 Xi Yuan
Xi Yuan Kaifeng Ma
Kaifeng Ma Man Zhang
Man Zhang Qixiang Zhang
Qixiang Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 15 December 2021
Sec. Genomics of Plants and the Phytoecosystem
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.779557
This article is part of the Research Topic The Development and Application of Multi-Omics Integration Approaches to Dissecting Complex Traits in Plants View all 25 articles
DNA methylation is a common epigenetic modification involved in regulating many biological processes. However, the epigenetic mechanisms involved in the formation of floral scent have rarely been reported within a famous traditional ornamental plant Prunus mume emitting pleasant fragrance in China. By combining whole-genome bisulfite sequencing and RNA-seq, we determined the global change in DNA methylation and expression levels of genes involved in the biosynthesis of floral scent in four different flowering stages of P. mume. During flowering, the methylation status in the “CHH” sequence context (with H representing A, T, or C) in the promoter regions of genes showed the most significant change. Enrichment analysis showed that the differentially methylated genes (DMGs) were widely involved in eight pathways known to be related to floral scent biosynthesis. As the key biosynthesis pathway of the dominant volatile fragrance of P. mume, the phenylpropane biosynthesis pathway contained the most differentially expressed genes (DEGs) and DMGs. We detected 97 DMGs participated in the most biosynthetic steps of the phenylpropane biosynthesis pathway. Furthermore, among the previously identified genes encoding key enzymes in the biosynthesis of the floral scent of P. mume, 47 candidate genes showed an expression pattern matching the release of floral fragrances and 22 of them were differentially methylated during flowering. Some of these DMGs may or have already been proven to play an important role in biosynthesis of the key floral scent components of P. mume, such as PmCFAT1a/1c, PmBEAT36/37, PmPAL2, PmPAAS3, PmBAR8/9/10, and PmCNL1/3/5/6/14/17/20. In conclusion, our results for the first time revealed that DNA methylation is widely involved in the biosynthesis of floral scent and may play critical roles in regulating the floral scent biosynthesis of P. mume. This study provided insights into floral scent metabolism for molecular breeding.
Floral fragrance is one of the most important ornamental traits of horticultural plants (Schiestl, 2010). More than 1700 floral scent substances have been identified, which are mainly biosynthesized through the terpenoid, phenylpropane, and fatty acid biosynthesis pathways. For example, Cananga odorata (Jin et al., 2015), Magnolia champaca (Dhandapani et al., 2017), and Syringa oblata (Zheng et al., 2015) have been found to have terpenoids as their main volatiles; Petunia hybrida and Damask rose (Karami et al., 2015) have phenylpropanes; and Hedychium coronarium (Yue et al., 2015) and Rosa hybrida have terpenoids and phenylpropanes. The diverse combinations of floral volatiles emitted confer the unique floral fragrances among different aromatic plants (Knudsen et al., 2006; Muhlemann et al., 2014). Genes related to the biosynthesis of substances conferring floral aromas have been comprehensively studied in model plants (Tzin and Galili, 2010), such as Clarkia breweri, Antirrhinum majus, Rosa ssp., and Petunia hybrida (Boatright et al., 2004; Fraser and Chapple, 2011; Dudareva et al., 2013; Muhlemann et al., 2014; Widhalm and Dudareva, 2015). Some important genes for the biosynthesis of phenylpropanes have been confirmed in petunia (Boatright et al., 2004; Moerkercke et al., 2009).
Prunus mume (mei) from Rosaceae, a traditional ornamental tree, was domesticated in China more than 3000 years ago (Chen, 1996). Numerous ornamental cultivars with colorful flowers, various types of branches and flower, variety of flowering periods, and characteristic floral scents have been bred (Sun et al., 2013; Xu et al., 2014; Zhang et al., 2018; Zhuo et al., 2021). As the only species within the Prunus genus to produce a strong floral scent (Zhang et al., 2019b), it is important to study the molecular mechanism behind the biosynthesis in P. mume’s floral scent. In previous studies, benzyl acetate and eugenol were revealed to be the main components of the headspace volatiles of the unique fragrances of different P. mume cultivars; additionally, benzyl alcohol and cinnamyl acetate were found to be the main components of the floral volatiles of the P. mume cultivar ‘Fenhong Zhusha’ (‘FZ’) (Zhang et al., 2019a; Bao et al., 2020). All of these volatiles were shown to be biosynthesized by the phenylpropane biosynthesis pathway (Hao R. et al., 2014; Hao R.-J. et al., 2014; Zhang et al., 2019a). Consistent with the pattern of emission of most floral scents (Fenske et al., 2015), the emission and biosynthesis of benzyl acetate and eugenol peak when P. mume is in full bloom (Hao R.-J. et al., 2014; Bao et al., 2019; Zhang et al., 2019b). The intracellular levels of metabolites of floral scent components in flower buds have also been identified in five hybrids of P. mume (Bao et al., 2020). The encoding genes, enzymatic activities, and functions of PmBEATs and PmBARs, which are closely related to the biosynthesis of benzyl acetate, have been determined within the P. mume genome (Zhao et al., 2017; Bao et al., 2019; Bao et al., 2020). The gene families PmEGSs and PmCFATs, which encode key functional enzymes involved in eugenol biosynthesis, have also been identified and characterized (Zhang et al., 2019b). However the epigenetic mechanisms involved in the biosynthesis of its floral scent have rarely been reported.
DNA methylation, one of the best-studied epigenetic modifications, plays a key role in genome stability, developmental regulation, gene expression regulation, transposon silencing, and environmental adaptation (Cubas et al., 1999; Zhu, 2009; Law and Jacobsen, 2010). In plants, DNA methylation mainly occurs on cytosine, including within segments with the sequence “CG,” “CHG,” or “CHH” (with H representing A, T, or C) (Finnegan and Kovac, 2000). The dynamic changes of DNA methylation level (ML) have been shown to regulate the expression of genes and ultimately lead to different phenotypes. The methylation status varies among different species, cultivars of the same species, and even tissues of the same plant (Vaughn et al., 2007; Suzuki and Bird, 2008). Evidence has shown that DNA methylation is involved in regulating the expression of key genes during the ripening of tomato fruit (Zhong et al., 2013); drought resistance (Xu et al., 2018) and flowering (Xing et al., 2019) of apple; peel color formation of apple (Bai et al., 2016; Li et al., 2019), red pear (Wang et al., 2013), and sweet orange (Huang et al., 2019); and the flower color formation of P. mume chimera (Jiang et al., 2020). However, the role it plays in the process of floral scent biosynthesis has not yet been reported.
Here, by using transcriptome sequencing (RNA-seq) and whole-genome bisulfite sequencing (WGBS), we discuss the methylation modification associated with floral scent biosynthesis in P. mume cv. ‘FZ’. The results provide new genome-wide evidence that deepens our understanding of the epigenetic regulatory mechanism underlying the floral scent trait.
The present study focused on ‘FZ’, the earliest flowering P. mume cultivar (Yuan et al., 2020) with a relatively long flowering time and pleasant smell, which was planted in Jiufeng International Mei Garden (40°03′53″N, 116°05′49″E, 132 m a.s.l.), Haidian, Beijing. To reflect the bud developmental state of the whole plant, all buds on one to two twigs of each branch from three different sides of each experimental tree were collected as one biological replicate. Three biological replicates (1, 2, and 3) constituted one sample. Samples were frozen in liquid nitrogen immediately after being obtained and stored at −80°C until sequencing. We selected four samples in total, and took the S2/S4/S6/S8 bud development stage as the dominant stage, from January 9th to March 13th in 2019 (Figure 1). The dominant stages were determined by the following reasons: some of the key floral components of P.mume began to evaporate/emit in the early stages of flowering and peak when P. mume is in full bloom (S8). Slightly earlier than that, the biosynthesis of eugenol can be extracted before blooming (S6) (Zhang et al., 2019b) and the expression of key enzyme-encoding genes closely related to floral scent biosynthesis (like PmCFATs and PmBEATs) in the budding phase (S4 or S5) (Bao et al., 2019; Zhang et al., 2019b). The sample (S2) on January 9th 2019 was also selected for sequencing, for considering that the buds of ‘FZ’ were already finished endo-dormancy on December 23th 2018, and the methylation modification may be earlier than the onset of gene expression (Yuan et al., 2020).

FIGURE 1. Definition of different flowering stages of P. mume: bud scale tight packed (S1), bud swelling (S2), sepal tips appeared (S3), sepals clearly visible (S4), petals appeared (S5), petals clearly visible (S6), beginning of blossom (S7), full blooming (S8), and petal drop (S9).
Genomic DNA of all four sample groups was extracted using Plant Genomic DNA Kit DP305 (Tiangen Biotech, Beijing, China) and then used for library construction using Illumina’s standard DNA methylation analysis protocol and the Accel-NGS®Methyl-Seq DNA Library Kit. The sequencing process was performed on the Illumina Hiseq 6000 platform to generate raw reads, which were preprocessed with Trimmomatic v0.36 software and FastQC analysis (Langmead and Salzberg, 2012). Finally, the clean reads were obtained for subsequent analysis.
Total RNA was extracted with the DP422 extraction kit (Tiangen Biotech). RNA quality and quantity were determined using 1% agarose gels and a NanoPhotometer spectrophotometer (Implen, Calabasas, CA, United States), respectively. An RNA Nano 6000 Assay kit of the Bioanalyzer 2100 system (Agilent, Carpinteria, CA, United States) was used to assess RNA integrity. A total of 3 μg of RNA was used as the input material for sequencing library construction. The NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, United States) was used for reverse transcription. The libraries were also sequenced on the Illumina Hiseq 6000 platform to generate 150 bp paired-end reads (clean bases > 9G).
Using the HISAT2 (https://daehwankimlab.github.io/hisat2/) and Bismark (https://github.com/FelixKrueger/Bismark.git) software to aligned the clean reads in transcriptome and methylome (Kim et al., 2015; Krueger and Andrews, 2011; Langmead and Salzberg, 2012; Wang et al., 2014) to the reference genome of P. mume (http://prunusmumegenome.bjfu.edu.cn/) (Zhang et al., 2012), respectivly. The sequencing depth and coverage were estimated according to duplicates that aligned to a unique genomic region (Krueger and Andrews, 2011; Wang et al., 2014). The non-conversion rate (r) of bisulfite treatment was defined as the rate of the number of sequenced cytosines at all cytosine reference positions divided by the number in the lambda genome.
mC sites were detected using Bismark (Krueger and Andrews, 2011) and defined using a binomial test, with thresholds of sequence depth ≥5 and q-value ≤ 0.01 (Lister et al., 2009; Gifford et al., 2013; Habibi et al., 2013). ML of C site was calculated as follows: ML = mC/(mC + umC) (C here indicate one single C site). The “mCG,” “mCHG,” and “mCHH” contexts (methylcytosine occurring at CG, CHG, and CHH regions, respectively) and their densities, as well as their distributions in each chromosome, were analyzed using previous methods (Chan et al., 2005; Krzywinski et al., 2009; Lister et al., 2009; Zhong et al., 2013). Between samples, the global ML and distributions were also tested (Song et al., 2013). DMRs and differentially methylated loci were identified using DSS software (Feng et al., 2014; Wu et al., 2015; Park and Wu, 2016). Among chomosomes, every 200bp were grouped in one bin. In different functional region of genes, each region is evenly divided into 50 bins. In genesbody and its up- and downstream, each region is evenly divided into 20 bins. The average ML (MLavg) and methylation density (MD) of each bin was calculated as follows: MD = mCX/CX (coverage > 5X, C here indicate all C site within each bin, CX means CG, CHG or CHH) (Wang et al., 2015).
Differentially methylated genes were defined as genes with DMRs within 2 kb of their promoter or gene body (from TSS to TES) region. To clarify the possible regulatory effects of methylation in the natural flowering process of P. mume, the differentially expressed genes (DEGs) between two samples were determined using the following criteria: false discovery rate (FDR) < 0.05 and |fold change| ≥0.
The GOseq R soft package was used for Gene Ontology (GO) enrichment analysis of DMR-related genes that were aligned with GO (p-value < 0.05) (Young et al., 2010). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was used to explore pathways of DMR-associated genes (Kanehisa et al., 2008). We also used KOBAS software to test DMR-related genes’ statistical enrichment in the KEGG pathways (Mao et al., 2005).
To show the involvement of methylation in the biosynthesis of P. mume’s flower scent, DMGs enriched in eight pathways that are related to floral scent biosynthesis were listed. We then summarized and drew the key pathway of floral scent biosynthesis, phenylpropanoid biosynthesis, and marked the processes involving DMGs with red asterisks. For further information about the methylation involved in regulating the expression of floral scent-related genes, the FPKM (Fragments Per Kilobase of exon model per Million mapped fragments) values of 145 summarized genes in previous reaserch that encoding the key enzymes in the biosynthesis of the dominant floral volatile of P. mume were used to make heat maps using the heatmap option and the normalized FPKM values of genes (row) in TBtools software. The DMGs were marked and classified according to region (promoter, gene body), context (CG, CHG, CHH), and ML change (hyper, hypo) properties on heatmap.
The qRT-PCR was performed on the CFX96 TouchTM Real-Time PCR Detection System (Bio-Rad, Hercules, California, United States) using the following parameters: 95°C for 5 min, 30 cycles of 95°C for 5 s, and 60°C for 30 s, concluding with a melting-curve stage for 10 s at 95°C, 5s at 65°C, and 5 s at 95°C. Each reaction consisted of 0.8 μL 1st strand cDNA, 10 μL SYBR Premix Ex Taq (Takara, Dalian, Japan), 0.8 μL each of 10 mM primer pairs, and 7.6 μL H2O. Each sample was assessed in three biological replicates for each sample and normalized using PmPP2A as an internal control. The transcription levels were determined using the 2-△△Ct method. The correlation coefficients between RNA-seq and qRT-PCR were calculated with the CORREL function in an Excel spreadsheet. The specific primers were designed online by Primer 3 plus (http://www.primer3plus.com/cgi-bin/dev/primer3plus.cgi) and are listed in Supplementary Table S9.
To investigate the modification of the biosynthesis of floral scent by DNA methylation during the natural flowering process of mei, we performed WGBS of P. mume buds at four different flowering stages: S2, S4, S6, and S8 (hereafter referred to as Fm1–Fm4) (Figure 1). Each stage was sequenced with three biological replicates. For each sample, at least 32 million clean reads were produced (Supplementary Table S2). Approximately 60% of the clean reads were mapped to the reference genome using Bismark (Krueger and Andrews, 2011) (Supplementary Table S2). The average genome coverage was 21.67 × (Supplementary Table S3). All of our sequenced methylated regions had ∼21 × average coverage per chromosome and ∼10 × average coverage per cytosine site, covering an average of 83.8% of the genome (Supplementary Figure S1; Supplementary Table S4).
Among the 82,368,087 C (cytosine) sites in the P. mume genome, the CHH context was the most common and the CG context was the least (Figure 2A). In each sample, ∼8% (7.32–8.46%) of the total C sites were methylated (Table 1). The mC sites mainly featured the mCG context (42.44–46.94%), with the mCHG (28.40–30.19%) and mCHH contexts (22.86–29.14%) comprising the rest (Figure 2B). Among the three contexts of C sites, 33.90%–35.72% of CG regions, 15.60%–16.97% of CHG regions, and 2.21–3.26% of CHH regions were methylated (Table 1). Meanwhile, it was shown that chromosome Pm3 was highly methylated (Supplementary Figure S2). In terms of ML, most mCG and mCHG regions had higher ML (80–100%), while most mCHH regions showed lower ML (10–40%) (Figure 2C). The analysis of methylation within chromosomes showed that both high MLavg and MD areas were enriched in pericentromeric regions, where the gene density is low (Supplementary Figure S3). The results are consistent with findings from the petals of P. mume cv. ‘FT’ (Jiang et al., 2020), apples, and Arabidopsis.
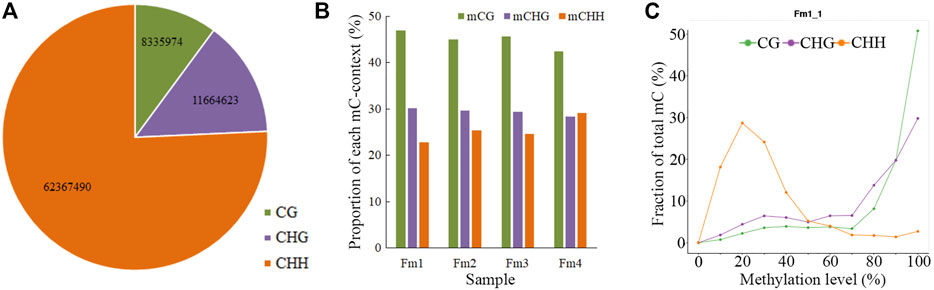
FIGURE 2. Proportions of the three different contexts (CG, CHG, CHH) in (A) total cytosine (C) sites, (B) total mC (methylated cytosine) sites, and (C) ML distribution. The mC distribution diagrams of all samples are shown in the Supporting Information (Figure S4).
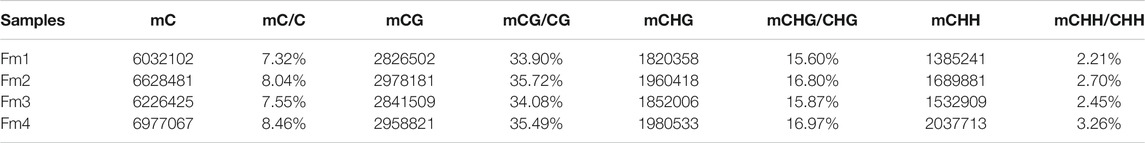
TABLE 1. Proportion of mC in the three different contexts (CG, CHG, CHH) of four different flowering stages of Prunus mume ‘Fenhong Zhusha’ whole genome.
The average values of ML in the mCG, mCHG, and mCHH contexts were 38.13% (36.29–39.32%), 17.25% (16.47–17.96%), and 1.85% (1.69–2.15%), respectively, while the average global ML in flower buds of P. mume was 8.35% during the flowering process (Table 2). Among different gene functional regions, all CX contexts had the highest ML in the TE region (Figure 3A). Among the gene body and 2 kb regions up- and downstream of it, the CG context had higher ML in the gene body, while the CHG and CHH contexts had lower ML in the gene body than in other regions (Figure 3B).
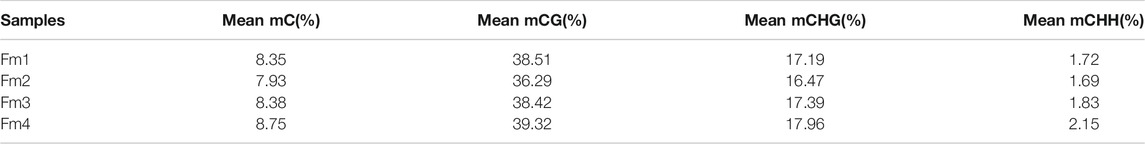
TABLE 2. Global mean ML of mC in the three different contexts (CG, CHG, CHH) of different flowering stages of Prunus mume ‘Fenhong Zhusha’.
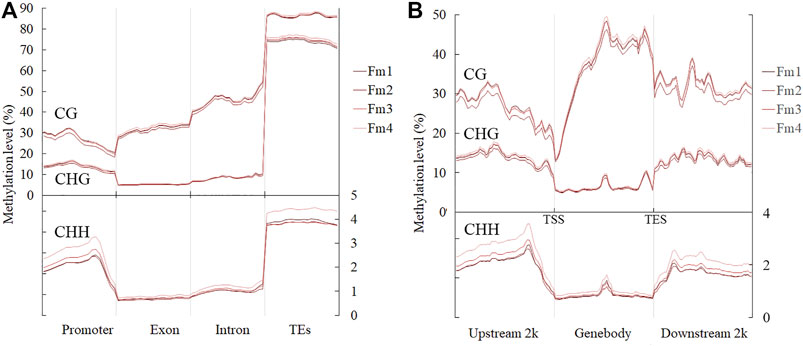
FIGURE 3. ML in (A) different gene functional regions and (B) gene body and regions 2 kb up- and downstream of it, during flowering. TSS and TES stand for transcription start and end sites, respectively.
In the search for DMR-related genes, we detected 5492 (3461 hyper- and hypomethylated), 4744 (2796 hyper- and 1948 hypomethylated), and 7677 (6320 hyper- and 1357 hypomethylated) DMRs within three methylome comparisons between F2v1, F3v2, and F4v3, respectively. During the flowering process, the number of hyper-DMRs was greater than that of hypo-DMRs (Figure 4A). As shown in this Figure 4B, the number of DMRs was mainly from the CHH context, which occurs in promoter regions, and the number of hyper-DMRs was larger than that of hypo-DMRs. The result is consistent with the gradual increase of ML in promoter regions in the CHH context during flowering (Figures 3A,B). In contrast, the number of hypo-DMRs in the final stage of flowering was mainly contributed to by hypomethylated CG-DMRs in promoter regions (Figure 4B).
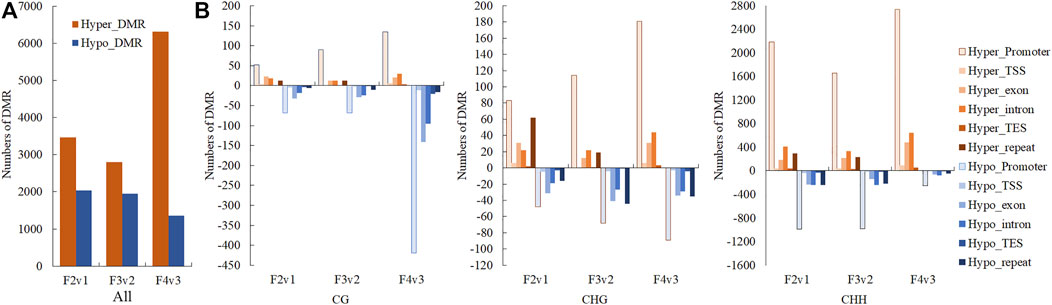
FIGURE 4. Numbers of hyper- and hypo-DMRs during flowering, shown in (A) all mC site and (B) three different contexts (CG, CHG, CHH) and different gene region (promoter, TSS, exon, intron, TES and repeat). The terms “hyper-” and “hypo-” represent DMRs that were hypermethylated and hypomethylated, respectively.
Next, we detected 4005 (promoter, 3178; gene body, 1006), 3515 (promoter, 2765; gene body, 881), and 5306 (promoter, 4123; gene body, 1533) DMGs, which were associated with DMRs, in comparisons of Fm2v1, Fm3v2 and Fm4v3 (“m” here indicate methylome). In brief, the results showed that the genes related to CHH context DMR within promoter region constituted the largest proportion of DMGs among all methylome comparisons (Figure 5), which is consistent with previous results (Supplementary Table S5). We revealed the functions of those DMGs by KEGG enrichment analysis. Remarkably, some crucial DMGs were enriched in eight KEGG pathways related to floral scent biosynthesis. Overall, 97 (66 promoter, 44 gene body) and 17 DMGs were enriched in phenylpropanoid biosynthesis (ko pmum00940) and its upstream pathway phenylalanine, tyrosine, and tryptophan biosynthesis (ko pmum00400), respectively. Moreover, 20, 11, 14, 4 and 45 DMGs were enriched in the pathways of terpenoid backbone biosynthesis (ko pmum00900), monoterpenoid biosynthesis (ko pmum00902), diterpenoid biosynthesis (ko pmum00904), sesquiterpenoid and triterpenoid biosynthesis (ko pmum00909), and phenylalanine metabolism (ko pmum00360). Furthermore, 371 DMGs were enriched in the biosynthesis of secondary metabolites (ko pmum01110) (Supplementary Table S5).
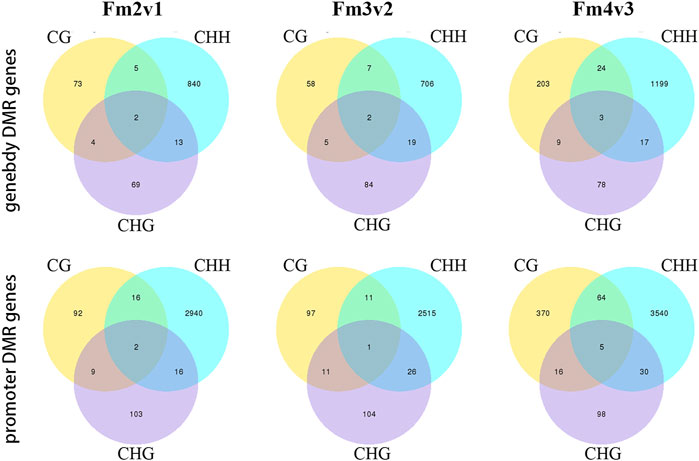
FIGURE 5. Numbers of genes related to DMRs within promoters and gene bodies, shown for three contexts.
Using the same samples of mei buds, we also performed RNA-seq to investigate the DEGs involved in floral scent biosynthesis during the natural flowering process. For each sample, at least 50 million (average 62 million, 9.32G over 30× coverage) clean reads were produced (Supplementary Table S6). Over 91% of the clean reads were mapped to the reference genome using Bismark (Supplementary Table S6) (Krueger and Andrews, 2011).
At the chromosome level, low ML and high gene expression levels were distributed in the same region on the chromosome. Higher gene density was also observed in this region (Figure 6A). All genes were divided into four groups according to their expression levels from low to high, and their average ML was determined in different gene regions (gene body and 2 kb up- and downstream of it, TSS, and TES). Among all methylated genes (MGs) at each flowering stage, a higher expression level of the gene was associated with a lower methylation level of the TSS region. Furthermore, higher average ML was detected in the promoter regions of the CG and CHG contexts of unexpressed genes (Figure 6B).

FIGURE 6. Correlation between ML and expression level of genes, shown in three different CX contexts. (A) The overall relationship between ML and expression level on chromosomes. (B) The MLs of gene body and regions 2 kb up- and downstream of it in different CX contexts of genes under different expression levels. FPKM_25% and FPKM_75% refer to values at the boundary of the 25th and 75th percentiles of expression levels, respectively. Each region of each gene was divided equally into 50 bins, and the MLs of all CX sites in each bin were averaged as the ML of the bin. (C) The expression level distribution frequencies of six groups of genes with different MLs from low to high, shown separately for gene body and promoter regions. DNA methylation levels were classified into five groups: group 1 (red; 0 < methylation level < level_20%); group 2 (yellow-green; level_20% ≤ methylation level < level_40%); group 3 (green; level_40% ≤ methylation level < level_60%); group 4 (turquoise; level_60% ≤ methylation level < level_80%); and group 5 (blue; methylation level ≥ level_80%). Level_20%, _40%, _60%, and _80% represent values at the boundaries of the 20th, 40th, 60th, and 80th percentiles of methylation levels. (D) Venn plots showing the numbers of DEGs that were up- or downregulated in each comparison. The terms “hyper-” and “hypo-” represent DMRs within DEGs that were hypermethylated and hypomethylated, respectively. The results are displayed according to DMRs within promoter or gene body regions.
The MGs were divided into five groups (groups 1–5) according to their ML from low to high, and the gene distribution frequencies (y-axis) at different expression levels (x-axis) were represented graphically for each group. In the promoter regions of the CG- and CHG-context MGs, a higher ML was associated with more genes being distributed in the low-expression region log2(FPKM+1) <1, and fewer genes being distributed in 2 < log2(FPKM+1) < 8. The CHH-context MGs also had similar distributions in frequency, although genes with the highest ML (group 5) appeared to have more genes in region 2 < log2(FPKM+1) < 8 than group 4 (Figure 6C). The results indicated that ML in the promoter regions was negatively correlated with expression level. In the gene body regions, the MG group with the highest ML was dominated by a low expression level in all CX contexts. From groups 1 to 3, a higher ML was associated with a higher frequency of genes in the expression level of region 2 < log2(FPKM+1) < 8. The results indicated that ML in gene body regions is positively correlated with expression level (Figure 6C).
The comparison of F2v1 in Figure 6D is here taken as an example. In the gene body region, there were 32 upregulated and 43 downregulated genes in hyper-DMG, and 28 upregulated and 41 downregulated genes in hypo-DMG. Meanwhile, in the promoter region, there were 253 upregulated and 268 downregulated genes in hyper-DMG, and 130 upregulated and 107 downregulated genes in hypo-DMG. Similar results were also found in F3v2 and F4v3. Some of the DMGs have both hyper- and hypomethylated regions (Figure 6D). These results showed that, when limiting the analysis to certain genes, the expression levels of many genes do not always conform to having a negative correlation with the ML in the promoter and a positive correlation with the ML in the gene body.
We detected 7706 (upregulated 3848; downregulated, 3858), 5016 (upregulated 2259; downregulated, 2457), and 12,451 (upregulated 5502; downregulated, 6949) DEGs in compairsion Fr2v1, Fr3v2 and Fr4v3, respectively (“r” here indicate transcriptome) (Figure 6D). The results of KEGG enrichment analysis showed that a total of 214 DEGs were involved in seven floral scent biosynthesis pathways. As the key pathway for biosynthesis of the characteristic aroma of P. mume, phenylpropanoid biosynthesis (ko pmum00940) was enriched the most for DEGs (96 DEGs). Meanwhile, its upstream pathway, phenylalanine, tyrosine, and tryptophan biosynthesis (ko pmum00400), featured 32 DEGs. Moreover, 21, 39, 13, 14, and 10 DEGs were involved in phenylalanine metabolism (ko pmum00360), terpenoid backbone biosynthesis (ko pmum00900), monoterpenoid biosynthesis (ko pmum00902), diterpenoid biosynthesis (ko pmum00904), and sesquiterpenoid and triterpenoid biosynthesis (ko pmum00909), respectively (Supplementary Table S7).
Over 90% of the volatile components of P. mume fragrance were shown to be synthesized by the phenylpropane biosynthetic pathway. We also integrated some genes that previously characterized in the biosynthesis of other characteristic fragrance components in P. mume into the phenylpropane biosynthetic pathway, and then marked the processes involving DMGs with red asterisks (Figure 7). The results clearly showed that DMGs were widely involved in almost all steps in the phenylpropanoid biosynthesis process, including not only the dominant and key components, but also the low (FPKM value < lower quartile) or none (FPKM value < 1) part in P. mume fragrance.
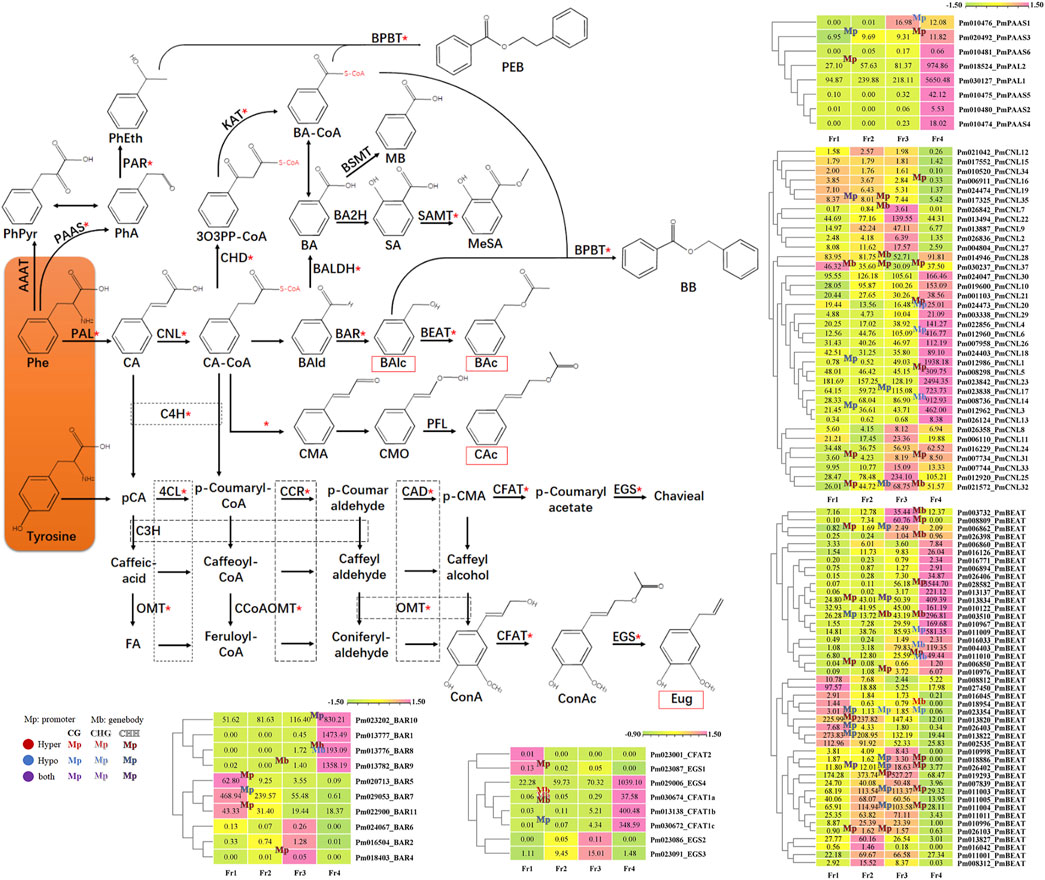
FIGURE 7. Methylation modification and the heat maps of DEGs encoding key enzymes in the pathway of floral scent biosynthesis in P. mume. The key floral scent biosynthetic pathway in P. mume is shown. The biological compounds on an orange background are the starting substrates of the phenylpropane pathway, synthesized by the upstream pathway. The red rectangles indicate the metabolites that were detected in the headspace. Red asterisks indicate that genes encoding enzymes have undergone differential methylation during the process. The dashed boxes indicate that the processes involved share the same enzyme. In the heat maps, pink and light-green represent high and low transcript expression, respectively. The methylation statement were also marked in the heat maps with “Mp” and “Mb” to indicate the differential methylation occurred in promoter and genebody region, respectively; Text color with red, blue and purple represent ML up (hypermethylated), down (hypomethylated) and both happened respectively; Different fonts refer to different CX context, as shown in the figure.
We further summarized 145 genes that were related to the key enzyme in the phenylpropane biosynthetic pathway, as identified in our published results of P. mume floral fragrance research (Bao et al., 2019; Bao et al., 2020; Zhang et al., 2019b; Zhao et al., 2017), including 2 PmPALs, 6 PmPAASs, 37 PmCNLs, 11 PmBARs, 81 PmBEATs, 4 PmCFATs, and 4 PmEGSs. According to the heatmap results, among 47 candidate genes that exhibited an expression pattern matching the pattern of floral scent emission, 22 were differentially methylated during the flowering process (Figure 7, Supplementary Table S10). Some of these genes were proven to encode key enzymes involved in the characteristic fragrance of P. mume in previous studies, such as PmCFAT1a/1c (Zhang et al., 2019b), PmBEAT36/37 (Bao et al., 2019), and PmBAR8/9/10 (Bao et al., 2020); or to have the same expression pattern as in other P. mume cultivars, such as PmPAL2, PmPAAS3, and PmCNL1/3/5/6/14/17/20 (Zhao et al., 2017; Bao et al., 2020) (Supplementary Table S8).
Among them, PmCFAT1a, which have been proved to produced eugenol through an enzyme assay (Zhang et al., 2019b), had four hyper-DMR in its genebody region during F1 to F2, involving all three CX contextsthe with ML changes from: 26.19–44.65% (CG), 20.05–39.34% (CHG), 2.43–5.64% and 1.06–3.45% (CHG). The ML of all hyper-DMR exceeded the average ML of the sample (Table 2; Figure 7). Pm011009 and Pm011010 (PmBEAT36 and PmBEAT37) have been reported to significantly positive affect the synthesis of benzyl acetate in the petal cells of P. mume when overexpressed (Bao et al., 2019). Pm011009 had CG context hypo-DMR (90.49–81.02%) in the promoter region while the gene expressing were significantly increased in F3 to F4. Pm011010, which have been reported to inhibited the expression of Pm011009 in protoplasts, had CHH context hyper-DMR (22.41–39.87%) in its promoter region and CG context hypo-DMR (41.82–17.63%) in its genebody region from F3 to F4, although the expressing were also increased (Figure 7). We randomly selected 6 genes to do qRT-PCR to verify the transcriptome results. The qRT-PCR results showed a similar trend to the FPKM value (Supplementary Figure S4), proving that the transcriptome results are reliable.
The modification of DNA sequences by methylation is one of the heritable types of gene expression that does not change the genetic background. Research on DNA methylation thus has advantages in explaining the different phenotypes caused by differential gene expression in the same or similar genetic backgrounds. Studies have also shown variations in floral scent among different P. mume cultivars, despite strong similarity in their genetic background. As the only species of Prunus that produces a floral fragrance (Zhang et al., 2019b), research on the molecular mechanism regulating floral scent biosynthesis in P. mume is important.
Our results showed that DMGs were widely involved in eight recognized floral scent biosynthesis pathways during the natural flowering process of P. mume. Among these, the phenylpropane biosynthesis pathway was found to have the most enriched DMGs, which is consistent with the fact that over 90% of the floral volatiles emitted by the P. mume cv. ‘FZ’ are synthesized by the phenylpropane biosynthesis pathway (Zhang et al., 2019a). These DMGs participate in most of the floral biosynthetic processes in the phenylpropane biosynthetic pathway. All of these results indicated that methylation plays a role in the process of floral scent biosynthesis.
We selected 47 candidate genes that exhibit an expression pattern matching the emission of floral scent, 22 of which were differentially methylated during flowering. Some of these genes have been proven to encode key enzymes involved in the characteristic floral scent of P. mume in previous studies, such as PmCFAT1a/1c (Zhang et al., 2019b) and PmBEAT36/37 (Bao et al., 2019), or have the same expression pattern in other P. mume cultivars, such as PmPAL2, PmPAAS3, PmBAR8/9/10, and PmCNL1/3/5/6/14/17/20 (Zhao et al., 2017; Bao et al., 2020). These lines of evidence indicate that the ML of DNA is involved in regulating the expression of genes encoding key enzymes in floral scent biosynthesis, and ultimately forms the unique floral scent of P. mume.
Notably, the DEGs involved in floral scent biosynthesis showed diverse patterns of methylation. Although our results were consistent with previous studies, hypermethylated regions were usually associated with segments showing low gene expression on chromosomes; and the gene expression level was found to be negatively and positively correlated with the ML of the promoter and gene body regions, respectively, among all methylated genes. However, when limiting the analysis to certain genes, the relationship between ML and gene expression did not always follow this rule. Similar result can be found in the result of previous research (Ma et al., 2018; Jiang et al., 2020). A more likely explanation was the complexity of the process of floral scent biosynthesis, so that methylation is not the only regulation factor. For example, the promoter of PmBEAT36/37 has elements that respond to temperature and light (Bao et al., 2019). In the future, more research should be carried out within DMRs of key genes involved in floral scent biosynthesis that exhibit expression patterns matching the patterns of volatile emission and that differ in their expression patterns among P. mume cultivars with different scents.
In conclusion, our results revealed that DNA methylation is widely involved in the process of floral scent biosynthesis and may play critical roles in regulating such biosynthesis in P. mume. This was revealed by a comprehensive analysis of the RNA-seq and WGBS data of flower buds at four different flowering stages of the P. mume cv. ‘FZ’. The results provide a new epigenetic perspective that deepens our understanding of the mechanism behind the biosynthesis of floral scent.
The original contributions presented in the study are publicly available in NCBI under accession numbers PRJNA765446 and PRJNA766216.
XY, KM, MZ, and QZ conceived and designed the experiments. XY, and MZ performed the experiments. XY, KM, MZ, and JW analyzed the data. XY and KM wrote the manuscript. All authors have read and approved the published version of the manuscript.
This research was funded by The National Key R&D Program of China (2020YFD1000500) and Special Fund for Beijing Common Construction Project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.779557/full#supplementary-material
Supplementary Table S10 | MR related to the 22 candidate genes.
Bai, S., Tuan, P. A., Saito, T., Honda, C., Hatsuyama, Y., Ito, A., et al. (2016). Epigenetic Regulation of MdMYB1 Is Associated with Paper Bagging-Induced Red Pigmentation of Apples. Planta 244 (3), 573–586. doi:10.1007/s00425-016-2524-4
Bao, F., Ding, A., Zhang, T., Luo, L., Wang, J., Cheng, T., et al. (2019). Expansion of PmBEAT Genes in the Prunus Mume Genome Induces Characteristic floral Scent Production. Hortic. Res. 6, 24. doi:10.1038/s41438-018-0104-4
Bao, F., Zhang, T., Ding, A., Ding, A., Yang, W., Wang, J., et al. (2020). Metabolic, Enzymatic Activity, and Transcriptomic Analysis Reveals the Mechanism Underlying the Lack of Characteristic floral Scent in Apricot Mei Varieties. Front. Plant Sci. 11, 574982. doi:10.3389/fpls.2020.574982
Boatright, J., Negre, F., Chen, X., Kish, C. M., Wood, B., Peel, G., et al. (2004). Understanding In Vivo Benzenoid Metabolism in Petunia Petal Tissue. Plant Physiol. 135 (4), 1993–2011. doi:10.1104/pp.104.045468
Chan, S. W.-L., Henderson, I. R., and Jacobsen, S. E. (2005). Gardening the Genome: DNA Methylation in Arabidopsis thaliana. Nat. Rev. Genet. 6 (5), 351–360. doi:10.1038/nrg1601
Cubas, P., Vincent, C., and Coen, E. (1999). An Epigenetic Mutation Responsible for Natural Variation in floral Symmetry. Nature 401 (6749), 157–161. doi:10.1038/43657
Dhandapani, S., Jin, J., Sridhar, V., Sarojam, R., Chua, N.-H., and Jang, I.-C. (2017). Integrated Metabolome and Transcriptome Analysis of Magnolia Champaca Identifies Biosynthetic Pathways for floral Volatile Organic Compounds. BMC Genomics 18 (1), 463. doi:10.1186/s12864-017-3846-8
Dudareva, N., Klempien, A., Muhlemann, J. K., and Kaplan, I. (2013). Biosynthesis, Function and Metabolic Engineering of Plant Volatile Organic Compounds. New Phytol. 198 (1), 16–32. doi:10.1111/nph.12145
Feng, H., Conneely, K. N., and Wu, H. (2014). A Bayesian Hierarchical Model to Detect Differentially Methylated Loci from Single Nucleotide Resolution Sequencing Data. Nucleic Acids Res. 42 (8), e69. doi:10.1093/nar/gku154
Fenske, M. P., Hewett Hazelton, K. D., Hempton, A. K., Shim, J. S., Yamamoto, B. M., Riffell, J. A., et al. (2015). Circadian Clock Gene LATE ELONGATED HYPOCOTYL Directly Regulates the Timing of floral Scent Emission in Petunia. Proc. Natl. Acad. Sci. USA 112 (31), 9775–9780. doi:10.1073/pnas.1422875112
Finnegan, E. J., and Kovac, K. A. (2000). Plant DNA Methyltransferases. Plant Mol. Biol. 43 (2-3), 189–201. doi:10.1023/a:1006427226972
Fraser, C. M., and Chapple, C. (2011). The Phenylpropanoid Pathway in Arabidopsis. The Arabidopsis Book 9, e0152. doi:10.1199/tab.0152
Gifford, C. A., Ziller, M. J., Gu, H., Trapnell, C., Donaghey, J., Tsankov, A., et al. (2013). Transcriptional and Epigenetic Dynamics during Specification of Human Embryonic Stem Cells. Cell 153 (5), 1149–1163. doi:10.1016/j.cell.2013.04.037
Habibi, E., Brinkman, A. B., Arand, J., Kroeze, L. I., Kerstens, H. H. D., Matarese, F., et al. (2013). Whole-genome Bisulfite Sequencing of Two Distinct Interconvertible DNA Methylomes of Mouse Embryonic Stem Cells. Cell Stem Cell 13 (3), 360–369. doi:10.1016/j.stem.2013.06.002
Hao, R.-J., Zhang, Q., Yang, W.-R., Wang, J., Cheng, T.-R., Pan, H.-T., et al. (2014b). Emitted and Endogenous floral Scent Compounds of Prunus Mume and Hybrids. Biochem. Syst. Ecol. 54, 23–30. doi:10.1016/j.bse.2013.12.007
Hao, R., Du, D., Wang, T., Yang, W., Wang, J., and Zhang, Q. (2014a). A Comparative Analysis of Characteristic floral Scent Compounds in Prunus Mume and Related Species. Biosci. Biotechnol. Biochem. 78 (10), 1640–1647. doi:10.1080/09168451.2014.936346
Huang, H., Liu, R., Niu, Q., Tang, K., Zhang, B., Zhang, H., et al. (2019). Global Increase in DNA Methylation during orange Fruit Development and Ripening. Proc. Natl. Acad. Sci. USA 116 (4), 1430–1436. doi:10.1073/pnas.1815441116
Jiang, L., Zhang, M., and Ma, K. (2020). Whole-Genome DNA Methylation Associated with Differentially Expressed Genes Regulated Anthocyanin Biosynthesis within Flower Color Chimera of Ornamental Tree Prunus Mume. Forests 11 (1), 90. doi:10.3390/f11010090
Jin, J., Kim, M. J., Dhandapani, S., Tjhang, J. G., Yin, J.-L., Wong, L., et al. (2015). The floral Transcriptome of Ylang Ylang (Cananga Odorata Var. Fruticosa) Uncovers Biosynthetic Pathways for Volatile Organic Compounds and a Multifunctional and Novel Sesquiterpene Synthase. J. Exp. Bot. 66 (13), 3959–3975. doi:10.1093/jxb/erv196
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., et al. (2008). KEGG for Linking Genomes to Life and the Environment. Nucleic Acids Res. 36, D480–D484. doi:10.1093/nar/gkm882
Karami, A., Niazi, A., Kavoosi, G., Khosh-Khui, M., and Salehi, H. (2015). Temporal Characterization of 2-phenylethanol in Strongly and Weakly Scented Genotypes of Damask Rose. Physiol. Mol. Biol. Plants 21 (1), 43–49. doi:10.1007/s12298-014-0274-y
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 12 (4), 357–360. doi:10.1038/nmeth.3317
Knudsen, J. T., Eriksson, R., Gershenzon, J., and Ståhl, B. (2006). Diversity and Distribution of floral Scent. Bot. Rev. 72 (1), 1–120. doi:10.1663/0006-8101(2006)72[1:DADOFS]2.0.CO;2
Krueger, F., and Andrews, S. R. (2011). Bismark: a Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinformatics 27 (11), 1571–1572. doi:10.1093/bioinformatics/btr167
Krzywinski, M., Schein, J., Birol, İ., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an Information Aesthetic for Comparative Genomics. Genome Res. 19 (9), 1639–1645. doi:10.1101/gr.092759.109
Langmead, B., and Salzberg, S. L. (2012). Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 9 (4), 357–359. doi:10.1038/nmeth.1923
Law, J. A., and Jacobsen, S. E. (2010). Establishing, Maintaining and Modifying DNA Methylation Patterns in Plants and Animals. Nat. Rev. Genet. 11 (3), 204–220. doi:10.1038/nrg2719
Li, W.-F., Ning, G.-X., Mao, J., Guo, Z.-G., Zhou, Q., and Chen, B.-H. (2019). Whole-genome DNA Methylation Patterns and Complex Associations with Gene Expression Associated with Anthocyanin Biosynthesis in Apple Fruit Skin. Planta 250 (6), 1833–1847. doi:10.1007/s00425-019-03266-4
Lister, R., Pelizzola, M., Dowen, R. H., Hawkins, R. D., Hon, G., Tonti-Filippini, J., et al. (2009). Human DNA Methylomes at Base Resolution Show Widespread Epigenomic Differences. Nature 462 (7271), 315–322. doi:10.1038/nature08514
Ma, K.-F., Zhang, Q.-X., Cheng, T.-R., Yan, X.-L., Pan, H.-T., and Wang, J. (2018). Substantial Epigenetic Variation Causing Flower Color Chimerism in the Ornamental Tree Prunus Mume Revealed by Single Base Resolution Methylome Detection and Transcriptome Sequencing. Ijms 19 (8), 2315. doi:10.3390/ijms19082315
Mao, X., Cai, T., Olyarchuk, J. G., and Wei, L. (2005). Automated Genome Annotation and Pathway Identification Using the KEGG Orthology (KO) as a Controlled Vocabulary. Bioinformatics 21 (19), 3787–3793. doi:10.1093/bioinformatics/bti430
Moerkercke, A. V., Schauvinhold, I., Pichersky, E., Haring, M. A., and Schuurink, R. C. (2009). A Plant Thiolase Involved in Benzoic Acid Biosynthesis and Volatile Benzenoid Production. Plant J. 60 (2), 292–302. doi:10.1111/j.1365-313X.2009.03953.x
Muhlemann, J. K., Klempien, A., and Dudareva, N. (2014). Floral Volatiles: from Biosynthesis to Function. Plant Cell Environ. 37 (8), 1936–1949. doi:10.1111/pce.12314
Park, Y., and Wu, H. (2016). Differential Methylation Analysis for BS-Seq Data under General Experimental Design. Bioinformatics 32 (10), 1446–1453. doi:10.1093/bioinformatics/btw026
Schiestl, F. P. (2010). The Evolution of floral Scent and Insect Chemical Communication. Ecol. Lett. 13 (5), 643–656. doi:10.1111/j.1461-0248.2010.01451.x
Song, Q.-X., Lu, X., Li, Q.-T., Chen, H., Hu, X.-Y., Ma, B., et al. (2013). Genome-wide Analysis of DNA Methylation in Soybean. Mol. Plant 6 (6), 1961–1974. doi:10.1093/mp/sst123
Sun, L., Yang, W., Zhang, Q., Cheng, T., Pan, H., Xu, Z., et al. (2013). Genome-wide characterization and linkage mapping of simple sequence repeats in mei (Prunus mume Sieb. et Zucc). PLoS One 8 (3), e59562. doi:10.1371/journal.pone.0059562
Suzuki, M. M., and Bird, A. (2008). DNA Methylation Landscapes: Provocative Insights from Epigenomics. Nat. Rev. Genet. 9 (6), 465–476. doi:10.1038/nrg2341
Tzin, V., and Galili, G. (2010). New Insights into the Shikimate and Aromatic Amino Acids Biosynthesis Pathways in Plants. Mol. Plant 3 (6), 956–972. doi:10.1093/mp/ssq048
Vaughn, M. W., Tanurdžić, M., Lippman, Z., Jiang, H., Carrasquillo, R., Rabinowicz, P. D., et al. (2007). Epigenetic Natural Variation in Arabidopsis thaliana. Plos Biol. 5 (7), e174. doi:10.1371/journal.pbio.0050174
Wang, L., Zhang, J., Duan, J., Gao, X., Zhu, W., Lu, X., et al. (2014). Programming and Inheritance of Parental DNA Methylomes in Mammals. Cell 157 (4), 979–991. doi:10.1016/j.cell.2014.04.017
Wang, M., Yuan, D., Tu, L., Gao, W., He, Y., Hu, H., et al. (2015). Long Noncoding RNA S and Their Proposed Functions in Fibre Development of Cotton (Gossypium spp). New Phytol. 207 (4), 1181–1197. doi:10.1111/nph.13429
Wang, Z., Meng, D., Wang, A., Li, T., Jiang, S., Cong, P., et al. (2013). The Methylation of the PcMYB10 Promoter Is Associated with Green-Skinned Sport in Max Red Bartlett Pear. Plant Physiol. 162 (2), 885–896. doi:10.1104/pp.113.214700
Widhalm, J. R., and Dudareva, N. (2015). A Familiar Ring to it: Biosynthesis of Plant Benzoic Acids. Mol. Plant 8 (1), 83–97. doi:10.1016/j.molp.2014.12.001
Wu, H., Xu, T., Feng, H., Chen, L., Li, B., Yao, B., et al. (2015). Detection of Differentially Methylated Regions from Whole-Genome Bisulfite Sequencing Data without Replicates. Nucleic Acids Res. 43 (21), gkv715. doi:10.1093/nar/gkv715
Xing, L., Li, Y., Qi, S., Zhang, C., Ma, W., Zuo, X., et al. (2019). Comparative RNA-Sequencing and DNA Methylation Analyses of Apple (Malus Domestica Borkh.) Buds with Diverse Flowering Capabilities Reveal Novel Insights into the Regulatory Mechanisms of Flower Bud Formation. Plant Cell Physiol. 60 (8), 1702–1721. doi:10.1093/pcp/pcz080
Xu, J., Zhou, S., Gong, X., Song, Y., van Nocker, S., Ma, F., et al. (2018). Single-base Methylome Analysis Reveals Dynamic Epigenomic Differences Associated with Water Deficit in Apple. Plant Biotechnol. J. 16 (2), 672–687. doi:10.1111/pbi.12820
Xu, Z., Zhang, Q., Sun, L., Du, D., Cheng, T., Pan, H., et al. (2014). Genome-wide Identification, Characterisation and Expression Analysis of the MADS-Box Gene Family in Prunus Mume. Mol. Genet. Genomics 289 (5), 903–920. doi:10.1007/s00438-014-0863-z
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol. 11 (2), R14. doi:10.1186/gb-2010-11-2-r14
Yuan, X., Ma, K. F., Zhang, M. H., Zhang, Z. Y., Wang, J., and Zhang, Q. X. (2020). How Does an Early Flowering Cultivar of Prunus Mume Arrange its Bud Development in Late winter? Acta Hortic. 1291, 15–20. doi:10.17660/ActaHortic.2020.1291.3
Yue, Y., Yu, R., and Fan, Y. (2015). Transcriptome Profiling Provides New Insights into the Formation of floral Scent in Hedychium Coronarium. BMC Genomics 16 (1), 470. doi:10.1186/s12864-015-1653-7
Zhang, Q., Chen, W., Sun, L., Zhao, F., Huang, B., Yang, W., et al. (2012). The Genome of Prunus Mume. Nat. Commun. 3 (1), 1318. doi:10.1038/ncomms2290
Zhang, Q., Zhang, H., Sun, L., Fan, G., Ye, M., Jiang, L., et al. (2018). The Genetic Architecture of floral Traits in the Woody Plant Prunus Mume. Nat. Commun. 9 (1), 1702. doi:10.1038/s41467-018-04093-z
Zhang, T., Bao, F., Yang, Y., Hu, L., Ding, A., Ding, A., et al. (2019a). A Comparative Analysis of Floral Scent Compounds in Intraspecific Cultivars of Prunus Mume with Different Corolla Colours. Molecules 25 (1), 145. doi:10.3390/molecules25010145
Zhang, T., Huo, T., Ding, A., Hao, R., Wang, J., Cheng, T., et al. (2019b). Genome-wide Identification, Characterization, Expression and Enzyme Activity Analysis of Coniferyl Alcohol Acetyltransferase Genes Involved in Eugenol Biosynthesis in Prunus Mume. PLoS One 14 (10), e0223974. doi:10.1371/journal.pone.0223974
Zhao, K., Yang, W., Zhou, Y., Zhang, J., Li, Y., Ahmad, S., et al. (2017). Comparative Transcriptome Reveals Benzenoid Biosynthesis Regulation as Inducer of Floral Scent in the Woody Plant Prunus Mume. Front. Plant Sci. 8, 319. doi:10.3389/fpls.2017.00319
Zheng, J., Hu, Z., Guan, X., Dou, D., Bai, G., Wang, Y., et al. (2015). Transcriptome Analysis of Syringa Oblata Lindl. Inflorescence Identifies Genes Associated with Pigment Biosynthesis and Scent Metabolism. PLoS One 10 (11), e0142542. doi:10.1371/journal.pone.0142542
Zhong, S., Fei, Z., Chen, Y.-R., Zheng, Y., Huang, M., Vrebalov, J., et al. (2013). Single-base Resolution Methylomes of Tomato Fruit Development Reveal Epigenome Modifications Associated with Ripening. Nat. Biotechnol. 31 (2), 154–159. doi:10.1038/nbt.2462
Zhu, J.-K. (2009). Active DNA Demethylation Mediated by DNA Glycosylases. Annu. Rev. Genet. 43, 143–166. doi:10.1146/annurev-genet-102108-134205
Zhuo, X., Zheng, T., Li, S., Zhang, Z., Zhang, M., Zhang, Y., et al. (2021). Identification of the PmWEEP Locus Controlling Weeping Traits in Prunus Mume through an Integrated Genome-wide Association Study and Quantitative Trait Locus Mapping. Hortic. Res. 8 (1), 131. doi:10.1038/s41438-021-00573-4
3O3PP-CoA 3-oxo-3-phenylpropionyl-CoA
4CL 4-coumarate CoA-ligase
BA benzoic acid
BAc Benzyl acetate
BA-CoA benzoyl-CoA
BAlc benzylalcohol
BAld benzaldehyde
BALDH benzaldehyde dehydrogenase
BB benzylbenzoate
BEAT acetyl-CoA:benzylalcohol acetyltransferase
BPBT benzoyl-CoA:benzylalcohol/2-phenylethanol benzoyltransferase
BSMT benzoic acid/salicylic acid carboxyl methyltransferase
C4H cinnamic acid 4-hydroxylase
CA cinnamic acid
CAc Cinnamyl acetate
CA-CoA cinnamoyl-CoA
CAld Cinnamyl dehyde
CCoAOMT caffeoyl-CoA O-methyltransferase
CFAT coniferylalcohol acetyltransferase
CHD cinnamoyl-CoA hydratase/dehydrogenase
CMA cinnamaldehyde
CMO cinnamyl alcohol
CNL cinnamoyl-CoA ligase
ConA coniferyl alcohol
EGS eugenol synthase
Eug eugenol
FA ferulic acid
KAT 3-ketoacyl-CoA thiolase
MB methylbenzoate
MeSA Methyl Salicylate
PAAS phenylacetaldehyde synthase
PAL phenylalanine ammonia-lyase
PAR phenylacetaldehyde reductase
pCA p-Coumaric acid
p-CMA p-Coumaryl alcohol
PhA phenylacetaldehyde
Phe L-phenylalanine
PhEth 2-phenylethanol
PhPyr phenylpyruvic acid
SA Salicylate
Tyr Tyrosine.
Keywords: floral scent, phenylpropane biosynthesis pathway, Prunus mume, cytosine methylation, transcriptome
Citation: Yuan X, Ma K, Zhang M, Wang J and Zhang Q (2021) Integration of Transcriptome and Methylome Analyses Provides Insight Into the Pathway of Floral Scent Biosynthesis in Prunus mume. Front. Genet. 12:779557. doi: 10.3389/fgene.2021.779557
Received: 19 September 2021; Accepted: 19 November 2021;
Published: 15 December 2021.
Edited by:
Shang-Qian Xie, Hainan University, ChinaReviewed by:
Xiao Han, Zhejiang Agriculture and Forestry University, ChinaCopyright © 2021 Yuan, Ma, Zhang, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaifeng Ma, bWFrYWlmZW5nQGJqZnUuZWR1LmNu; Qixiang Zhang, enF4YmpmdUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.