 Sher Alam Khan1
Sher Alam Khan1 Saadullah Khan1*
Saadullah Khan1* Noor Muhammad1Zia Ur Rehman1Muhammad Adnan Khan2
Noor Muhammad1Zia Ur Rehman1Muhammad Adnan Khan2 Abdul Nasir3
Abdul Nasir3 Umm-e- Kalsoom4Anwar Kamal Khan1Hassan Khan1
Umm-e- Kalsoom4Anwar Kamal Khan1Hassan Khan1 Naveed Wasif5,6*
Naveed Wasif5,6*- 1Department of Biotechnology and Genetic Engineering, Kohat University of Science and Technology (KUST), Kohat, Pakistan
- 2Dental Material, Institute of Basic Medical Sciences, Khyber Medical University Peshawar, Peshawar, Pakistan
- 3Department of Molecular Science and Technology, Ajou University, Suwon, South Korea
- 4Department of Biochemistry, Hazara University, Mansehra, Pakistan
- 5Institute of Human Genetics, University of Ulm, Ulm, Germany
- 6Institute of Human Genetics, University Hospital Schleswig-Holstein, Campus Kiel, Kiel, Germany
Background: The syndromic and non-syndromic congenital missing teeth phenotype is termed tooth agenesis. Since tooth agenesis is a heterogeneous disorder hence, the patients show diverse absent teeth phenotypes. Thus identifying novel genes involved in the morphogenesis of ectodermal appendages, including teeth, paves the way for establishing signaling pathways.
Methods and Results: We have recruited an autosomal recessive non-syndromic tooth agenesis family with two affected members. The exome sequencing technology identified a novel missense sequence variant c.1421T > C; p.(Ile474Thr) in a regulatory factor X (RFX) family member (RFX2, OMIM: 142,765). During the data analysis eight rare variants on various chromosomal locations were identified, but the co-segregation analysis using Sanger sequencing confirmed the segregation of only two variants RFX2: c.1421T > C; p.(Ile474Thr), DOHH: c.109C > G; p.(Pro37Ala) lying in a common 7.1 MB region of homozygosity on chromosome 19p13.3. Furthermore, the online protein prediction algorithms and protein modeling analysis verified the RFX2 variant as a damaging genetic alteration and ACMG pathogenicity criteria classified it as likely pathogenic. On the other hand, the DOHH variant showed benign outcomes.
Conclusion: RFX2 regulates the Hedgehog and fibroblast growth factor signaling pathways, which are involved in the epithelial and mesenchymal interactions during tooth development. Prior animal model studies have confirmed the expression of rfx2 at a developmental stage governing mouth formation. Moreover, its regulatory role and close association with ciliary and non-ciliary genes causing various dental malformations makes it a potential candidate gene for tooth agenesis phenotype. Further studies will contribute to exploring the direct role of RFX2 in human tooth development.
Introduction
Tooth agenesis (TA) is a craniofacial malformation characterized by the absence of one or more teeth due to failure in the early stages of odontogenesis (Letra et al., 1993). TA is one of the most commonly occurring orofacial congenital anomalies in humans, with a prevalence of ∼3–11% depending upon the ethnicity and geography of the population (Larmour et al., 2005; Shimizu and Maeda, 2009). Population-based studies have shown that the incidence of third molar agenesis (20%) is the most commonly known missing teeth phenotype. Excluding third molars, the prevalence of permanent teeth agenesis is higher (1.6–9.6%) than the primary dentition (0.5–0.9%) (Vastardis, 2000). Based on the number of missing teeth, TA can be classified into three categories: hypodontia (<six missing teeth), oligodontia (>six missing teeth), and anodontia (congenital absence of all primary and permanent teeth) (Stockton et al., 2000; De Coster et al., 2009). Hypodontia is relatively a common condition (1.6–6.9%) as compared to oligodontia (0.14%), while anodontia is extremely rare (Schalk-van der Weide et al., 1992; Polder et al., 2004; Al-Ani et al., 2017). Environmental (infections, trauma, chemotherapy, and radiotherapy) and genetic factors have been reported to cause TA phenotypes, but the latter is the most common cause of TA (Chhabra et al., 2014). Tooth agenesis shows autosomal recessive/dominant (Murakami et al., 2017; Park et al., 2019) and X-linked recessive/dominant patterns of inheritances (Rasool et al., 2008; Sarkar et al., 2014). Formerly, the monogenic inheritance was considered for TA in the previous studies, but recently several studies have proposed oligogenic patterns (Dinckan et al., 2018; Du et al., 2018), supporting the concept of mutational load in human genetic disorders (Posey et al., 2017).
Tooth development is accomplished in a highly coordinated and genetically controlled fashion (Kapadia et al., 2007; Brook, 2009) and is regulated via some molecules involved in the signaling cascades (Kapadia et al., 2007), initiating a series of reciprocal interactions between the epithelium and underlying mesenchyme (Thesleff, 2006). The regular expression of several hundred genes is essential for tooth development (Thesleff, 2014). Genetic alterations in numerous genes have been reported to arrest tooth development in mice and/or humans (Kapadia et al., 2007; Fleischmannova et al., 2008; Brook, 2009; Thesleff, 2014). Four signaling pathways (FGF, BMP, Wnt, and SHH) lead the human tooth formation. In addition, a large number of genes encoding various components of these pathways are either directly or indirectly involved in various tooth conditions (Neubüser et al., 1997; Jussila and Thesleff, 2012). Their loss of function or gain of function may distort the signaling cascades and cause various orofacial anomalies (Nieminen, 2009).
Based on the clinical manifestations, TA can be divided into two sub-categories: congenital non-syndromic tooth agenesis (NSTA) and syndromic tooth agenesis (STA) (Yu et al., 2019). In addition, TA is a prominent feature of more than 300 syndromes (Dinckan et al., 2018), involving primarily in oro-facial cleft and ectodermal dysplasia (Nieminen, 2009), including Witkop syndrome (OMIM: 189,500) (Jumlongras et al., 2001), hypohidrotic ectodermal dysplasia (HED) (Parveen et al., 2019; Khan et al., 2020), Ellis-van Creveld syndrome (EVC, OMIM: 225,500) (Niceta et al., 2018), odonto-onycho-dermal dysplasia (OODD, OMIM: 257,980) (Adaimy et al., 2007) and Bloch-Sulzberger syndrome (OMIM: 308,300) (Si and Liu, 2018).
The genetic etiology of tooth agenesis is heterogeneous. To date, approximately nine genes have been identified to cause different NSTA conditions. Based on clinical phenotypes of TA in the literature and OMIM database (https://www.omim.org/), it can be generalized that some of the genes cause only NSTA, but others are involved in both the NSTA and STA. For instance, GREM2 (OMIM: 608,832) cause non-syndromic hypodontia (NSH). Similarly, LRP6 (OMIM: 603,507) cause non-syndromic oligodontia (NSO), while EDARADD (OMIM: 606,603) and AXIN2 (OMIM: 604,025) may cause both the NSO and STA. Four genes including, MSX1 (OMIM: 142,983), EDAR (OMIM: 606,095), WNT10A (OMIM: 606,268) and EDA (OMIM: 300,451) may cause NSH, NSO and STA, while PAX9 (OMIM: 167,416) is involved in causing only NSH and NSO (Fournier et al., 2018).
We aimed to characterize the clinical features of an autosomal recessive Pakistani family displaying tooth agenesis and to identify an underlying genetic cause. We present a comprehensive clinical investigation followed by exome sequencing analysis. The exome sequencing data revealed a novel biallelic variant c.1421T > C; p.(Ile474Thr) in 13-exon of RFX2.
Materials and Methods
Family Recruitment, Pedigree Construction, and Blood Collection
A four-generation consanguineous sixteen membered family with two affected individuals (IV-4 and IV-5) with missing teeth phenotype was recruited from the southern region of Khyber Pakhtunkhwa province, Pakistan. Pedigree was constructed based on the information provided by the father (III-1) of the affected members (Figure 1A). Informed written consent was taken from all the participants. The Research and Ethical Committee (REC) of Kohat University of Science and Technology (KUST), Kohat, Pakistan, approved the study protocols, strictly following the recommendations of the Declarations of Helsinki.
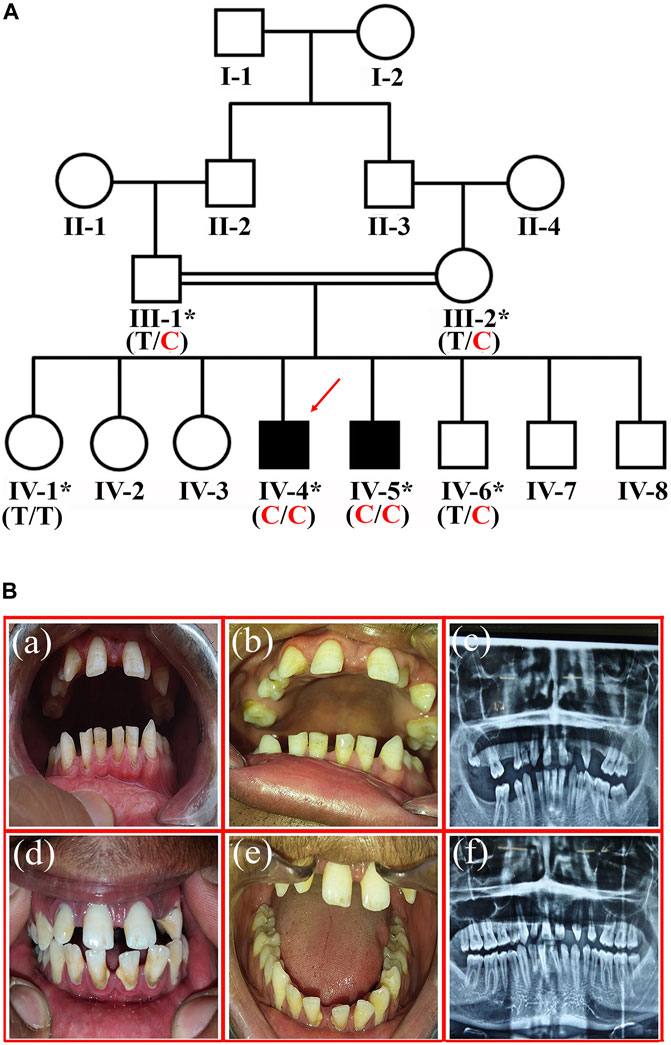
FIGURE 1. (A) Pedigree of the family showing segregation of RFX2 variant c.1421T > C; p.(Ile474Thr) in an autosomal recessive fashion. The asterisks show participating individuals. The red arrow indicates the index patient subjected for exome sequencing. C shows the disease-allele, while T is the wild-type allele (B). The representation of tooth agenesis. (a), and (b) are the clinical features of patient IV-4 showing generalized spacing in the anterior region of maxilla and mandible. (c) the Orthopantomogram (OPG) of patient IV-4 shows the absence of permanent maxillary lateral incisors, left maxillary first premolar, and all four 2nd premolars (mandibular and maxillary). It also shows the missing right maxillary second molar, left maxillary third molar, right mandibular second and third molars. (d), and (e) are the clinical photographs of patient IV-5, showing the generalized spacing in the anterior region of the maxilla and mandible. (f) is the Orthopantomogram (OPG) of patient IV-5, showing the absence of permanent maxillary lateral incisors, maxillary second premolars, and right maxillary first premolar.
DNA Extraction
Genomic DNA was isolated from the whole peripheral blood of the patients (IV-4 and IV-5) and other family members (III-1, III-2, IV-1 and IV-6) by using the GenElute™ blood genomic DNA kit (Sigma-Aldrich MO, United States). Qubit Fluorometer (ThermoFisher Scientific, United States) was used for the quantification of DNA.
Exome Sequencing, Alignment and Variant Calling
A 100 ng/μl DNA of two members (IV-1 and IV-4) was used for exome sequencing. The sequencing libraries of the DNA were prepared with the SeqCap EZ human exome library v2.0 kit. The sequencing was done on Illumina HiSeq 4000 sequencing machine via a paired-end 100-bp protocol (Hussain et al., 2013). The filtration of primary data was carried out by the Illumina real-time analysis (RTA) software v1.8. Afterward, the mapping of the reads to the human reference genome build GRCh37/hg19 (http://www.genome.ucsc.edu/) was performed using the BWA-SW alignment algorithm. Picard tools were used to improve the read quality, and Genome Analysis Toolkit (GATK) was used for realignment and base quality score recalibration. The calling of single nucleotide polymorphisms (SNPs) and short insertions/deletions (INDELs) was performed by Platypus, Haplotype Caller, and Mpileup programs and further filtration was carried out through variant quality score calibration (VQSR) using GATK. The ALLEGRO program identified several large runs of homozygosity (ROH) based on multipoint linkage analysis. For the coverage analysis of CNV detection, CNMOPS and ExomeDepth algorithms were utilized. In addition, the COMBINE and FUNC algorithms were used to combine the data and the annotation of functional variants.
Variant Search, Classification and Sanger Sequencing
The Varbank pipeline v2.26 (https://varbank.ccg.uni-koeln.de/) of Cologne Center for Genomics (CCG), Cologne, Germany, was utilized for the exome data analysis. The mean coverage of the data was 85%, while at 20X and 10X, the coverage of the targeted bases was 93.6 and 96.6%, respectively. A panel of genes, including MSX1, PAX9, AXIN2, FGFR1, IRF6, LRP6, WNT10A, WNT10B, EDAR, and EDARADD, underlying various dental malformations, was also filtered out to exclude the involvement of any copy number variations, missense, nonsense, or compound heterozygous variants in these genes.
Considering the consanguinity among the parents, ROH in the affected members (≥5 Mb) were identified. Later, a variant search was carried out in the ROH to find out rare homozygous variants. Furthermore, an exome-wide search irrespective of ROH was also executed to search for rare biallelic variants. Based on the autosomal recessive inheritance pattern, allele read frequency (75%-100%) for homozygous changes and allele frequency (<1%) for recessive variants was used. VarSome (Kopanos et al., 2019), Human Gene Mutation Database (HGMD) Professional 2019.4, and Database of Single Nucleotide Polymorphisms (dbSNP) were utilized for the evaluation of variants. Genome Aggregation Database v.2.1.1 (gnomAD; https://gnomad.broadinstitute.org/) was consulted to establish the minor allele frequency (MAF; value < 0.01) of the variants. An in-house database of 511 exomes of patients with diverse phenotype and another dataset of 21-exomes of ethically matched Pakhtun patients along with 90-exomes of other Pakistani patients of various ethnic backgrounds were employed as a control. ACMG guidelines were consulted for the classification of pathogenic, likely pathogenic, uncertain significance and benign variants (Richards et al., 2015).
In addition, a thorough search was performed on the variant data of chromosome X, considering the gender of the affected members in the pedigree diagram, but no bonafide variant, neither in EDA nor in other X-linked genes, was identified.
After performing the exome data analysis, all the gene variants were filtered and validated in various databases including dbSNP (https://www.ncbi.nlm.nih.gov/snp/), 1000 genome project (https://www.internationalgenome.org/), EVS (https://evs.gs.washington.edu/EVS/), and gnomAD (https://gnomad.broadinstitute.org/).
The reference sequences were obtained from the University of California Santa Cruz (UCSC) genome database browser (http://genome.ucsc.edu/cgi-bin/hgGateway). Amplifx version 2.1 (https://inp.univ-amu.fr/en/amplifx-manage-test-and-design-your-primers-for-pcr) was used for designing the primers for the amplification of the regions of interest. First, a genomic sequence of 700 bp up-and-downstream from the position of the rare variant was scanned to design an appropriate primer pair. Then, the PCR amplification of the regions of interest was carried out, and the Exo-Sap protocol (https://www.thermofisher.com) was used for purifying the PCR products. Next, the DNA sequencing was performed on the ABI3730 genetic analyzer with BigDye chemistry v3.1. For aligning the sequences against the reference sequence, a sequence alignment tool, BioEdit version 6.0.7 (http://www.mbio.ncsu.edu/BioEdit/bioedit.htm) was utilized.
Pathogenicity Prediction and Protein Sequence Alignment
In silico analysis was performed using different pathogenicity prediction tools including, 1) Mutation Taster (http://www.mutationtaster.org/), 2) PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), 3) PROVEAN (http://provean.jcvi.org/seq_submit.php), 4) PhD-SNP (https://snps.biofold.org/phd-snp/phd-snp.html), 5) I-Mutant2.0 (https://folding.biofold.org/i-mutant/i-mutant2.0.html), 6) SIFT (https://sift.bii.a-star.edu.sg/), 7) MutPred2 (http://mutpred.mutdb.org/), 8) FATHMM (http://fathmm.biocompute.org.uk/inherited.html) 9) VarSome (https://varsome.com/) (10) CADD (https://cadd.gs.washington.edu/snv) and 11) GERP (https://bio.tools/gerp).
Clustal Omega tool (https://www.ebi.ac.uk/Tools/msa/clustalo/) was used for the multiple sequence alignment of RFX2 proteins among different species including human (ENST00000303657.10), Chimpanzee (ENSPTRT00000080337.1), Gorilla (ENSGGOT00000060572.1), Gibbon (ENSNLET00000048810.1), Guinea (ENSCPOT00000022084.2), Opossum (ENSMODT00000058801.1), Koala (ENSPCIT00000013029.2), Cat (ENSFCAT00000038434.3), Panda (ENSAMET00000008566.1), Donkey (ENSEAST00005036789.1) and Dog (ENSCAFT00000029731.4).
Protein Structure Prediction
The homology modeling technique was utilized to construct the three-dimensional structures of RFX2 and DOHH proteins using I-TASSER (Iterative Threading ASSEmbly Refinement) server (Yang et al., 2015). For wild-type and mutant (p.Ile474Thr) RFX2 modelling, we used PDB (Protein Data Bank) ID: IDP7 (intrinsically disordered protein-7) structure as a template with an 84% structure homology. In the case of DOHH-p.Pro37Ala, we used the crystal structure of human DOHH (PDB ID: 4D50). A similar energy minimization strategy was utilized for the wild-type and mutant structures. Finally, structures were visualized with PyMOL software (www.pymol.org) (Delano, 2002).
Results
Clinical Report
The affected individuals (IV-4, 29-years, and IV-5, 24-years) showing variable missing teeth conditions were recruited at the dentistry department, Khyber Medical University, Peshawar, Khyber Pakhtunkhwa (KP), Pakistan. A dental specialist performed a clinical evaluation of the patients. The unaffected members (III-1, III-2, IV-1, IV-6) available for the study (Figure 1A) were also evaluated at the same clinic. The patients showed spacing in the upper jaw between the two central incisors. In addition, intra-oral examination showed the generalized spacing in the anterior region of the maxilla and mandible. During diagnosis, the missing third molars were excluded. In the case of patient-IV-4, a total of nine permanent teeth were missing (maxillary lateral incisors, left maxillary first premolar, all 4 s premolars, the right maxillary second molar, and right mandibular second molar) (Figure 1B a, b). However, in patient-IV-5, five permanent teeth including, maxillary lateral incisors, maxillary second premolars, and right maxillary first premolar, were missing (Figure 1B d, e).
Maxillary central incisors were proclined labially in both patients with patchy chalky white discoloration on the labial surface and around the root of the deciduous first molar. All maxillary teeth, except the central incisors, were small in size, and there was a lower arch spacing due to missing mandibular molars on both sides. Deep attrition in the case of maxillary incisors, canines, and premolars with enamel loss from their incisal surfaces and a taurodontism in the left mandibular third molar was observed. In addition, generalized horizontal bone loss and deep biting were also discerned. Furthermore, there was a clinical midline diastema due to the missing lateral incisors and a thin enamel covering over the dentine, especially in the lower incisor and lower premolars. Periapical radiolucency around the root of the deciduous first molar was also reported (Figure 1B c, f). The unaffected members showed normal permanent dentition in both arches (maxillary and mandibular).
The patients were of standard heights (IV-4 = 152.4 cm, IV-5 = 153.6 cm), and they did not show any neurological, skeletal, and visceral organ defects upon medical examinations. Likewise, the abnormalities of other ectodermal appendages, including hypotrichosis, hyponychia/anonychia, cleft lip/palate, hypo/hyperpigmentation, and hypohidrosis, were ruled out by the medical consultants during the physical investigations of these patients. In addition, the patients did not record any complaints of muscular weakness or any progressive physical lethargies.
Variant Identification and Co-segregation Analysis
During the exome analysis, several ROH, including a 42.7 MB ROH on chromosome 1p31.1–12, a 19.8 MB ROH on chromosome 7q31.32-q34, a 13.8 MB ROH on chromosome 6p21.32-q23.3, and a 7.1 MB ROH on chromosome 19p13.3–13.2, were identified. These chromosomal locations carried eight rare homozygous variants, including c.1891C > T; p.(Leu631Phe) (NM_003,594.4) in TTF2 (1p13.1), c.980C > T; p.(Thr327Met) (NM_207,163.3) in LMOD2 (7q31.32), c.3732G > T; p.(Leu1244Phe) (NM_173,569.4) in UBN2 (7q34), c.406C > A; p.(Pro136Thr) (NM_053,278.2) in TAAR8 (6q23.2), c.122T > G; p.(Phe41Cys) (NM_002,123.4) in HLA-DQB1 (6p21.32), c.266T > C; p.(Ile89Thr) (NM_152,990.3) in PXT1 (6p21.31), c.1421T > C; p.(Ile474Thr) (NM_000,635.4) in RFX2 (19p13.3), and c.109C > G; p.(Pro37Ala) (NM_031,304.4) in DOHH (19p13.3) (Table 1). The exome-wide search regardless of ROH identified ∼120 homozygous variants on various chromosomal locations. Upon applying the variant validation criteria described in Materials and Methods, we could not find any rare variant other than the variants mentioned in Table 1. Therefore, these variants were considered for the co-segregation analysis.
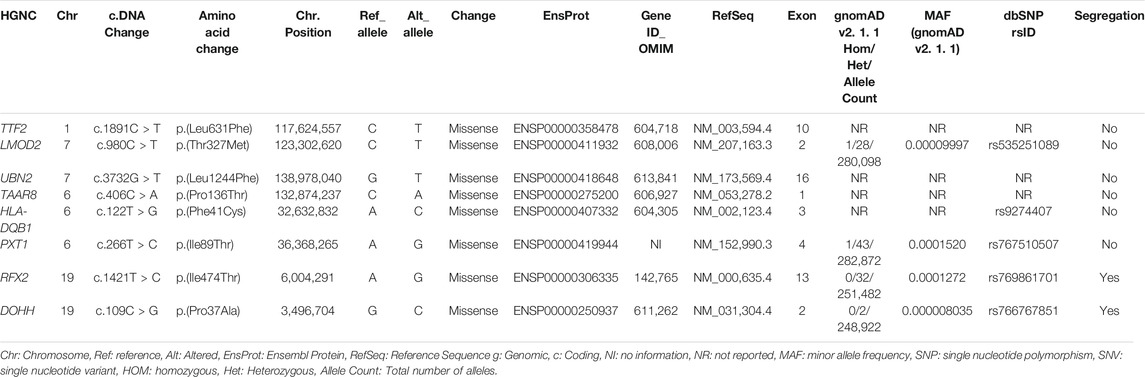
TABLE 1. Rare coding variants prioritized from exome sequencing data, which were tested for segregation.
The fine-mapped variants were sequenced via Sanger sequencing for the validation and confirmation of segregation. During the co-segregation analysis, the variants in RFX2 and DOHH, located in a common ROH of 7.1 MB on chromosome 19 segregated in the family (Figures 1A, 2C). The DOHH variant c.109C > G; p.(Pro37Ala) was predicted benign and tolerated by the online prediction tools, while the variant c.1421T > C; p.(Ile474Thr) in RFX2 was predicted damaging. Moreover, the RFX2 variant achieved highly significant CADD (27.3) and GERP (5.1399) scores (Table 2). The ACMG variant classification criteria classified the RFX2 variant as likely pathogenic, while the DOHH variant was benign (Table 2). Hence, the RFX2 variant is the most likely candidate causing non-syndromic tooth agenesis. An allele frequency (AF) (0.0001272) of this variant has been calculated in gnomAD. This variant is 32 times monoallelically identified in South Asian (17), European (non-Finnish, 13), Latino/Admixed Americans (1), and in a minor population (1), where the total number of alleles is 251,482 while the number of homozygous alleles is zero. From the South Asian population, 30,616 alleles are included with an AF of 0.0005553 in this database. A dbSNP ID rs769861701 has been assigned to this variant. According to our knowledge, this is the first report of a biallelic variation at c.1421T > C in RFX2, which is likely to cause human absent teeth phenotype (Figures 2A–C).
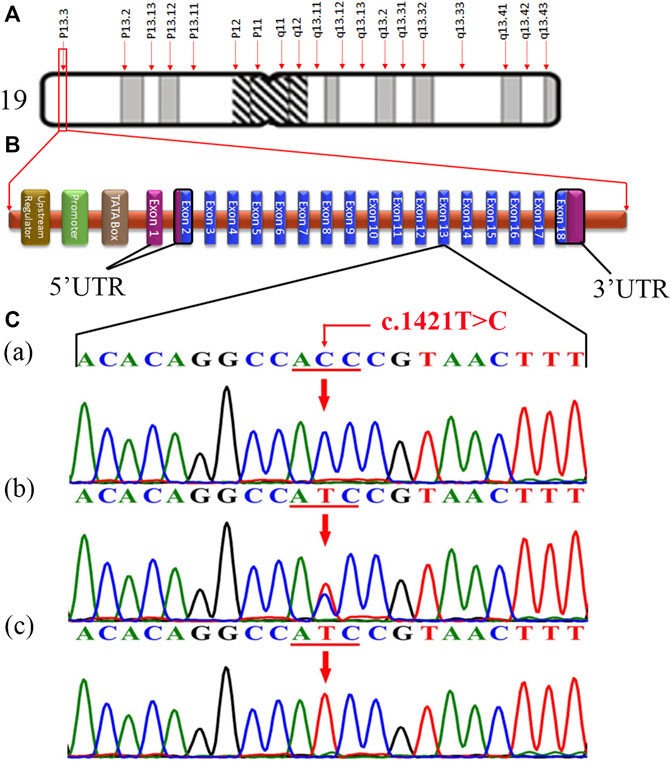
FIGURE 2. (A) Structure of chromosome 19, showing the RFX2 locus (p13.3) (http://atlasgeneticsoncology.org/ISCN09/Chrom19ISCN09.html). (B) Typical structure of RFX2 containing 18 exons, showing the position of genetic alteration c.1421T > C in 13-exon in the present study (red) (generated manually using the information from ENSEMBL Genome Browser). (C) Chromatogram of an unaffected individual (IV-1) in the upper panel (a), a carrier/heterozygous parent (III-1) in the middle panel (b), and a homozygous affected individual (IV-4) in the lower panel (c).
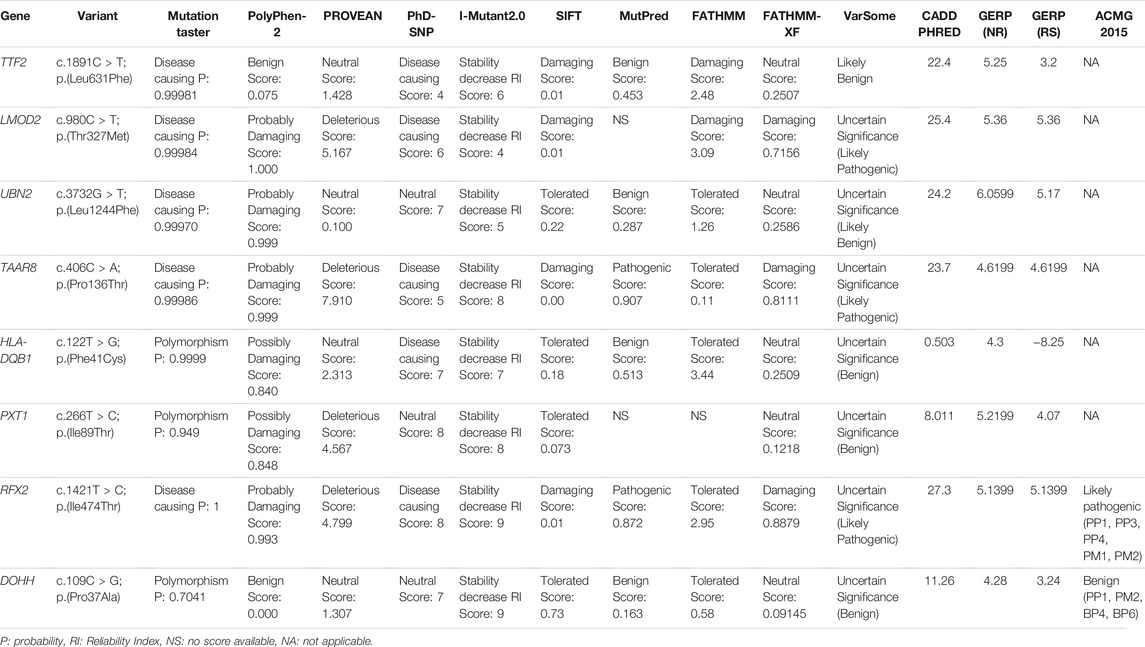
TABLE 2. In silico analysis of the coding variants with scores by using the various online pathogenicity prediction tools.
Protein Structure Prediction
Multiple sequence alignment revealed conservation of the RFX2-Isoleucine-474 residue, as shown in Figure 3A, and the conserved residue is essential for protein structure determination, stability, and functionality. The structural relevance of the highly conserved Isoleucine residue was determined using the homology modeling technique. We examined and studied both wild-type and mutant structures to discover the structural differences, depicted in Figure 3B i and ii. As illustrated in the zoomed-up view, we detected differences in intramolecular distance among residues in the immediate mutation vicinity. Because of the variation in bonding, there is a local difference in the structure, as indicated by the arrows. We also noticed a difference in surface charge distribution between the two forms of the RFX2 protein, as indicated by the arrows (Figure 3B iii and iv).
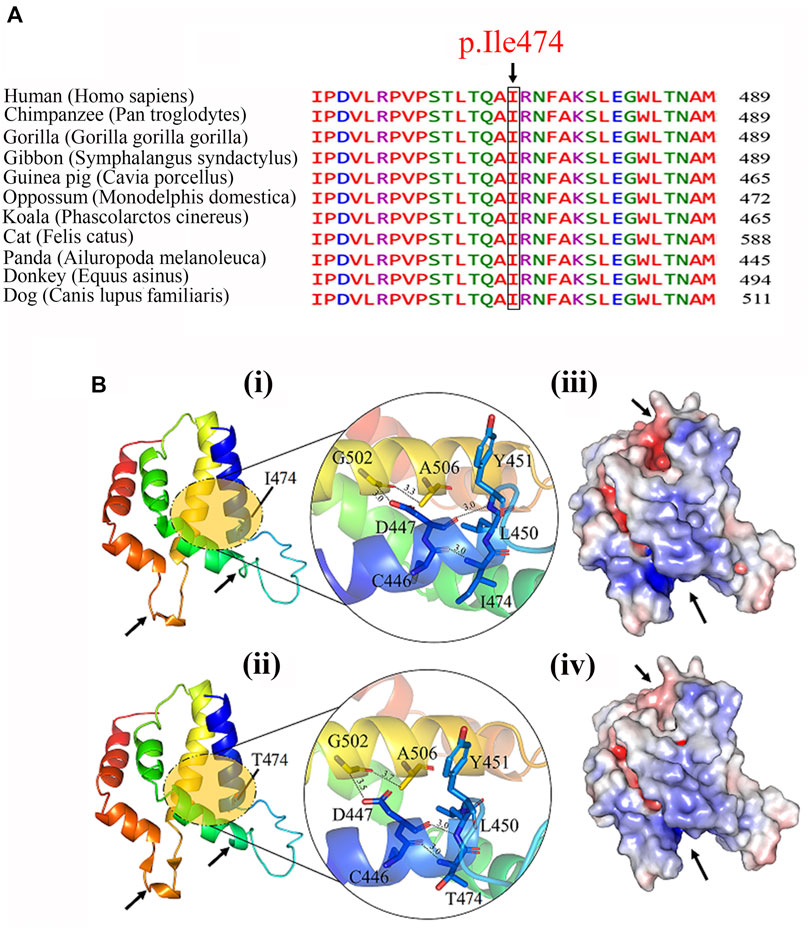
FIGURE 3. (A) Multiple sequence alignment of Ile474 with its mammalian orthologs, showing conserved wild-type residue (Isoleucine) at this position. (B) The predicted structure of wild (i) and mutant (ii) of RFX2 protein. Zoom-up view of interaction pattern of the nearby residues of wild-type and mutant protein. Possible structural effects of substituting conserved isoleucine into a threonine at residue 474 are highlighted with an arrow and surface charge distribution of (iii) wild-type and (iv) mutant- RFX2.
On the other hand, Pro37 and Ala37 are located on the surface of wild and mutant DOHH proteins, respectively, suggesting the DOHH variant is substantially insignificant (Figures 4A,B). Furthermore, electrostatic analysis of wild-type and mutant DOHH showed that the residue is not present at the interface of DOHH dimeric form and is not involved in any inter-protein and intra-protein interactions (Figures 4C,D).
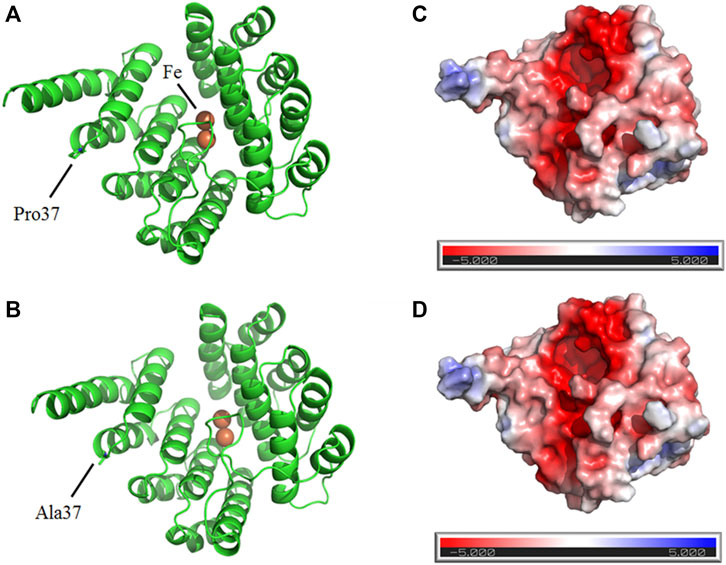
FIGURE 4. Predicted structures of wild (A) and mutant (B) DOHH proteins show the surface location Pro37 and Ala37, respectively. The electrostatic analysis of wild-type (C) and mutant (D) DOHH protein.
Discussion
In this study, we have investigated the genetic cause of the NSTA in a Pakistani family through exome and Sanger sequencing. The affected individuals manifested a generalized spacing in the upper jaw between the two central incisors in addition to congenitally missing permanent maxillary lateral incisors, left and right maxillary first premolars, and mandibular and maxillary second premolars. Besides, the exome data revealed a novel homozygous sequence variant c.1421T > C; p.(Ile474Thr) in RFX2, which lies in an extended dimerization domain C of RFX2 protein. According to the ACMG guidelines (2015), this variant has been categorized as a likely pathogenic variant (Table 2).
RFX2 comprises 18 exons (Figures 2A,B), encoding a 723 amino acids protein (RFX2), and has two isoforms, P48378 and P48378-2 (https://www.uniprot.org/uniprot/). In humans, eight RFX genes (RFX1-RFX8) have been characterized (Aftab et al., 2008; Sugiaman-Trapman et al., 2018). RFX genes encode transcription factors (TFs) termed RFX transcription factors (RFX-TFs), performing multiple functions in different organisms and have been argued to be involved in the regulation of various cellular and developmental mechanisms (Zaim et al., 2005; Garg et al., 2015). As the RFX genes have essential activities in development hence, the alterations in their sequence may lead to severe pathogenic consequences (Senti and Swoboda, 2008; Choksi et al., 2014). According to Human Gene Mutation Database (HGMD) Professional 2021.2, five missense variants associated with autism (Sanders et al., 2012; Kosmicki et al., 2017; Satterstrom et al., 2020), cerebral palsy (McMichael et al., 2015), and congenital heart defect (Jin et al., 2017) have already been reported in RFX2. Here we report a novel sequence variant c.1421T > C; p.(Ile474Thr) in this gene in an autosomal recessive family exhibiting tooth agenesis. As predicted by the bioinformatics tools, this missense variant results in structural deviations in RFX2 protein, leading to significant perturbations or abolishing its function.
rfx2 expresses in various ciliated tissues in Xenopus, including the neural tube, which leads to the development of the brain, spinal cord and epidermis, gastrocoel roof plate, inner ear, and renal tissues (Chung et al., 2014). In addition, severe cilia-defective embryonic phenotypes were observed in rfx2-/- Xenopus (Chung et al., 2014). Rfx-TFs are involved in the hearing mechanism of mice (Elkon et al., 2015). rfx2 is highly expressed in testes (Reith et al., 1994), and Wu et al. (2016) have studied the defective spermatogenesis and severe growth retardation in rfx2-/- mice (Wu et al., 2016). In a mouse model study, Bisgrove et al. (2012) have observed the expression of rfx2 at the late headfold stage (Bisgrove et al., 2012). The headfold stage governs the formation of the heart (Varner et al., 2010) and an oral membrane in the jawed vertebrates, which leads to mouth morphogenesis, including the development of jaws and teeth (Soukup et al., 2013). Considering the role of RFX2 as a transcriptional factor and its significant expression in various tissues, we hypothesize that it regulates the morphogenesis of multiple tissues and organs in animals and humans, including the formation of craniofacial tissues. Besides, the sequence alterations of RFX2 may cause various intellectual disabilities, heart anomalies, renal defects, otologic disorders, and abnormal growth of ectodermal appendages, including teeth.
RFX proteins play a central role in the morphogenesis of cilia on the surface of polarized cells (Senti and Swoboda, 2008; Senti et al., 2009). In humans and animals, the primary cilia are involved in dental development, located in the epithelial and mesenchymal tissues during initial stages, differentiation, and formation of complex tissues during tooth development (Kero et al., 2014; Hampl et al., 2017). As the primary cilia are the important signaling centers during vertebrate development (Goetz and Anderson, 2010), any genetic or experimental alteration in their structure or function may lead to defective odontogenesis (Liu et al., 2014), resulting in a change in the tooth formula, size, morphology, position (Hardcastle et al., 1998; Lan et al., 2014), and odontoblast/ameloblast differentiation (Bei, 2009). Moreover, several ciliopathies, including Bardet-Biedl syndrome (BBS), Ellis-van Creveld syndrome (EVC; OMIM: 225,500), Weyers acrofacial dysostosis (WAD; OMIM: 183,530), have been reported in the literature displaying syndromic genetic anomalies, including the dental defects, hence, demonstrating the principal role of primary cilia in tooth morphogenesis (Hampl et al., 2017). Recently, it has been studied that Rfx2 regulates the expression of genes encoding axonemal dynein subunits, proteins involved in epithelial-mesenchymal transition, and the BBSome elements (Chung et al., 2014). These genes have been reported in human ciliopathies (BBS, EVC, WAD) (Sharma et al., 2008; Reiter et al., 2012). Furthermore, Rfx2 directly influences the downstream expression of 911 genes, and 180 genes out of those are only the ciliary genes (Chung et al., 2014).
RFX2 is essential for the expression of numerous genes, regulating the Hedgehog (Hh) signaling (Chung et al., 2012; Chung et al., 2014). Sonic hedgehog (Shh) is expressed in the dental epithelium and regulates the formation of enamel, dentin, cementum, and soft tissues (Hosoya et al., 2020). A recent study by Neugebauer et al. (2009) revealed that the disruption of FGF signaling through fgfr1 reduces the expression of ift88 and two ciliogenic transcription factors foxj1 and rfx2 (Neugebauer et al., 2009). An ift88 null mice exhibit skeletal defects and hypoplastic maxilla, mandible, and supernumerary teeth (Zhang et al., 2003; Ohazama et al., 2009). The pathogenic genetic changes in fgfr1 also cause phenotypes consistent with those seen in ift88. The pathogenic variants in human FGFR1 have been reported to cause various tooth malformations (Dodé et al., 2003; Pitteloud et al., 2006). In human ciliated cells, IFT88 (OMIM: 600,595) co-localizes with OFD1 (OMIM: 300,170), and genetic alterations in OFD1 cause Orofaciodigital Syndrome-1 (Singla et al., 2010). As the expression of the genes discussed above is dependent on each other and they govern the same signaling pathway, we firmly believe that IFT88, FOXJ1 (OMIM: 602,291), and RFX2 are the strong candidate genes associated with human development and the genetic variations in these genes may lead to skeletal, oral and maxillofacial anomalies.
Conclusion
In this study, we report a clinical and molecular diagnosis of a consanguineous NSTA Pakistani family. Exome sequencing and Sanger sequencing identified a novel homozygous missense variant c.1421T > C; p.(Ile474Thr) in a novel candidate gene RFX2 causing NSTA in this family. We claim that RFX2, based on its critical regulatory role in ciliary development, SHH, and FGF signaling, is an essential molecular player in human odontogenesis.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: ClinVar, SCV001934213.1.
Ethics Statement
The studies involving human participants were reviewed and approved by the Research and Ethical Committee (REC) of Kohat University of Science and Technology (KUST), Kohat, Pakistan. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
SAK, NM, ZR, UK, AK, and HK contributed to study design and synopsis writing. MK did a clinical evaluation of the patients and wrote a report about it. NW and SAK contributed to data generation and analysis. AN has performed the bioinformatics analysis. SAK, SK, NM, ZR, MK, AN, UK, AK, HK, and NW have critically reviewed clinical and molecular data, and agreed upon writing a manuscript. SAK, SK, and NW contributed to writing, revising, and finalizing the draft.
Funding
NW got the research support from the George Forster Fellowship (2015–2018) of the Alexander von Humboldt Foundation, Bonn, Germany. The Higher Education Commission (HEC), Islamabad, Pakistan, supported SK through the NRPU project (No. 4857/NRPU/R&D/HEC2014).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adaimy, L., Chouery, E., Mégarbané, H., Mroueh, S., Delague, V., Nicolas, E., and et al, (2007). Mutation in WNT10A is Associated with an Autosomal Recessive Ectodermal Dysplasia: the Odonto-Onycho-Dermal Dysplasia. Am. J. Hum. Genet. 81 (4), 821–828. doi:10.1086/520064
Aftab, S., Semenec, L., Chu, J. S.-C., and Chen, N. (2008). Identification and Characterization of Novel Human Tissue-specific RFX Transcription Factors. BMC Evol. Biol. 8 (1), 226. doi:10.1186/1471-2148-8-226
Al-Ani, A. H., Antoun, J. S., Thomson, W. M., Merriman, T. R., and Farella, M. (2017). Hypodontia: An Update on its Etiology, Classification, and Clinical Management. Biomed. Res. Int. 2017, 1–9. doi:10.1155/2017/9378325
Bei, M. (2009). Molecular Genetics of Ameloblast Cell Lineage. J. Exp. Zool B Mol. Dev. Evol. 312b (5), 437–444. doi:10.1002/jez.b.21261
Bisgrove, B. W., Makova, S., Yost, H. J., and Brueckner, M. (2012). RFX2 is Essential in the Ciliated Organ of Asymmetry and an RFX2 Transgene Identifies a Population of Ciliated Cells Sufficient for Fluid Flow. Developmental Biol. 363 (1), 166–178. doi:10.1016/j.ydbio.2011.12.030
Brook, A. H. (2009). Multilevel Complex Interactions between Genetic, Epigenetic and Environmental Factors in the Aetiology of Anomalies of Dental Development. Arch. Oral Biol. 54 (Suppl. 1), S3–S17. doi:10.1016/j.archoralbio.2009.09.005
Chhabra, N., Goswami, M., and Chhabra, A. (2014). Genetic Basis of Dental Agenesis - Molecular Genetics Patterning Clinical Dentistry. Med. Oral Patol Oral Cir Bucal 19 (2), e112–e119. doi:10.4317/medoral.19158
Choksi, S. P., Lauter, G., Swoboda, P., and Roy, S. (2014). Switching on Cilia: Transcriptional Networks Regulating Ciliogenesis. Development 141 (7), 1427–1441. doi:10.1242/dev.074666
Chung, M.-I., Kwon, T., Tu, F., Brooks, E. R., Gupta, R., Meyer, M., et al. (2014). Coordinated Genomic Control of Ciliogenesis and Cell Movement by RFX2. Elife 3, e01439. doi:10.7554/eLife.01439
Chung, M.-I., Peyrot, S. M., LeBoeuf, S., Park, T. J., McGary, K. L., Marcotte, E. M., et al. (2012). RFX2 is Broadly Required for Ciliogenesis during Vertebrate Development. Developmental Biol. 363 (1), 155–165. doi:10.1016/j.ydbio.2011.12.029
De Coster, P. J., Marks, L. A., Martens, L. C., and Huysseune, A. (2009). Dental Agenesis: Genetic and Clinical Perspectives. J. Oral Pathol. Med. 38 (1), 1–17. doi:10.1111/j.1600-0714.2008.00699.x
Dinckan, N., Du, R., Petty, L. E., Coban-Akdemir, Z., Jhangiani, S. N., Paine, I., et al. (2018). Whole-Exome Sequencing Identifies Novel Variants for Tooth Agenesis. J. Dent Res. 97 (1), 49–59. doi:10.1177/0022034517724149
Dodé, C., Levilliers, J., Dupont, J.-M., De Paepe, A., Le Dû, N., Soussi-Yanicostas, N., et al. (2003). Loss-of-function Mutations in FGFR1 Cause Autosomal Dominant Kallmann Syndrome. Nat. Genet. 33 (4), 463–465. doi:10.1038/ng1122
Du, R., Dinckan, N., Song, X., Coban-Akdemir, Z., Jhangiani, S. N., Guven, Y., et al. (2018). Identification of Likely Pathogenic and Known Variants in TSPEAR, LAMB3, BCOR, and WNT10A in Four Turkish Families with Tooth Agenesis. Hum. Genet. 137 (9), 689–703. doi:10.1007/s00439-018-1907-y
Elkon, R., Milon, B., Morrison, L., Shah, M., Vijayakumar, S., Racherla, M., et al. (2015). RFX Transcription Factors Are Essential for Hearing in Mice. Nat. Commun. 6, 8549. doi:10.1038/ncomms9549
Fleischmannova, J., Matalova, E., Tucker, A. S., and Sharpe, P. T. (2008). Mouse Models of Tooth Abnormalities. Eur. J. Oral Sci. 116 (1), 1–10. doi:10.1111/j.1600-0722.2007.00504.x
Fournier, B. P., Bruneau, M. H., Toupenay, S., Kerner, S., Berdal, A., Cormier-Daire, V., et al. (2018). Patterns of Dental Agenesis Highlight the Nature of the Causative Mutated Genes. J. Dent Res. 97 (12), 1306–1316. doi:10.1177/0022034518777460
Garg, A., Futcher, B., and Leatherwood, J. (2015). A New Transcription Factor for Mitosis: in Schizosaccharomyces pombe, the RFX Transcription Factor Sak1 Works with Forkhead Factors to Regulate Mitotic Expression. Nucleic Acids Res. 43 (14), 6874–6888. doi:10.1093/nar/gkv274
Goetz, S. C., and Anderson, K. V. (2010). The Primary Cilium: a Signalling centre during Vertebrate Development. Nat. Rev. Genet. 11 (5), 331–344. doi:10.1038/nrg2774
Hampl, M., Cela, P., Szabo-Rogers, H. L., Bosakova, M. K., Dosedelova, H., Krejci, P., et al. (2017). Role of Primary Cilia in Odontogenesis. J. Dent Res. 96 (9), 965–974. doi:10.1177/0022034517713688
Hardcastle, Z., Mo, R., Hui, C. C., and Sharpe, P. T. (1998). The Shh Signalling Pathway in Tooth Development: Defects in Gli2 and Gli3 Mutants. Development 125 (15), 2803–2811. doi:10.1242/dev.125.15.2803
Hosoya, A., Shalehin, N., Takebe, H., Shimo, T., and Irie, K. (2020). Sonic Hedgehog Signaling and Tooth Development. Ijms 21 (5), 1587. doi:10.3390/ijms21051587
Hussain, M. S., Baig, S. M., Neumann, S., Peche, V. S., Szczepanski, S., Nürnberg, G., et al. (2013). CDK6 Associates with the Centrosome during Mitosis and is Mutated in a Large Pakistani Family with Primary Microcephaly. Hum. Mol. Genet. 22 (25), 5199–5214. doi:10.1093/hmg/ddt374
Jin, S. C., Homsy, J., Zaidi, S., Lu, Q., Morton, S., DePalma, S. R., et al. (2017). Contribution of Rare Inherited and De Novo Variants in 2,871 Congenital Heart Disease Probands. Nat. Genet. 49 (11), 1593–1601. doi:10.1038/ng.3970
Jumlongras, D., Bei, M., Stimson, J. M., Wang, W.-F., DePalma, S. R., Seidman, C. E., et al. (2001). A Nonsense Mutation in MSX1 Causes Witkop Syndrome. Am. J. Hum. Genet. 69 (1), 67–74. doi:10.1086/321271
Jussila, M., and Thesleff, I. (2012). Signaling Networks Regulating Tooth Organogenesis and Regeneration, and the Specification of Dental Mesenchymal and Epithelial Cell Lineages. Cold Spring Harbor Perspect. Biol. 4 (4), a008425. doi:10.1101/cshperspect.a008425
Kapadia, H., Mues, G., and D'Souza, R. (2007). Genes Affecting Tooth Morphogenesis. Orthod. Craniofac. Res. 10 (4), 237–244. doi:10.1111/j.1601-6343.2007.00407.x
Kero, D., Novakovic, J., Vukojevic, K., Petricevic, J., Kalibovic Govorko, D., Biocina-Lukenda, D., et al. (2014). Expression of Ki-67, Oct-4, γ-tubulin and α-tubulin in Human Tooth Development. Arch. Oral Biol. 59 (11), 1119–1129. doi:10.1016/j.archoralbio.2014.05.025
Khan, S. A., Rukan, A., Ullah, A., Bibi, N., Humayun, M., Ullah, W., et al. (2020). Homozygous Variants of EDAR Underlying Hypohidrotic Ectodermal Dysplasia in Three Consanguineous Families. Eur. J. Dermatol. 30 (4), 408–416. doi:10.1684/ejd.2020.3844
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). VarSome: the Human Genomic Variant Search Engine. Bioinformatics 35 (11), 1978–1980. doi:10.1093/bioinformatics/bty897
Kosmicki, J. A., Samocha, K. E., Howrigan, D. P., Sanders, S. J., Slowikowski, K., Lek, M., et al. (2017). Refining the Role of De Novo Protein-Truncating Variants in Neurodevelopmental Disorders by Using Population Reference Samples. Nat. Genet. 49 (4), 504–510. doi:10.1038/ng.3789
Lan, Y., Jia, S., and Jiang, R. (2014). Molecular Patterning of the Mammalian Dentition. Semin. Cel Developmental Biol. 25-26, 61–70. doi:10.1016/j.semcdb.2013.12.003
Larmour, C. J., Mossey, P. A., Thind, B. S., Forgie, A. H., and Stirrups, D. R. (2005). Hypodontia--a Retrospective Review of Prevalence and Etiology. Part I. Quintessence Int. 36 (4), 263–270.
Letra, A., Chiquet, B., Hansen-Kiss, E., Menezes, S., and Hunter, E. (1993). in Nonsyndromic Tooth Agenesis OverviewGeneReviews(®). Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, G. Mirzaaet al. (Seattle (WA): University of Washington, Seattle Copyright © 1993-2021University of Washington, Seattle) (GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.
Liu, B., Chen, S., Cheng, D., Jing, W., and Helms, J. A. (2014). Primary Cilia Integrate Hedgehog and Wnt Signaling during Tooth Development. J. Dent Res. 93 (5), 475–482. doi:10.1177/0022034514528211
McMichael, G., Bainbridge, M. N., Haan, E., Corbett, M., Gardner, A., Thompson, S., et al. (2015). Whole-exome Sequencing Points to Considerable Genetic Heterogeneity of Cerebral Palsy. Mol. Psychiatry 20 (2), 176–182. doi:10.1038/mp.2014.189
Murakami, A., Yasuhira, S., Mayama, H., Miura, H., Maesawa, C., and Satoh, K. (2017). Characterization of PAX9 Variant P20L Identified in a Japanese Family with Tooth Agenesis. PLoS One 12 (10), e0186260. doi:10.1371/journal.pone.0186260
Neubüser, A., Peters, H., Balling, R., and Martin, G. R. (1997). Antagonistic Interactions between FGF and BMP Signaling Pathways: a Mechanism for Positioning the Sites of Tooth Formation. Cell 90 (2), 247–255. doi:10.1016/s0092-8674(00)80333-5
Neugebauer, J. M., Amack, J. D., Peterson, A. G., Bisgrove, B. W., and Yost, H. J. (2009). FGF Signalling during Embryo Development Regulates Cilia Length in Diverse Epithelia. Nature 458 (7238), 651–654. doi:10.1038/nature07753
Niceta, M., Margiotti, K., Digilio, M. C., Guida, V., Bruselles, A., Pizzi, S., et al. (2018). Biallelic Mutations in DYNC2LI1 Are a Rare Cause of Ellis-van Creveld Syndrome. Clin. Genet. 93 (3), 632–639. doi:10.1111/cge.13128
Nieminen, P. (2009). Genetic Basis of Tooth Agenesis. J. Exp. Zool. 312b (4), 320–342. doi:10.1002/jez.b.21277
Ohazama, A., Haycraft, C. J., Seppala, M., Blackburn, J., Ghafoor, S., Cobourne, M., et al. (2009). Primary Cilia Regulate Shh Activity in the Control of Molar Tooth Number. Development 136 (6), 897–903. doi:10.1242/dev.027979
Park, H., Song, J.-S., Shin, T. J., Hyun, H.-K., Kim, Y.-J., and Kim, J.-W. (2019). WNT10A Mutations Causing Oligodontia. Arch. Oral Biol. 103, 8–11. doi:10.1016/j.archoralbio.2019.05.007
Parveen, A., Khan, S. A., Mirza, M. U., Bashir, H., Arshad, F., Iqbal, M., et al. (2019). Deleterious Variants in WNT10A, EDAR, and EDA Causing Isolated and Syndromic Tooth Agenesis: A Structural Perspective from Molecular Dynamics Simulations. Ijms 20 (21), 5282. doi:10.3390/ijms20215282
Pitteloud, N., Acierno, J. S., Meysing, A., Eliseenkova, A. V., Ma, J., Ibrahimi, O. A., et al. (2006). Mutations in Fibroblast Growth Factor Receptor 1 Cause Both Kallmann Syndrome and Normosmic Idiopathic Hypogonadotropic Hypogonadism. Proc. Natl. Acad. Sci. 103 (16), 6281–6286. doi:10.1073/pnas.0600962103
Polder, B. J., Van't Hof, M. A., Van der Linden, F. P. G. M., and Kuijpers-Jagtman, A. M. (2004). A Meta-Analysis of the Prevalence of Dental Agenesis of Permanent Teeth. Commun. Dent Oral Epidemiol. 32 (3), 217–226. doi:10.1111/j.1600-0528.2004.00158.x
Posey, J. E., Harel, T., Liu, P., Rosenfeld, J. A., James, R. A., Coban Akdemir, Z. H., et al. (2017). Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 376 (1), 21–31. doi:10.1056/NEJMoa1516767
Rasool, M., Schuster, J., Aslam, M., Tariq, M., Ahmad, I., Ali, A., et al. (2008). A Novel Missense Mutation in the EDA Gene Associated with X-Linked Recessive Isolated Hypodontia. J. Hum. Genet. 53 (10), 894–898. doi:10.1007/s10038-008-0323-x
Reiter, J. F., Blacque, O. E., and Leroux, M. R. (2012). The Base of the Cilium: Roles for Transition Fibres and the Transition Zone in Ciliary Formation, Maintenance and Compartmentalization. EMBO Rep. 13 (7), 608–618. doi:10.1038/embor.2012.73
Reith, W., Ucla, C., Barras, E., Gaud, A., Durand, B., Herrero-Sanchez, C., et al. (1994). RFX1, a Transactivator of Hepatitis B Virus Enhancer I, Belongs to a Novel Family of Homodimeric and Heterodimeric DNA-Binding Proteins. Mol. Cel. Biol. 14 (2), 1230–1244. doi:10.1128/mcb.14.2.1230-1244.1994
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De Novo mutations Revealed by Whole-Exome Sequencing Are Strongly Associated with Autism. Nature 485 (7397), 237–241. doi:10.1038/nature10945
Sarkar, T., Bansal, R., and Das, P. (2014). Whole Genome Sequencing Reveals Novel Non-synonymous Mutation in Ectodysplasin A (EDA) Associated with Non-syndromic X-Linked Dominant Congenital Tooth Agenesis. PLoS One 9 (9), e106811. doi:10.1371/journal.pone.0106811
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., De Rubeis, S., An, J.-Y., et al. (2020). Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180 (3), 568–584.e523. doi:10.1016/j.cell.2019.12.036
Schalk-van der Weide, Y., Steen, W. H., and Bosman, F. (1992). Distribution of Missing Teeth and Tooth Morphology in Patients with Oligodontia. ASDC J. Dent Child. 59 (2), 133–140.
Senti, G., Ezcurra, M., Löbner, J., Schafer, W. R., and Swoboda, P. (2009). Worms with a Single Functional Sensory Cilium Generate Proper Neuron-specific Behavioral Output. Genetics 183 (2), 595–605. doi:10.1534/genetics.109.105171
Senti, G., and Swoboda, P. (2008). Distinct Isoforms of the RFX Transcription Factor DAF-19 Regulate Ciliogenesis and Maintenance of Synaptic Activity. MBoC 19 (12), 5517–5528. doi:10.1091/mbc.e08-04-0416
Sharma, N., Berbari, N. F., and Yoder, B. K. (2008). Chapter 13 Ciliary Dysfunction in Developmental Abnormalities and Diseases. Curr. Top. Dev. Biol. 85, 371–427. doi:10.1016/s0070-2153(08)00813-2
Shimizu, T., and Maeda, T. (2009). Prevalence and Genetic Basis of Tooth Agenesis. Jpn. Dental Sci. Rev. 45 (1), 52–58. doi:10.1016/j.jdsr.2008.12.001
Si, D., and Liu, J. (2018). Comments on "NEMO Gene Mutations in Chinese Patients with Incontinentia Pigmenti". J. Formos. Med. Assoc. 117 (10), 946. doi:10.1016/j.jfma.2018.06.003
Singla, V., Romaguera-Ros, M., Garcia-Verdugo, J. M., and Reiter, J. F. (2010). Ofd1, a Human Disease Gene, Regulates the Length and Distal Structure of Centrioles. Developmental Cel 18 (3), 410–424. doi:10.1016/j.devcel.2009.12.022
Soukup, V., Horácek, I., and Cerny, R. (2013). Development and Evolution of the Vertebrate Primary Mouth. J. Anat. 222 (1), 79–99. doi:10.1111/j.1469-7580.2012.01540.x
Stockton, D. W., Das, P., Goldenberg, M., D'Souza, R. N., and Patel, P. I. (2000). Mutation of PAX9 is Associated with Oligodontia. Nat. Genet. 24 (1), 18–19. doi:10.1038/71634
Sugiaman-Trapman, D., Vitezic, M., Jouhilahti, E.-M., Mathelier, A., Lauter, G., Misra, S., et al. (2018). Characterization of the Human RFX Transcription Factor Family by Regulatory and Target Gene Analysis. BMC Genomics 19 (1), 181. doi:10.1186/s12864-018-4564-6
Thesleff, I. (2014). Current Understanding of the Process of Tooth Formation: Transfer from the Laboratory to the Clinic. Aust. Dent J. 59 (Suppl. 1), 48–54. doi:10.1111/adj.12102
Thesleff, I. (2006). The Genetic Basis of Tooth Development and Dental Defects. Am. J. Med. Genet. 140A (23), 2530–2535. doi:10.1002/ajmg.a.31360
Varner, V. D., Voronov, D. A., and Taber, L. A. (2010). Mechanics of Head Fold Formation: Investigating Tissue-Level Forces during Early Development. Development 137 (22), 3801–3811. doi:10.1242/dev.054387
Vastardis, H. (2000). The Genetics of Human Tooth Agenesis: New Discoveries for Understanding Dental Anomalies. Am. J. Orthod. Dentofacial Orthopedics 117 (6), 650–656. doi:10.1016/s0889-5406(00)70173-9
Wu, Y., Hu, X., Li, Z., Wang, M., Li, S., Wang, X., and et al, (2016). Transcription Factor RFX2 Is a Key Regulator of Mouse Spermiogenesis. Sci. Rep. 6, 20435. doi:10.1038/srep20435
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 12 (1), 7–8. doi:10.1038/nmeth.3213
Yu, M., Wong, S. W., Han, D., and Cai, T. (2019). Genetic Analysis: Wnt and Other Pathways in Nonsyndromic Tooth Agenesis. Oral Dis. 25 (3), 646–651. doi:10.1111/odi.12931
Zaim, J., Speina, E., and Kierzek, A. M. (2005). Identification of New Genes Regulated by the Crt1 Transcription Factor, an Effector of the DNA Damage Checkpoint Pathway in Saccharomyces cerevisiae. J. Biol. Chem. 280 (1), 28–37. doi:10.1074/jbc.M404669200
Keywords: tooth agenesis, non-syndromic, RFX2, missense variant, hedgehog and fibroblast growth factor signaling pathways
Citation: Khan SA, Khan S, Muhammad N, Rehman ZU, Khan MA, Nasir A, Kalsoom U-e-, Khan AK, Khan H and Wasif N (2022) The First Report of a Missense Variant in RFX2 Causing Non-Syndromic Tooth Agenesis in a Consanguineous Pakistani Family. Front. Genet. 12:782653. doi: 10.3389/fgene.2021.782653
Received: 24 September 2021; Accepted: 07 December 2021;
Published: 25 January 2022.
Edited by:
Noor Ahmad Shaik, King Abdulaziz University, Saudi ArabiaReviewed by:
Ehsan Ullah, National Eye Institute (NEI), United StatesAbubakar Moawia, University of Cologne, Germany
Copyright © 2022 Khan, Khan, Muhammad, Rehman, Khan, Nasir, Kalsoom, Khan, Khan and Wasif. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saadullah Khan, c2FhZEBrdXN0LmVkdS5waw==; Naveed Wasif, bmF2ZWVkd2FzaWZAZ21haWwuY29t