variant calling. Variants with lower quality (read depth < 10×, allele fraction < 30%) were eliminated. Variant annotation and interpretation were conducted by ANNOVAR (Wang et al., 2010) and the Enliven® Variants Annotation Interpretation System authorized by Berry Genomics. All variants were filtered through population databases including the 1,000 Genomes Project (1000G), Exome Aggregation Consortium (ExAC), and gnomAD, only those variants with population frequencies less than 1/1,000 in all databases were counted. Variant pathogenicity/deleteriousness prediction was evaluated using SIFT, Poly-Phen_2, Mutation Taster, REVEL, FATHMM, CADD. Prediction of variant impact on splicing was evaluated by dbscSNV, Human Splicing Finder (HSF), and SpliceAI. To maximize clinically diagnostic yield, the known pathogenic variants from Human Gene Mutation Database and ClinVar (Landrum et al., 2016; Stenson et al., 2020) were also retained for further evaluation. The variants were classified to five categories “pathogenic,” “likely pathogenic,” “uncertain significance,” “likely benign,” and “benign”--according to the American College of Medical Genetics and Genomics (ACMG) guidelines for interpretation of genetic variants (Richards et al., 2015). The suspected SNV/Indels were validated using Sanger sequencing.
The three-dimensional (3D) structure of CYCS protein of the missense variant was analyzed by Swiss-PDBviewer (PDB:3ZOO).
3 Results
3.1 Genetic Analysis and Co-Segregation in the Family
A heterozygous variant in the CYCS (NM_018947.6: c.79C > T (p.His27Tyr) was identified for the proband by WES. Sanger sequencing further confirmed that all affected members (IV-2, IV-1, III-4, II-4, II-5, III-5, IV-4, IV-5) carried the same heterozygous variant, while other studied members (III-3, III-6, IV-3) did not carry it (Figure 1C). So, the variant segregated with the disorder in this family (PP1_Strong). This novel variant has not been reported in previous literature, and the c.79C > T substitution was not seen in gnomAD database (PM2_Supporting). Sequence alignment of CYCS protein among multiple species showed that p.His27 is highly conserved across evolution (Figure 2A), suggesting that His27 could play a vital role in maintaining the stability and function of proteins. The results of multiple statistical methods (REVEL) predicted that the variant could cause detrimental effect on gene function (PP3). According to the ACMG guidelines, c.79C > T (p.His27Tyr) is defined as “Likely Pathogenic”. This novel variant has been submitted to ClinVar with the variation ID: 1210164.
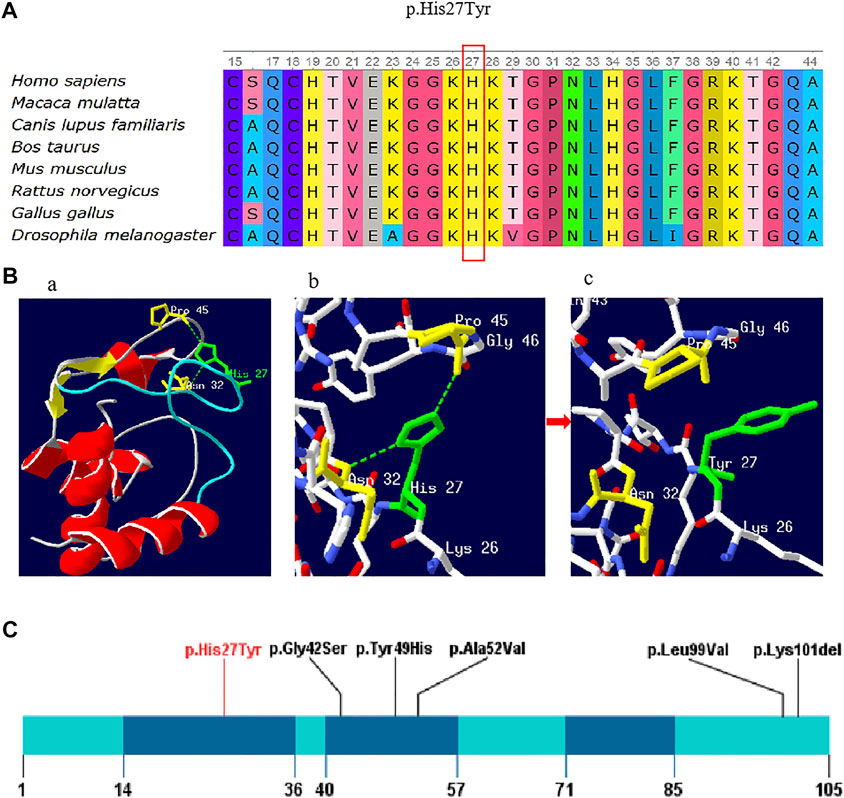
FIGURE 2. (A) Conservation of the p. His27Tyr variant across various species. (B) Amino acid and conformation changes of the p.His27Tyr polypeptide wild-type and mutant type. His27 is located in the random coil domain of CYCS by colored green (Ba); Stick models shows the amino acids around His27 and the selected side chains, wild-type His27 forms a hydrogen bond (green dotted line) with Asn32 and Pro45 (colored yellow) respectively (Bb), and the hydrogen bond was lost in mutant type Tyr27 (Bc). (C) Schematic presentation of linear CYCS protein (NM_018947.6) with all variants, red font indicate reported variant in the present study. The dark blue indicates three Ω-loops.
3.2 Structure-Function Correlations of CYCS Variants
The impact of this missense variant was also evaluated by examining the 3D structural viewpoint of the variant protein. Our study mapped the mutation position onto the crystal structure of human cytochrome c (PDB:3ZOO). As shown in Figure 2B, p.His27Tyr variant is located in the random coil domain of CYCS (Figure 2Ba), which may affect the global conformation and activity of protein. When His27 is replaced by Tyr, the hydrogen bonds of His27 interacting with Asn32 and Pro45 were broken, and the amino acid side chain is changed from imidazole to benzene (Figures 2Bb,Bc).
4 Discussion
In this study, we firstly reported a 3-generation family with autosomal dominant non-syndromic thrombocytopenia in China. The whole exome sequencing identified a novel missense heterozygous variant c.79C > T (p.His27Tyr) of CYCS gene in the pedigree with eight patients (IV-2, IV-1,III-4, II-4, II-5, III-5, IV-4, IV-5). The clinical features of the studied patients were consistent with that from other reports (Morison et al., 2008; De Rocco et al., 2014; Johnson et al., 2016). More, they had normal fertility and longevity, with no evidence of mitochondriopathies (Finsterer, 2004), while other family members (III-3, III-6, IV-3) of the study did not carry the variant. It was assumed that the variation p. His27Tyr of CYCS could be the genetic etiology of autosomal dominant non-syndromic thrombocytopenia in this family case. The finding enriches the mutation spectrum of CYCS gene and laid a foundation for future studies on the correlation between genotype and phenotype. This conclusion may help patients to prevent the disease risk associated with thrombocytopenia and help clinicians to do differential diagnoses including acquired and inherited forms.
In 2008, Morison et al. reported the first variant of the CYCS gene identified in a family with thrombocytopenia (Morison et al., 2008). Globally, total five variants have been reported to be associated with thrombocytopenia, including four missense variant (p.Gly42Ser, p.Tyr49His, p.Ala52Val, and p.Leu99Val) and one small deletion variant p.Lys301del (in-frame) (De Rocco et al., 2014; Johnson et al., 2016; Uchiyama et al., 2018; Turro et al., 2020). The p. His27Tyr variant found in this study was the fifth missense mutation. To date, multiple studies has investigated the missense mutation effect on physical structure and bio-function of CYCS protein. Liptak et al. demonstrated that the proapoptotic Gly42Ser mutation altered the heme electronic structure and increased the rate of electron self-exchange, resulting in the enhanced proapoptotic activity of Gly42Ser (Liptak et al., 2011). De Rocco et al. reported that the p. Gly42Ser and p.Tyr49His variants in yeast and mouse cellular models were responsible of the diminished respiratory level and increased apoptotic rate (De Rocco et al., 2014). Lei et al. showed that the Ala51Val variant enhanced peroxidase activity by destabilizing the native state of Cyt-c, and all three variants Gly42Ser, Tyr49His, and Ala51Val had reduced global and local stability than that of wild type Cyt-c (Lei and Bowler, 2019). Moreover, Uchiyama et al. provided that the mutation of p.Lys301del could significantly reduced cytochrome c protein expression and cause functional defects in the mitochondrial respiratory chain (Uchiyama et al., 2018). The existing findings are not enough to explain the molecular mechanism of thrombocytopenia, however, they could be used as scientific evidence for thrombocytopenia diagnosis.
Cyt-c, a highly evolved protein in different species (St-evens, 2011), contains three highly conserved Ω-loops including residues 14–36, 40–57, and 71–85 (Karsisiotis et al., 2016). Ω-loops play an important role in maintaining the protein function and stability (Fetrow, 1995). Of the five reported missense variants, three variants p.Gly42Ser, p.Tyr49His, and p.Ala52Val were located in the second Ω-loop domain (residues 40–57), two variants p.Leu99Val, and p.Lys301del were located in the C-terminal of the protein. The p.His27Tyr variant found in this study was in the first Ω-loop (residues 14–36) (Figure 2C), which may affect the stability of Cyt-c. Previous studies have reported that His27 in the wild type Cyt-c interacted with Pro45 through an hydrogen bond which is essential for sustaining the orientation of the heme conformation and the α-helices, inflicting the cardiolipin binding to cyt-c and subsequent apoptotic events (Balakrishnan et al., 2012). Additionally, the 3D structure of CYCS protein was predicted and it showed that the amino acid substitution (His27Tyr) could lead to the cleavage of hydrogen bonds between His27 and Pro45, causing the instability of the protein structure and destruction of its biological function. For mutant protein, the positive charged Histidine was replaced by the neutral Tyrosine, which negatively influence the electron transport in mitochondrial oxidative respiratory chain. Hence, we assumed that the variant p.His27Tyr could be the genetic etiology of the thrombocytopenia in the family case. Nevertheless, the genetic effect of this variant needs to be verified in future study.
In summary, with the rapid development of molecular biotechnology, genetic analysis has been widely used in rare disease diagnosis. Our findings demonstrated that a new missense variant of the CYCS gene was associated with non-syndromic thrombocytopenia identified by WES. Further research is required to understand the impact of CYCS variants on changing platelet production. The outcome could be beneficial for thrombocytopenia diagnosis and prognostic management.
Data Availability Statement
The datasets presented in this article are not readily available due to ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Xi’an Children’s Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
FC, JZ, and YY contributed to conception and design of the study. YZ organized the database. ZW and LZ performed the statistical analysis. FC wrote the first draft of the article. YY and JZ wrote sections of the article. All authors contributed to article revision, read, and approved the submitted version.
Funding
This work was supported by Shaanxi Provincial Science and Technology Project (No. 2019KJXX-055) and Xi’an Children’s Hospital research project (No. 2020A07).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank all of the participation of the family members in this study. And to thank the Berry Gnomics Co., for the technical support.
References
Balakrishnan, G., Hu, Y., and Spiro, T. G. (2012). His26 Protonation in Cytochrome C Triggers Microsecond β-sheet Formation and Heme Exposure: Implications for Apoptosis. J Am Chem Soc 134 (46), 19061–19069. doi:10.1021/ja307100a
De Rocco, D., Cerqua, C., Goffrini, P., Russo, G., Pastore, A., Meloni, F., et al. (2014). Mutations of Cytochrome C Identified in Patients with Thrombocytopenia THC4 Affect Both Apoptosis and Cellular Bioenergetics. Biochim Biophys Acta 1842 (2), 269–274. doi:10.1016/j.bbadis.2013.12.002
Fetrow, J. S. (1995). Omega Loops: Nonregular Secondary Structures Significant in Protein Function and Stability. FASEB J 9 (9), 708–717. doi:10.1096/fasebj.9.9.7601335
Finsterer, J. (2004). Mitochondriopathies. Eur J Neurol 11 (3), 163–186. doi:10.1046/j.1351-5101.2003.00728.x
Franchini, M., Veneri, D., and Lippi, G. (2017). Thrombocytopenia and Infections. Expert Rev Hematol 10 (1), 99–106. doi:10.1080/17474086.2017.1271319
Greenberg, E. M., and Kaled, E. S. (2013). Thrombocytopenia. Crit Care Nurs Clin North Am 25 (4), 427–434. doi:10.1016/j.ccell.2013.08.003
Johnson, B., Lowe, G. C., Futterer, J., Lordkipanidzé, M., MacDonald, D., Simpson, M. A., et al. (2016). Whole Exome Sequencing Identifies Genetic Variants in Inherited Thrombocytopenia with Secondary Qualitative Function Defects. Haematologica 101 (10), 1170–1179. doi:10.3324/haematol.2016.146316
Kagan, V. E., Tyurin, V. A., Jiang, J., Tyurina, Y. Y., Ritov, V. B., Amoscato, A. A., et al. (2005). Cytochrome C Acts as a Cardiolipin Oxygenase Required for Release of Proapoptotic Factors. Nat Chem Biol 1 (4), 223–232. doi:10.1038/nchembio727
Karsisiotis, A. I., Deacon, O. M., Wilson, M. T., Macdonald, C., Blumenschein, T. M., Moore, G. R., et al. (2016). Increased Dynamics in the 40-57Ω-Loopof the G41S Variant of Human Cytochrome C Promote its Pro -apoptoticconformation. Sci Rep 6, 30447. doi:10.1038/srep30447
Landrum, M. J., Lee, J. M., Benson, M., Brown, G., Chao, C., Chitipiralla, S., et al. (2016). ClinVar: Public Archive of Interpretations of Clinically Relevant Variants. Nucleic Acids Res 44, D862–D868. doi:10.1093/nar/gkv1222
Lee, E. J., and Lee, A. I. (2016). Thrombocytopenia. Prim Care 43 (4), 543–557. doi:10.1016/j.pop.2016.07.008
Lei, H., and Bowler, B. E. (2019). Naturally Occurring A51V Variant of Human Cytochrome C Destabilizes the Native State and Enhances Peroxidase Activity. J Phys Chem B 123 (42), 8939–8953. doi:10.1021/acs.jpcb.9b05869
Li, Y., Park, J. S., Deng, J. H., and Bai, Y. (2006). Cytochrome C Oxidase Subunit IV Is Essential for Assembly and Respiratory Function of the Enzyme Complex. J Bioenerg Biomembr 38 (5-6), 283–291. doi:10.1007/s10863-006-9052-z
Liptak, M. D., Fagerlund, R. D., Ledgerwood, E. C., Wilbanks, S. M., and Bren, K. L. (2011). The Proapoptotic G41S Mutation to Human Cytochrome C Alters the Heme Electronic Structure and Increases the Electron Self-Exchange Rate. J Am Chem Soc 133 (5), 1153–1155. doi:10.1021/ja106328k
Lee, M., and Xu, J. X. (2007). Detoxifying Function of Cytochrome C against Oxygen Toxicity. Mitochondrion 7 (1-2), 13–16. doi:10.1016/j.mito.2006.11.011
Morison, I. M., Cramer Bordé, E. M., Cheesman, E. J., Cheong, P. L., Holyoake, A. J., Fichelson, S., et al. (2008). A Mutation of Human Cytochrome C Enhances the Intrinsic Apoptotic Pathway but Causes Only Thrombocytopenia. Nat Genet 40 (4), 387–389. doi:10.1038/ng.103
Nurden, A. T., and Nurden, P. (2020). Inherited Thrombocytopenias: History, Advances and Perspectives. Haematologica 105 (8), 2004–2019. doi:10.3324/haematol.2019.233197
Ow, Y. P., Green, D. R., Hao, Z., and Mak, T. W. (2008). Cytochrome C: Functions beyond Respiration. Nat Rev Mol Cell Biol 9 (7), 532–542. doi:10.1038/nrm2434
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17 (5), 405–424. doi:10.1038/gim.2015.30
Santucci, R., Sinibaldi, F., Cozza, P., Polticelli, F., and Fiorucci, L. (2019). Cytochrome C: An Extreme Multifunctional Protein with a Key Role in Cell Fate. Int J Biol Macromol 136, 1237–1246. doi:10.1016/j.ijbiomac.2019.06.180
Stenson, P. D., Mort, M., Ball, E. V., Chapman, M., Evans, K., Azevedo, L., et al. (2020). The Human Gene Mutation Database (HGMD®): Optimizing its Use in a Clinical Diagnostic or Research Setting. Hum Genet 139 (10), 1197–1207. Epub 2020 Jun 28. doi:10.1007/s00439-020-02199-3
Stevens, J. M. (2011). Cytochrome C as an Experimental Model Protein. Metallomics 3 (4), 319–322. doi:10.1039/c0mt00089b
Turro, E., Astle, W. J., Megy, K., Gräf, S., Greene, D., Shamardina, O., et al. (2020). Whole-genome Sequencing of Patients with Rare Diseases in a National Health System. Nature 583 (7814), 96–102. doi:10.1038/s41586-020-2434-2
Uchiyama, Y., Yanagisawa, K., Kunishima, S., Shiina, M., Ogawa, Y., Nakashima, M., et al. (2018). A Novel CYCS Mutation in the α-helix of the CYCS C-Terminal Domain Causes Non-syndromic Thrombocytopenia. Clin Genet 94 (6), 548–553. doi:10.1111/cge.13423
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: Functional Annotation of Genetic Variants from Next-Generation Sequencing Data. Nucleic Acids Res 38 (16), e164. Epub 2010 Jul 3. doi:10.1093/nar/gkq603
Wang, Z. B., Li, M., Zhao, Y., and Xu, J. X. (2003). Cytochrome C Is a Hydrogen Peroxide Scavenger in Mitochondria. Protein Pept Lett 10 (3), 247–253. doi:10.2174/0929866033479013
Keywords: CYCS gene, platelet counts, cytochrome c, mutation, thrombocytopenia
Citation: Che F, Zhao J, Zhao Y, Wang Z, Zhang L and Yang Y (2022) A Novel Heterozygous Pathogenic Variation in CYCS Gene Cause Autosomal Dominant Non-Syndromic Thrombocytopenia 4 in a Large Chinese Family. Front. Genet. 12:783455. doi: 10.3389/fgene.2021.783455
Received: 26 September 2021; Accepted: 06 December 2021;
Published: 18 January 2022.
Edited by:
Kathleen M. Gorman, Temple Street Children’s University Hospital, IrelandReviewed by:
Stefano Castellana, Casa Sollievo della Sofferenza (IRCCS), ItalyAlejandro Ferrer, Mayo Clinic, United States
Copyright © 2022 Che, Zhao, Zhao, Wang, Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Yang, eXlpbmcxOTMwQDE2My5jb20=