 In Seok Moon
In Seok Moon Andrew R. Grant
Andrew R. Grant Varun Sagi5,6
Varun Sagi5,6 Heidi L. Rehm
Heidi L. Rehm Konstantina M. Stankovic
Konstantina M. Stankovic
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 19 November 2021
Sec. Human and Medical Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.780874
This article is part of the Research TopicRecent Advances in the Prediction of Functional Effects of Non-Coding Variants and their Clinical ApplicationsView all 12 articles
Objective: To identify and report novel variants in the TMPRSS3 gene and their clinical manifestations related to hearing loss as well as intervention outcomes. This information will be helpful for genetic counseling and treatment planning for these patients.
Methods: Literature review of previously reported TMPRSS3 variants was conducted. Reported variants and associated clinical information was compiled. Additionally, cohort data from 18 patients, and their families, with a positive result for TMPRSS3-associated hearing loss were analyzed. Genetic testing included sequencing and copy number variation (CNV) analysis of TMPRSS3 and the Laboratory for Molecular Medicine’s OtoGenome-v1, -v2, or -v3 panels. Clinical data regarding patient hearing rehabilitation was interpreted along with their genetic testing results and in the context of previously reported cochlear implant outcomes in individuals with TMPRSS3 variants.
Results: There have been 87 previously reported TMPRSS3 variants associated with non-syndromic hearing loss in more than 20 ancestral groups worldwide. Here we report occurrences of known variants as well as one novel variant: deletion of Exons 1–5 and 13 identified from our cohort of 18 patients. The hearing impairment in many of these families was consistent with that of previously reported patients with TMPRSS3 variants (i.e., typical down-sloping audiogram). Four patients from our cohort underwent cochlear implantation.
Conclusion: Bi-allelic variants of TMPRSS3 are associated with down-sloping hearing loss regardless of ancestry. The outcome following cochlear implantation in patients with variants of TMPRSS3 is excellent. Therefore, cochlear implantation is strongly recommended for hearing rehabilitation in these patients.
Autosomal recessive non-syndromic hearing loss (ARNSHL) is the most common form of hereditary hearing loss. It accounts for about 70–80% of congenital hereditary hearing loss. ARNSHL is an extremely heterogenous condition as more than 98 loci have been mapped and 77 causative genes have been identified to date (http://hereditaryhearingloss.org/).
The TMPRSS3 gene encodes a type III transmembrane serine protease that is structurally defined by four functional domains: a transmembrane domain, low density lipoprotein receptor A domain, scavenger receptor cysteine rich domain, and a carboxyl terminal serine protease domain (Südhof et al., 1985; van Driel et al., 1987; Sarrias et al., 2004; Rawlings et al., 2010). The TMPRSS3 gene is expressed in inner hair cells, spiral ganglion neurons (SGNs), the stria vascularis, and cochlear aqueducts of fetal cochlea (Guipponi et al., 2002). Four alternatively spliced transcripts have been described (DiStefano et al., 2018). The transmembrane serine protease 3 protein is thought to be involved in the development and maintenance of the inner ear, perilymph, endolymph and SGNs (Guipponi et al., 2002). While the function of the TMPRSS3 gene in the auditory system is not fully understood, its alteration has been linked with non-syndromic genetic hearing loss (DiStefano et al., 2018).
The incidence of TMPRSS3-associated ARNSHL is variable amongst different ancestral backgrounds but TMPRSS3 is a significant contributor in some populations. Pathogenic TMPRSS3 variants account for 0.7% of Japanese (Miyagawa et al., 2015), 3% of Pakistani (Ben-Yosef et al., 2001), 4.6% of Chinese (Gao et al., 2017), 5–6% of Tunisian (Masmoudi et al., 2001), 5.9% of Korean (Chung et al., 2014), and 11% of Turkish (Wattenhofer et al., 2005) ARNSHL cases. However, this gene has been reported in less than 1% of non-syndromic genetic deafness in White individuals (Wattenhofer et al., 2002). In contrast, pathogenic variants in the GJB2 gene are found in up to 50% of patients with ARNSHL. Despite the relatively low proportion of ARNSHL cases attributed to TMPRSS3, the gene remains a prime candidate for post lingual progressive ARNHSL in North European populations once GJB2 variants are ruled out (Seligman et al., 2021).
Patients with pathogenic variants in the TMPRSS3 gene have been described as having one of two discrete hearing phenotypes: severe, prelingual or progressive, post-lingual hearing loss. Weegerink et al. (2011) proposed that the phenotypic outcome of hearing loss is dependent on the combination and severity of TMPRSS3 variants (i.e., mild or severe). They assert that having two “severe” pathogenic variants leads to profound deafness with prelingual onset (DFNB10), whereas a single ‘severe’ pathogenic variant in trans with a milder TMPRSS3 pathogenic variant yields an initially less severe, but progressive and post-lingual onset hearing loss (DFNB8) (Weegerink et al., 2011). The TMPRSS3 gene encodes for a transmembrane serine protease which is expressed in SGNs (Guipponi et al., 2002). Therefore, the differential hearing phenotype may reflect the extent of loss of protease activity from a given variant.
In this study, we compile previously reported TMPRSS3 variants and present a novel variant along with their associated hearing phenotypes. We also aggregate reported outcomes and present new findings regarding the therapeutic effects of cochlear implantation (CI) in patients with pathogenic TMPRSS3 variants. Together, this information may assist with genetic counseling and treatment planning for patients with TMPRSS3 variants.
Literature databases were searched using different combinations of keywords such as “transmembrane serine protease 3,” “TMPRSS3,” “ear,” “hearing loss,” “non-syndromic hearing loss,” and “cochlear implantation.” The databases searched were PubMed, Google Scholar, and two selected gene database websites (https://hereditaryhearingloss.org; https://www.ncbi.nlm.nih.gov/clinvar/). The titles and abstracts were screened using following inclusion criteria: 1) written in English, 2) dealing with non-syndromic hearing loss, and 3) reporting human data.
Based on the search strategy, 39 TMPRSS3-associated papers published from May 2000 to Aug 2021 were reviewed and summarized (Figure 1; Table 1). Among those 39 studies, eleven studies described patients who underwent cochlear implantation (Table 2).
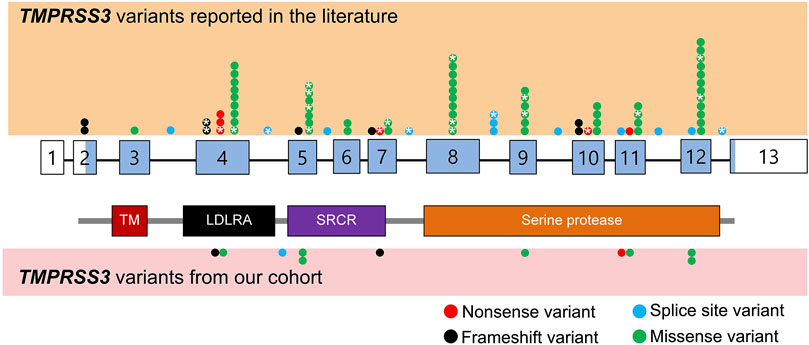
FIGURE 1. Schematic representation of TMPRSS3 variants identified from literature review as well as our cohort. Benign variants are not displayed. The exons are numbered with coding sequence shaded blue and untranslated regions unshaded. *: variants with reported cochlear implant outcomes. TM, transmembrane domain; LDLRA, LDL receptor-like domain; SRCR, scavenger receptor cysteine-rich domain.
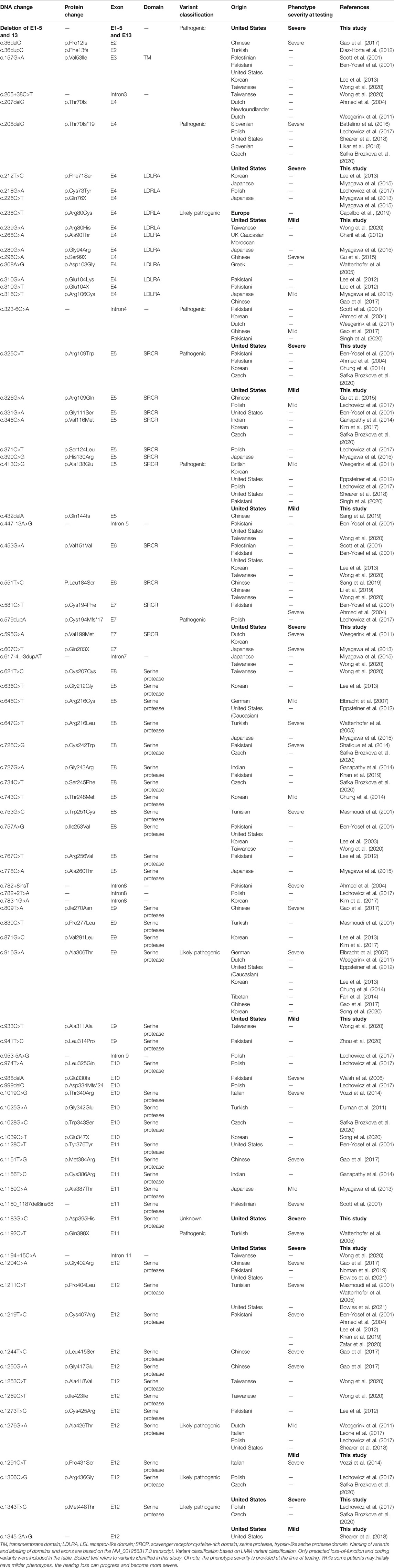
TABLE 1. Overview of TMPRSS3 variants resulting in non-syndromic hearing loss, including those identified in the present study.
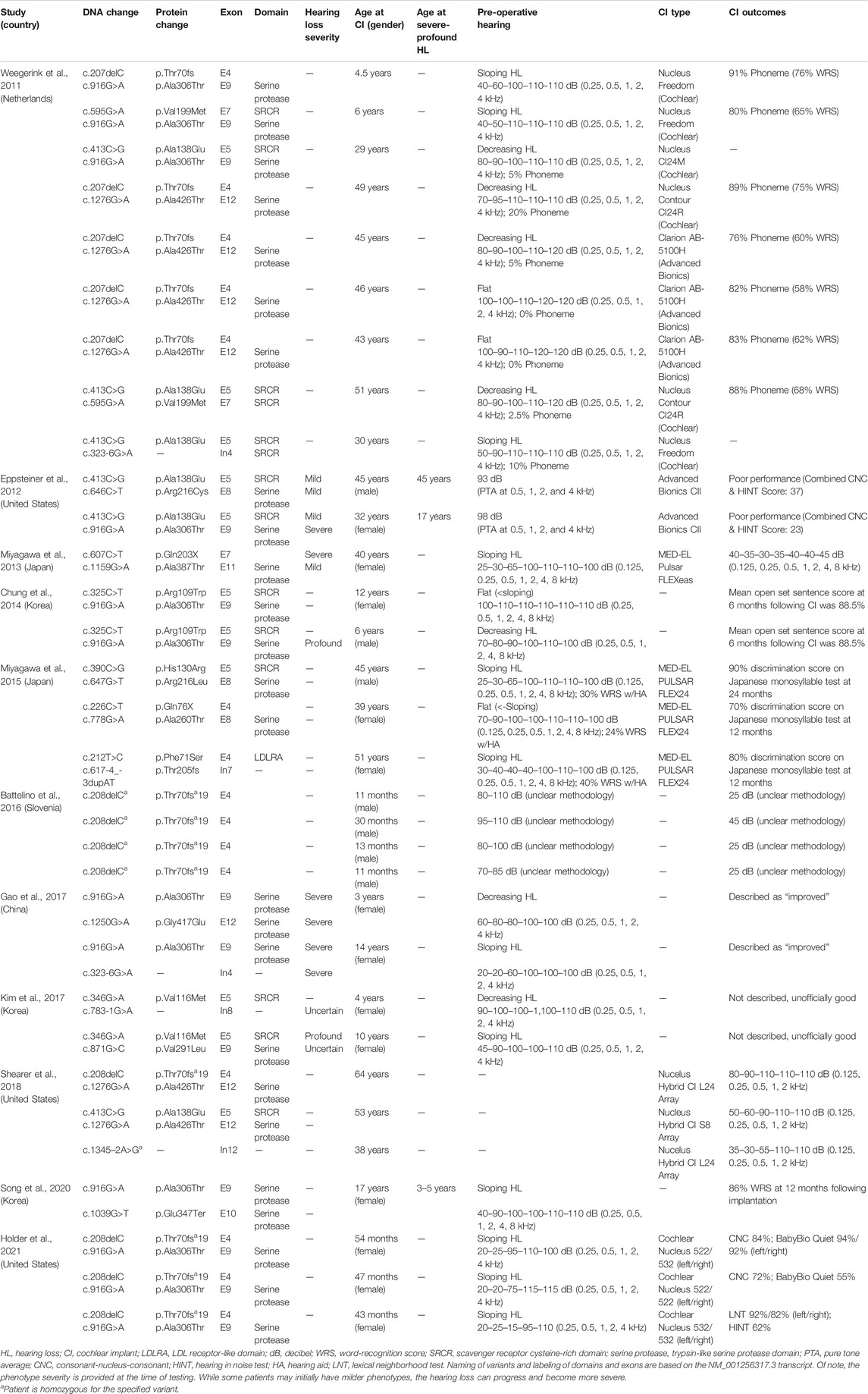
TABLE 2. Overview of clinical characteristics and genotypes of patients with TMPRSS3 variants who have received cochlear implantation.
Previously reported variants and their associated hearing phenotypes and clinical outcomes following CI, when available, were compiled. Additionally, our own cohort of patients was genetically screened as described below.
Our study included genetic and phenotypic data from 18 patients and their family members (when available), who were largely White, though Family A was a consanguineous White Egyptian family, Family B was “mixed,” and Families M and I were of Hispanic or Latino ethnicity. Of the patients with characterized hearing loss, the severity ranged from moderate to profound with some individuals experiencing congenital onset and others experiencing a childhood onset or an onset in the second decade of life. Patients were referred to the Laboratory for Molecular Medicine (LMM) at Mass General Brigham Personalized Medicine (Cambridge, MA, United States) from 2009 to 2017. Patients were referred from various clinics and hospitals across the United States. The LMM collected information pertinent to the nature of the hearing loss in the patients (if available) including family history of hearing loss and/or disease, audiological testing, temporal bone CT/MRI results, and CI status. Further information was requested through physicians via the Mass General Brigham Human Research Committee’s IRB protocol for the study of the genetics of hearing loss. Patients were selected based on whether they received a positive result for TMPRSS3-associated hearing loss with the intent of follow up of the outcome of CI, if received.
Patient DNA was extracted from whole blood from patients who were referred to the LMM for hearing-loss genetic testing. Our cohort contains patients from 2009 to 2017. The genetic testing varied for each patient based on the judgment of the ordering physician and the nature of the patient’s hearing loss. Testing was performed by single gene sequencing that included TMPRSS3, or LMM’s OtoGenome-v1,-v2, or -v3 panels.
The LMM’s bioinformatics pipeline for targeted next generation sequencing (NGS) panels has been described previously (Pugh et al., 2016). Patients with hearing loss who underwent genetic testing between 2010 and 2014 were tested with the Otogenome-v1 which included the following 71 genes: ACTG1, ATP6V1B1, BSND, CCDC50, CDH23, CLDN14, CLRN1, COCH, COL11A2, CRYM, DFNA5, DFNB31, DFNB59, DIAPH1, ESPN, ESRRB, EYA1, EYA4, GIPC3, GJB2, GJB3, GJB6, GPR98, GPSM2, GRHL2, GRXCR1, HGF, ILDR1, KCNE1, KCNQ1, KCNQ4, LHFPL5, LOXHD1, LRTOMT, MARVELD2, MIR183, MIR96, MSRB3, MTRNR1 (12S rRNA), MTTS1 (tRNAser(UCN)), MYH14, MYH9, MYO15A, MYO1A, MYO3A, MYO6, MYO7A, OTOA, OTOF, PCDH15, PDZD7, POU3F4, POU4F3, PRPS1, RDX, SERPINB6, SLC17A8, SLC26A4 (PDS), SLC26A5, TECTA, TIMM8A, TJP2, TMC1, TMIE, TMPRSS3, TPRN, TRIOBP, USH1C, USH1G, USH2A, and WFS1.
OtoGenome-v2 was used in patients who underwent testing at the LMM from 2014 to 2015. For this iteration, PDZD7 and SLC26A5 genes were removed and the STRC gene was added. In addition, copy number variant (CNV) detection was added using VisCap as previously described (Pugh et al., 2016; Tayoun et al., 2016).
OtoGenome-v3, used from 2015 to 2017, included 87 genes but did not include the following genes included in v2: CRYM, GJB3, MIR182, MYO1A, SLC17A8, and TJP2. The following 23 genes were added CACNA1D, CATSPER2, CEACAM16, CIB2, CLPP, DIABLO, EDN3, EDNRB, HARS2, HSD17B4, KARS, LARS2, MITF, OTOG, OTOGL, P2RX2, PAX3, SIX1, SMPX, SOX10, SYNE4, TBC1D24, and TSPEAR. Parents and other unaffected/affected family members, when available, were tested for detected variants. Variants were confirmed via Sanger sequencing for single-nucleotide variants (SNVs), or droplet digital PCR for CNVs called by VisCap (Pugh et al., 2016; Tayoun et al., 2016).
The LMM’s early variant classification methods are as previously described (Duzkale et al., 2013) and were subsequently updated to conform to more recent professional guidelines (Richards et al., 2015). Data used to classify variants included that from population databases (e.g., Exome Aggregation Consortium (ExAC); gnomAD), internal or external disease databases (e.g., ClinVar, LOVD, HGMD), the literature, functional studies, segregation, allelic observations and in silico missense and splicing prediction tools. Variants were classified as pathogenic (P), likely pathogenic (LP), of uncertain significance (VUS), likely benign, or benign. The VUS category was further subdivided into VUS-5, -4, and -3 where VUS-5 indicated leaning towards pathogenic, and VUS-3 indicated leaning towards benign. Likely benign and benign variants are not reported in this article but were submitted to ClinVar (www.ncbi.nlm.nih.gov/clinvar/) along with all other variants observed at the LMM.
We reviewed the type, position, origin, and variant classification of 87 previously reported TMPRSS3 variants and present one novel variant identified from our cohort (Figure 1; Table 1). Compiled variants are associated with non-syndromic hearing loss in more than 20 ancestral groups worldwide. Fourteen of the identified variants were predicted loss-of-function (pLOF) (frameshift, stop-codon, or splice-site variants) with either prematurely terminated protein products or nonsense-mediated decay of mRNA. Fifty-eight of the identified variants were missense variants. Nearly all variants were predicted to disrupt the proteolytic activity of the protein. Both prelingual and post lingual hearing impairment was reported, with most patients showing a typical ski-slope audiogram configuration. CI outcomes were reported for 32 patients with bi-allelic variants in TMPRSS3 across 11 different studies (Table 2) (Weegerink et al., 2011; Eppsteiner et al., 2012; Miyagawa et al., 2013; Chung et al., 2014; Miyagawa et al., 2015; Battelino et al., 2016; Gao et al., 2017; Kim et al., 2017; Shearer et al., 2018; Song et al., 2020; Holder et al., 2021). While degree of hearing improvement varied between patients, the majority of those who underwent CI had positive outcomes.
Our cohort included 18 patients—7 females and 11 males—with ages ranging from 3 months to 36 years (Figure 2). 15 patients were White with the remaining 3 identifying as Hispanic/Latino or mixed. We identified 12 different TMPRSS3 variants of which 1 has not been previously reported: deletion of Exons 1–5 and 13 (Table 3). This novel variant was classified as pathogenic as it met the criteria outlined by previous professional guidelines (Richards et al., 2015) with specifications provided by ClinGen (https://clinicalgenome.org/working-groups/sequence-variant-interpretation), specifically the combination of PVS1 (predicted loss of function), PM2 (absence in gnomAD), and PM3 (homozygous observation in an individual with phenotype matching the gene). The most commonly identified variants were p.Thr70fs*19 and p.Ala138Glu. Eight patients had congenital hearing loss, four of whom had biallelic pLOF variants.
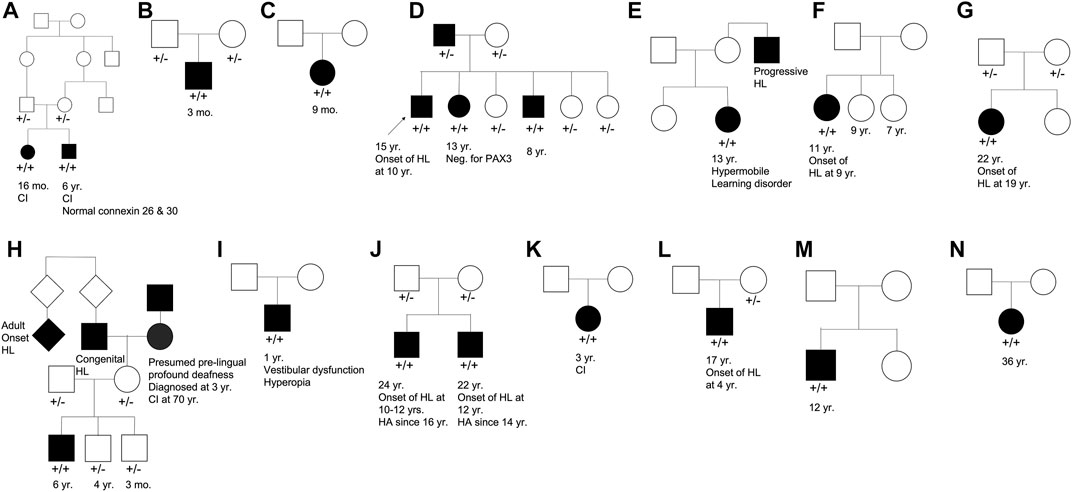
FIGURE 2. Pedigree chart for enrolled patient families (A–N). Age at genetic testing, age at onset of hearing loss, and other relevant clinical information is provided, when available, for patients and family members. CI, cochlear implant; HL, hearing loss; HA, hearing aid.
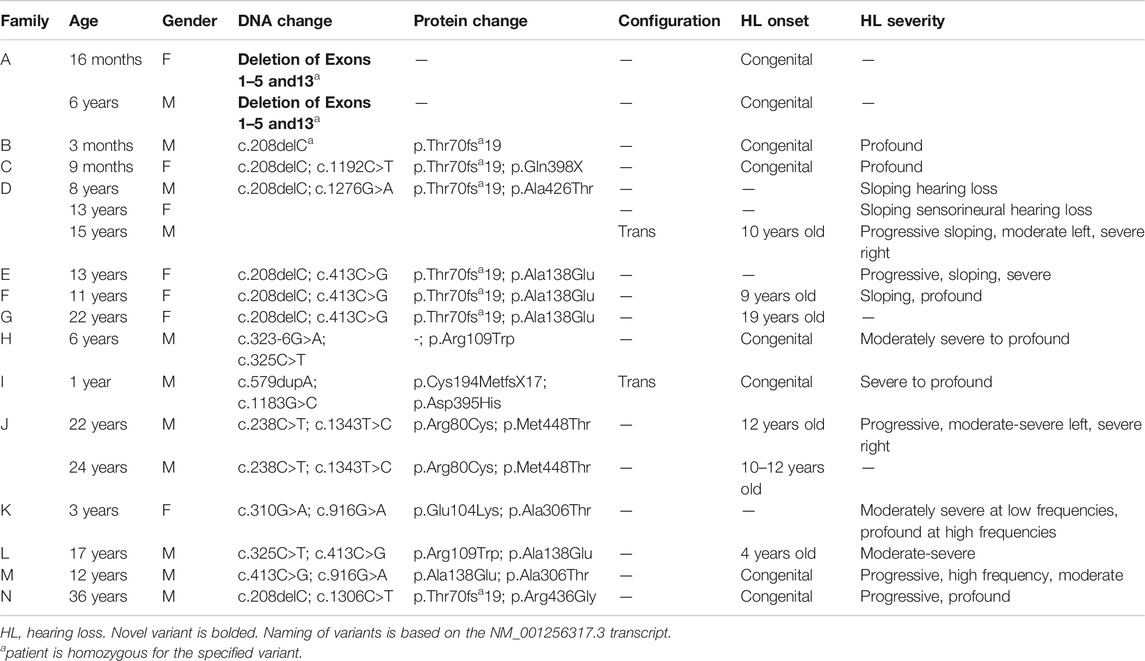
TABLE 3. Genotype and phenotype overview of our patient cohort.
Four patients in our cohort underwent CI, and outcome information was available for two patients. The first patient, from family B, was found to have congenital profound hearing loss and was homozygous for p.Thr70fs*19. It is unclear when the patient underwent CI. However, at a follow up at 4 years of age, the patient had functional speech. Clinical records indicated that the patient had ongoing articulation errors and required speech therapy but was able to maintain adequate hearing. The second patient, from family K, was compound heterozygous for p.Glu104Lys and p.Ala306Thr. Clinical records have suggested positive CI outcome for her moderate-profound hearing loss. The remaining two patients who underwent CI were the siblings from family A who both had profound congenital hearing loss and were homozygous for a deletion of Exons 1–5 and 13. Their current hearing status is unknown.
The genotype-phenotype correlations of TMPRSS3 variants have not been well characterized. It has been previously shown that the frequency of TMPRSS3-induced ARSNHL was low in White individuals (Wattenhofer et al., 2002). However, a recent epidemiological study of patients undergoing CI revealed that 10% (13) of patients with positive genetic testing had TMPRSS3 gene variants (Seligman et al., 2021). As adoption of genetic testing in clinical practice continues to grow, it is important to be aware of common TMPRSS3 variants and associated phenotypes to best counsel patients.
In our cohort of 18 patients, 15 of whom were White, the most frequently observed variants were p.Thr70fs*19 and p.Ala138Glu implying that those were either hot spots or founder variants. The combination of the p.Thr70fs*19 frameshift variant with a missense variant appeared to cause sloping hearing loss that varied in severity. Biallelic pLOF variants appeared to cause congenital profound hearing loss. This phenotype information is valuable when trying to understand potential patient prognosis based on genetic testing results.
Previous studies on the role of CI in patients with TMPRSS3 variants have reported variable results. In one study, poor outcomes following CI in patients with TMPRSS3 variants were attributed to the expression of the TMPRSS3 gene in SGNs as opposed to other locations in the cochlea such as the membranous labyrinth (Eppsteiner et al., 2012). These authors also suggested that patients with pathogenic TMPRSS3 variants may have continued loss of SGNs over time which could contribute to ongoing hearing deterioration even after CI. However, recent studies have shown predominantly positive outcomes following CI in patients with TMPRSS3 variants (Weegerink et al., 2011; Miyagawa et al., 2013; Chung et al., 2014; Miyagawa et al., 2015; Battelino et al., 2016; Gao et al., 2017; Shearer et al., 2018; Song et al., 2020; Holder et al., 2021). This discrepancy might be related to the large duration of deafness and older age of the two patients in Eppsteiner et al. (2012) and Holder et al. (2021). In addition, a study of CI outcomes in pediatric patients with TMPRSS3 variants reported positive outcomes with no evidence of SGN degeneration leading to decreased performance over time (Holder et al., 2021). Furthermore, it was suggested that even if SGN degeneration does contribute to a longitudinal decline in performance, early CI may help slow or reverse this process (Holder et al., 2021). Even so, many clinics do not implant patients with precipitously sloping hearing loss as they do not meet labeled indications for CI. However, off-label implantation has been shown to be beneficial and is being employed much more frequently at major academic medical centers (Carlson et al., 2015; Leigh et al., 2016; Carlson et al., 2018).
Taken together with the positive clinical outcomes following CI in two patients from our cohort, it is evident that CI is a promising treatment strategy for patients with TMPRSS3 variants. Active intervention with CI is likely to be beneficial, particularly in patients in whom residual hearing is preserved. It is imperative that the benefits of CI are made clear when counseling patients on their potential treatment options.
The evidence for all variants classified by the authors is included in submissions to ClinVar by the Laboratory for Molecular Medicine (Organization ID: 21766). All other data supporting the conclusions of this article, if not directly included in the paper, will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the Mass General Brigham Human Research Committee’s IRB. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
IM and AG co-wrote the manuscript and prepared the tables and figures, VS edited the manuscript and prepared the tables and figures for submission, HR edited the manuscript and provided technical feedback, KS conceived, designed, and supervised the manuscript writing and editing.
We gratefully acknowledge support from the National Institutes of Health grant R01 DC015824 (KMS) and Jennifer and Louis Hernandez (KMS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ahmed, Z. M., Cindy Li, X., Powell, S. D., Riazuddin, S., Young, T.-L., Ramzan, K., et al. (2004). Characterization of a New Full Length TMPRSS3 Isoform and Identification of Mutant Alleles Responsible for Nonsyndromic Recessive Deafness in Newfoundland and Pakistan. BMC Med. Genet. 5, 24. doi:10.1186/1471-2350-5-24
Battelino, S., Klancar, G., Kovac, J., Battelino, T., and Trebusak Podkrajsek, K. (2016). TMPRSS3 Mutations in Autosomal Recessive Nonsyndromic Hearing Loss. Eur. Arch. Otorhinolaryngol. 273 (5), 1151–1154. doi:10.1007/s00405-015-3671-0
Ben-Yosef, T., Wattenhofer, M., Riazuddin, S., Ahmed, Z. M., Scott, H. S., Kudoh, J., et al. (2001). Novel Mutations of TMPRSS3 in Four DFNB8/B10 Families Segregating Congenital Autosomal Recessive Deafness. J. Med. Genet. 38 (6), 396–400. doi:10.1136/jmg.38.6.396
Bowles, B., Ferrer, A., Nishimura, C. J., Pinto E Vairo, F., Rey, T., Leheup, B., et al. (2021). TSPEAR Variants Are Primarily Associated with Ectodermal Dysplasia and Tooth Agenesis but Not Hearing Loss: A Novel Cohort Study. Am. J. Med. Genet. 185 (8), 2417–2433. doi:10.1002/ajmg.a.62347
Capalbo, A., Valero, R. A., Jimenez-Almazan, J., Pardo, P. M., Fabiani, M., Jiménez, D., et al. (2019). Optimizing Clinical Exome Design and Parallel Gene-Testing for Recessive Genetic Conditions in Preconception Carrier Screening: Translational Research Genomic Data from 14,125 Exomes. Plos Genet. 15 (10), e1008409. doi:10.1371/journal.pgen.1008409
Carlson, M. L., Sladen, D. P., Gurgel, R. K., Tombers, N. M., Lohse, C. M., and Driscoll, C. L. (2018). Survey of the American Neurotology Society on Cochlear Implantation: Part 1, Candidacy Assessment and Expanding Indications. Otol. Neurotol. 39 (1), e12–e19. doi:10.1097/MAO.0000000000001632
Carlson, M. L., Sladen, D. P., Haynes, D. S., Driscoll, C. L., DeJong, M. D., Erickson, H. C., et al. (2015). Evidence for the Expansion of Pediatric Cochlear Implant Candidacy. Otol. Neurotol. 36 (1), 43–50. doi:10.1097/MAO.0000000000000607
Charif, M., Abidi, O., Boulouiz, R., Nahili, H., Rouba, H., Kandil, M., et al. (2012). Molecular Analysis of the TMPRSS3 Gene in Moroccan Families with Non-syndromic Hearing Loss. Biochem. Biophysical Res. Commun. 419 (4), 643–647. doi:10.1016/j.bbrc.2012.02.066
Chung, J., Park, S. M., Chang, S. O., Chung, T., Lee, K. Y., Kim, A. R., et al. (2014). A Novel Mutation of TMPRSS3 Related to Milder Auditory Phenotype in Korean Postlingual Deafness: a Possible Future Implication for a Personalized Auditory Rehabilitation. J. Mol. Med. 92 (6), 651–663. doi:10.1007/s00109-014-1128-3
Diaz-Horta, O., Duman, D., Foster, J., Sırmacı, A., Gonzalez, M., Mahdieh, N., et al. (2012). Whole-exome Sequencing Efficiently Detects Rare Mutations in Autosomal Recessive Nonsyndromic Hearing Loss. PLoS One 7 (11), e50628. doi:10.1371/journal.pone.0050628
DiStefano, M. T., Hemphill, S. E., Cushman, B. J., Bowser, M. J., Hynes, E., Grant, A. R., et al. (2018). Curating Clinically Relevant Transcripts for the Interpretation of Sequence Variants. J. Mol. Diagn. 20 (6), 789–801. doi:10.1016/j.jmoldx.2018.06.005
Duman, D., Sirmaci, A., Cengiz, F. B., Ozdag, H., and Tekin, M. (2011). Screening of 38 Genes Identifies Mutations in 62% of Families with Nonsyndromic Deafness in Turkey. Genet. Test. Mol. Biomarkers 15 (1-2), 29–33. doi:10.1089/gtmb.2010.0120
Duzkale, H., Shen, J., McLaughlin, H., Alfares, A., Kelly, M., Pugh, T., et al. (2013). A Systematic Approach to Assessing the Clinical Significance of Genetic Variants. Clin. Genet. 84 (5), 453–463. doi:10.1111/cge.12257
Elbracht, M., Senderek, J., Eggermann, T., Thurmer, C., Park, J., Westhofen, M., et al. (2007). Autosomal Recessive Postlingual Hearing Loss (DFNB8): Compound Heterozygosity for Two Novel TMPRSS3 Mutations in German Siblings. J. Med. Genet. 44 (6), e81. doi:10.1136/jmg.2007.049122
Eppsteiner, R. W., Shearer, A. E., Hildebrand, M. S., Deluca, A. P., Ji, H., Dunn, C. C., et al. (2012). Prediction of Cochlear Implant Performance by Genetic Mutation: the Spiral Ganglion Hypothesis. Hearing Res. 292 (1-2), 51–58. doi:10.1016/j.heares.2012.08.007
Fan, D., Zhu, W., Li, D., Ji, D., and Wang, P. (2014). Identification of a Novel Homozygous Mutation, TMPRSS3: c.535G>A, in a Tibetan Family with Autosomal Recessive Non-syndromic Hearing Loss. PLoS One 9 (12), e114136. doi:10.1371/journal.pone.0114136
Ganapathy, A., Pandey, N., Srisailapathy, C. R. S., Jalvi, R., Malhotra, V., Venkatappa, M., et al. (2014). Non-syndromic Hearing Impairment in India: High Allelic Heterogeneity Among Mutations in TMPRSS3, TMC1, USHIC, CDH23 and TMIE. PLoS One 9 (1), e84773. doi:10.1371/journal.pone.0084773
Gao, X., Huang, S.-S., Yuan, Y.-Y., Xu, J.-C., Gu, P., Bai, D., et al. (2017). Identification ofTMPRSS3as a Significant Contributor to Autosomal Recessive Hearing Loss in the Chinese Population. Neural Plasticity 2017, 1–8. doi:10.1155/2017/3192090
Gu, X., Guo, L., Ji, H., Sun, S., Chai, R., Wang, L., et al. (2015). Genetic Testing for Sporadic Hearing Loss Using Targeted Massively Parallel Sequencing Identifies 10 Novel Mutations. Clin. Genet. 87 (6), 588–593. doi:10.1111/cge.12431
Guipponi, M., Vuagniaux, G., Wattenhofer, M., Shibuya, K., Vazquez, M., Dougherty, L., et al. (2002). The Transmembrane Serine Protease (TMPRSS3) Mutated in Deafness DFNB8/10 Activates the Epithelial Sodium Channel (ENaC) In Vitro. Hum. Mol. Genet. 11 (23), 2829–2836. doi:10.1093/hmg/11.23.2829
Holder, J. T., Morrel, W., Rivas, A., Labadie, R. F., and Gifford, R. H. (2021). Cochlear Implantation and Electric Acoustic Stimulation in Children with TMPRSS3 Genetic Mutation. Otol. Neurotol. 42 (3), 396–401. doi:10.1097/MAO.0000000000002943
Khan, A., Han, S., Wang, R., Ansar, M., Ahmad, W., and Zhang, X. (2019). Sequence Variants in Genes Causing Nonsyndromic Hearing Loss in a Pakistani Cohort. Mol. Genet. Genomic Med. 7 (9), e917. doi:10.1002/mgg3.917
Kim, A., Chung, J., Kim, N., Lee, C., Park, W.-Y., Oh, D.-Y., et al. (2017). The Analysis of A Frequent TMPRSS3 Allele Containing P.V116M and P.V291L in A Cis Configuration Among Deaf Koreans. Ijms 18 (11), 2246. doi:10.3390/ijms18112246
Lechowicz, U., Gambin, T., Pollak, A., Podgorska, A., Stawinski, P., Franke, A., et al. (2017). Iterative Sequencing and Variant Screening (ISVS) as a Novel Pathogenic Mutations Search Strategy - Application for TMPRSS3 Mutations Screen. Sci. Rep. 7 (1), 2543. doi:10.1038/s41598-017-02315-w
Lee, J., Baek, J.-I., Choi, J. Y., Kim, U.-K., Lee, S.-H., and Lee, K.-Y. (2013). Genetic Analysis of TMPRSS3 Gene in the Korean Population with Autosomal Recessive Nonsyndromic Hearing Loss. Gene 532 (2), 276–280. doi:10.1016/j.gene.2013.07.108
Lee, K., Khan, S., Islam, A., Ansar, M., Andrade, P. B., Kim, S., et al. (2012). Novel TMPRSS3 Variants in Pakistani Families with Autosomal Recessive Non-syndromic Hearing Impairment. Clin. Genet. 82 (1), 56–63. doi:10.1111/j.1399-0004.2011.01695.x
Lee, Y. J., Park, D., Kim, S. Y., and Park, W. J. (2003). Pathogenic Mutations but Not Polymorphisms in Congenital and Childhood Onset Autosomal Recessive Deafness Disrupt the Proteolytic Activity of TMPRSS3. J. Med. Genet. 40 (8), 629–631. doi:10.1136/jmg.40.8.629
Leigh, J. R., Moran, M., Hollow, R., and Dowell, R. C. (2016). Evidence-based Guidelines for Recommending Cochlear Implantation for Postlingually Deafened Adults. Int. J. Audiol. 55 (2), S3–S8. doi:10.3109/14992027.2016.1146415
Leone, M. P., Palumbo, P., Ortore, R., Castellana, S., Palumbo, O., Melchionda, S., et al. (2017). Putative TMPRSS3/GJB2 Digenic Inheritance of Hearing Loss Detected by Targeted Resequencing. Mol. Cell Probes 33, 24–27. doi:10.1016/j.mcp.2017.03.001
Li, X., Tan, B., Wang, X., Xu, X., Wang, C., Zhong, M., et al. (2019). Identification of a Complex Genomic Rearrangement in TMPRSS3 by Massively Parallel Sequencing in Chinese Cases with Prelingual Hearing Loss. Mol. Genet. Genomic Med. 7 (6), e685. doi:10.1002/mgg3.685
Likar, T., Hasanhodžić, M., Teran, N., Maver, A., Peterlin, B., and Writzl, K. (2018). Diagnostic Outcomes of Exome Sequencing in Patients with Syndromic or Non-syndromic Hearing Loss. PLoS One 13 (1), e0188578. doi:10.1371/journal.pone.0188578
Masmoudi, S., Antonarakis, S. E., Schwede, T., Ghorbel, A. M., Gratri, M. h., Pappasavas, M.-P., et al. (2001). Novel Missense Mutations of TMPRSS3 in Two Consanguineous Tunisian Families with Non-syndromic Autosomal Recessive Deafness. Hum. Mutat. 18 (2), 101–108. doi:10.1002/humu.1159
Miyagawa, M., Nishio, S.-y., Ikeda, T., Fukushima, K., and Usami, S.-i. (2013). Massively Parallel DNA Sequencing Successfully Identifies New Causative Mutations in Deafness Genes in Patients with Cochlear Implantation and EAS. PLoS One 8 (10), e75793. doi:10.1371/journal.pone.0075793
Miyagawa, M., Nishio, S.-y., Sakurai, Y., Hattori, M., Tsukada, K., Moteki, H., et al. (2015). The Patients Associated with TMPRSS3 Mutations Are Good Candidates for Electric Acoustic Stimulation. Ann. Otol. Rhinol. Laryngol. 124 (1), 193S–204S. doi:10.1177/0003489415575056
Noman, M., Ishaq, R., Bukhari, S. A., Ahmed, Z. M., and Riazuddin, S. (2019). Delineation of Homozygous Variants Associated with Prelingual Sensorineural Hearing Loss in Pakistani Families. Genes 10 (12), 1031. doi:10.3390/genes10121031
Pugh, T. J., Amr, S. S., Bowser, M. J., Gowrisankar, S., Hynes, E., Mahanta, L. M., et al. (2016). VisCap: Inference and Visualization of Germ-Line Copy-Number Variants from Targeted Clinical Sequencing Data. Genet. Med. 18 (7), 712–719. doi:10.1038/gim.2015.156
Rawlings, N. D., Barrett, A. J., and Bateman, A. (2010). MEROPS: the Peptidase Database. Nucleic Acids Res. 38, D227–D233. doi:10.1093/nar/gkp971
Richards, S., Aziz, N., Aziz, N., Bale, S., Bick, D., Das, S., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–423. doi:10.1038/gim.2015.30
Safka Brozkova, D., Poisson Marková, S., Mészárosová, A. U., Jenčík, J., Čejnová, V., Čada, Z., et al. (2020). Spectrum and Frequencies of Non GJB2 Gene Mutations in Czech Patients with Early Non‐syndromic Hearing Loss Detected by Gene Panel NGS and Whole‐exome Sequencing. Clin. Genet. 98 (6), 548–554. doi:10.1111/cge.13839
Sang, S., Ling, J., Liu, X., Mei, L., Cai, X., Li, T., et al. (2019). Proband Whole-Exome Sequencing Identified Genes Responsible for Autosomal Recessive Non-syndromic Hearing Loss in 33 Chinese Nuclear Families. Front. Genet. 10, 639. doi:10.3389/fgene.2019.00639
Sarrias, M. R., Gronlund, J., Padilla, O., Madsen, J., Holmskov, U., and Lozano, F. (2004). The Scavenger Receptor Cysteine-Rich (SRCR) Domain: an Ancient and Highly Conserved Protein Module of the Innate Immune System. Crit. Rev. Immunol. 24 (1), 1–38. doi:10.1615/critrevimmunol.v24.i1.10
Scott, H. S., Kudoh, J., Wattenhofer, M., Shibuya, K., Berry, A., Chrast, R., et al. (2001). Insertion of β-satellite Repeats Identifies a Transmembrane Protease Causing Both Congenital and Childhood Onset Autosomal Recessive Deafness. Nat. Genet. 27 (1), 59–63. doi:10.1038/83768
Seligman, K. L., Shearer, A. E., Frees, K., Nishimura, C., Kolbe, D., Dunn, C., et al. (2021). Genetic Causes of Hearing Loss in a Large Cohort of Cochlear Implant Recipients. Otolaryngol. Head Neck Surg., 019459982110213. doi:10.1177/01945998211021308
Shafique, S., Siddiqi, S., Schraders, M., Oostrik, J., Ayub, H., Bilal, A., et al. (2014). Genetic Spectrum of Autosomal Recessive Non-syndromic Hearing Loss in Pakistani Families. PLoS One 9 (6), e100146. doi:10.1371/journal.pone.0100146
Shearer, A. E., Tejani, V. D., Brown, C. J., Abbas, P. J., Hansen, M. R., Gantz, B. J., et al. (2018). In Vivo Electrocochleography in Hybrid Cochlear Implant Users Implicates TMPRSS3 in Spiral Ganglion Function. Sci. Rep. 8 (1), 14165. doi:10.1038/s41598-018-32630-9
Singh, K., Bijarnia-Mahay, S., Ramprasad, V. L., Puri, R. D., Nair, S., Sharda, S., et al. (2020). NGS-based Expanded Carrier Screening for Genetic Disorders in North Indian Population Reveals Unexpected Results - a Pilot Study. BMC Med. Genet. 21 (1), 216. doi:10.1186/s12881-020-01153-4
Song, M. H., Jung, J., Rim, J. H., Choi, H. J., Lee, H. J., Noh, B., et al. (2020). Genetic Inheritance of Late-Onset, Down-Sloping Hearing Loss and its Implications for Auditory Rehabilitation. Ear Hear 41 (1), 114–124. doi:10.1097/aud.0000000000000734
Südhof, T. C., Goldstein, J. L., Brown, M. S., and Russell, D. W. (1985). The LDL Receptor Gene: a Mosaic of Exons Shared with Different Proteins. Science 228 (4701), 815–822. doi:10.1126/science.2988123
Tayoun, A. N. A., Mason-Suares, H., Frisella, A. L., Bowser, M., Duffy, E., Mahanta, L., et al. (2016). Targeted Droplet-Digital PCR as a Tool for Novel Deletion Discovery at the DFNB1 Locus. Hum. Mutat. 37 (1), 119–126. doi:10.1002/humu.22912
van Driel, I. R., Goldstein, J. L., Südhof, T. C., and Brown, M. S. (1987). First Cysteine-Rich Repeat in Ligand-Binding Domain of Low Density Lipoprotein Receptor Binds Ca2+ and Monoclonal Antibodies, but Not Lipoproteins. J. Biol. Chem. 262 (36), 17443–17449. doi:10.1016/s0021-9258(18)45399-9
Vozzi, D., Morgan, A., Vuckovic, D., D'Eustacchio, A., Abdulhadi, K., Rubinato, E., et al. (2014). Hereditary Hearing Loss: a 96 Gene Targeted Sequencing Protocol Reveals Novel Alleles in a Series of Italian and Qatari Patients. Gene 542 (2), 209–216. doi:10.1016/j.gene.2014.03.033
Walsh, T., Rayan, A. A., Sa'ed, J. A., Shahin, H., Shepshelovich, J., Lee, M. K., et al. (2006). Genomic Analysis of a Heterogeneous Mendelian Phenotype: Multiple Novel Alleles for Inherited Hearing Loss in the Palestinian Population. Hum. Genomics 2 (4), 203–211. doi:10.1186/1479-7364-2-4-203
Wattenhofer, M., Di Iorio, M., Rabionet, R., Dougherty, L., Pampanos, A., Schwede, T., et al. (2002). Mutations in the TMPRSS3 Gene Are a Rare Cause of Childhood Nonsyndromic Deafness in Caucasian Patients. J. Mol. Med. 80 (2), 124–131. doi:10.1007/s00109-001-0310-6
Wattenhofer, M., Sahin-Calapoglu, N., Andreasen, D., Kalay, E., Caylan, R., Braillard, B., et al. (2005). A Novel TMPRSS3 Missense Mutation in a DFNB8/10 Family Prevents Proteolytic Activation of the Protein. Hum. Genet. 117 (6), 528–535. doi:10.1007/s00439-005-1332-x
Weegerink, N. J. D., Schraders, M., Oostrik, J., Huygen, P. L. M., Strom, T. M., Granneman, S., et al. (2011). Genotype-phenotype Correlation in DFNB8/10 Families with TMPRSS3 Mutations. Jaro 12 (6), 753–766. doi:10.1007/s10162-011-0282-3
Wong, S.-H., Yen, Y.-C., Li, S.-Y., and Yang, J.-J. (2020). Novel Mutations in the TMPRSS3 Gene May Contribute to Taiwanese Patients with Nonsyndromic Hearing Loss. Ijms 21 (7), 2382. doi:10.3390/ijms21072382
Zafar, S., Shahzad, M., Ishaq, R., Yousaf, A., Shaikh, R. S., Akram, J., et al. (2020). Novel Mutations in CLPP, LARS2, CDH23, and COL4A5 Identified in Familial Cases of Prelingual Hearing Loss. Genes 11 (9), 978. doi:10.3390/genes11090978
Keywords: TMPRSS3, cochlear implantation, sensorineural hearing loss, genetic counseling, hereditary hearing loss
Citation: Moon IS, Grant AR, Sagi V, Rehm HL and Stankovic KM (2021) TMPRSS3 Gene Variants With Implications for Auditory Treatment and Counseling. Front. Genet. 12:780874. doi: 10.3389/fgene.2021.780874
Received: 21 September 2021; Accepted: 18 October 2021;
Published: 19 November 2021.
Edited by:
Gavin R. Oliver, Mayo Clinic, United StatesReviewed by:
Katarina Trebušak Podkrajšek, University of Ljubljana, SloveniaCopyright © 2021 Moon, Grant, Sagi, Rehm and Stankovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Konstantina M. Stankovic, a3N0YW5rb3ZpY0BzdGFuZm9yZC5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.