 Jingwen Li1†
Jingwen Li1† Jun Yang
Jun Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 03 January 2022
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.777926
Hydrocephalus is a neurological condition due to the aberrant circulation and/or obstruction of cerebrospinal fluid (CSF) flow with consequent enlargement of cerebral ventricular cavities. However, it is noticed that a lot of patients may still go through symptomatic progression despite standard shunting procedures, suggesting that hydrocephalus is far more complicated than a simple CSF circulative/obstructive disorder. Growing evidence indicates that genetic factors play a fundamental role in the pathogenesis of some hydrocephalus. Although the genetic research of hydrocephalus in humans is limited, many genetic loci of hydrocephalus have been defined in animal models. In general, the molecular abnormalities involved in the pathogenesis of hydrocephalus include brain development and ependymal cell dysfunction, apoptosis, inflammation, free radical generation, blood flow, and cerebral metabolism. Moreover, recent studies have indicated that the molecular abnormalities relevant to aberrant cerebral glymphatic drainage turn into an attractive subject in the CSF circulation disorder. Furthermore, the prevalent risk factors could facilitate the development of hydrocephalus. In this review, we elicited some possible fundamental molecular mechanisms and facilitating risk factors involved in the pathogenesis of hydrocephalus, and aimed to widen the diagnosis and therapeutic strategies for hydrocephalus management. Such knowledge could be used to improve patient care in different ways, such as early precise diagnosis and effective therapeutic regimens.
Hydrocephalus is defined as active distension of brain’s ventricular system, resulting from inadequate passage of cerebrospinal fluid from its point of production within the cerebral ventricles to its point of absorption into the systemic circulation (Rekate, 2008; Kousi and Katsanis, 2016). It is characterized by ventriculomegaly due to an imbalance between CSF production and its absorption. However, its precise causes remain largely unknown. Although the causes of obstructive hydrocephalus are not difficult to explore, the etiological factors of congenital and/or communicating hydrocephalus remain unclear. Moreover, up to 78% of hydrocephalus patients treated with functional shunting continue to suffer from cognitive function disorders, such as intellectual disability, memory loss, learning disabilities, and spasticity, suggesting that the etiology of hydrocephalus may be related to multiple factors (Villani et al., 1995; Silverberg et al., 2002; Zhang et al., 2020; Kundishora et al., 2021; Thavarajasingam et al., 2021). Accumulating evidence suggests that many molecular changes are involved in the pathogenesis of hydrocephalus, and about 40% of hydrocephalus may have a possible genetic etiology (Haverkamp et al., 1999; Zhang et al., 2006; Kundishora et al., 2021). Based on the genetic predisposition, acquired risk factors could expedite the progression of ventricular enlargement. Therefore, a clear understanding of the fundamental molecular and acquired risk factors of hydrocephalus is of critical importance for the study of its mechanism and adequate preventive measures. More importantly, a precise etiological diagnosis could facilitate optimal options for specific treatments of hydrocephalus; for example, CSF shunting vs. endoscopy vs. pharmacologic therapies (Figure 1; Table 1) (Koleva and De Jesus, 2021). Moreover, regarding a subset of hydrocephalus caused by impaired neurogenesis rather than active CSF accumulation, it is worth considering whether surgical CSF shunting is the therapeutic option for all hydrocephalus patients. In this review, we focused on the studies of the genetic and facilitating risk factors for hydrocephalus development, in animal as well as human studies, and examined the contributions of these molecular and risk factors in the pathogenesis and progression of hydrocephalus.
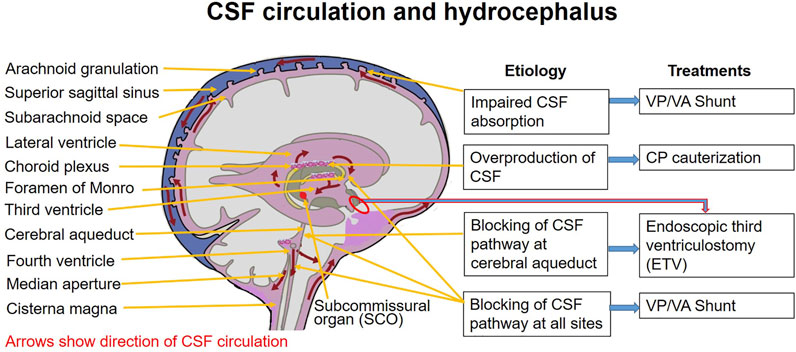
FIGURE 1. Hydrocephalus and therapeutic options. CP: choroid plexus; CSF: cerebrospinal fluid; EVT: endoscopic third ventriculostomy; VA: ventriculoatrial; VP: ventriculoperitoneal.

TABLE 1. Etiology and management options for hydrocephalus.
The genetic and histopathological similarities to human hydrocephalus made the animal model as a valuable tool to explore the pathogenesis of hydrocephalus. Most animal congenital hydrocephalus has been documented as genetic diseases, and many hydrocephalus-related loci have been identified (Table 2).
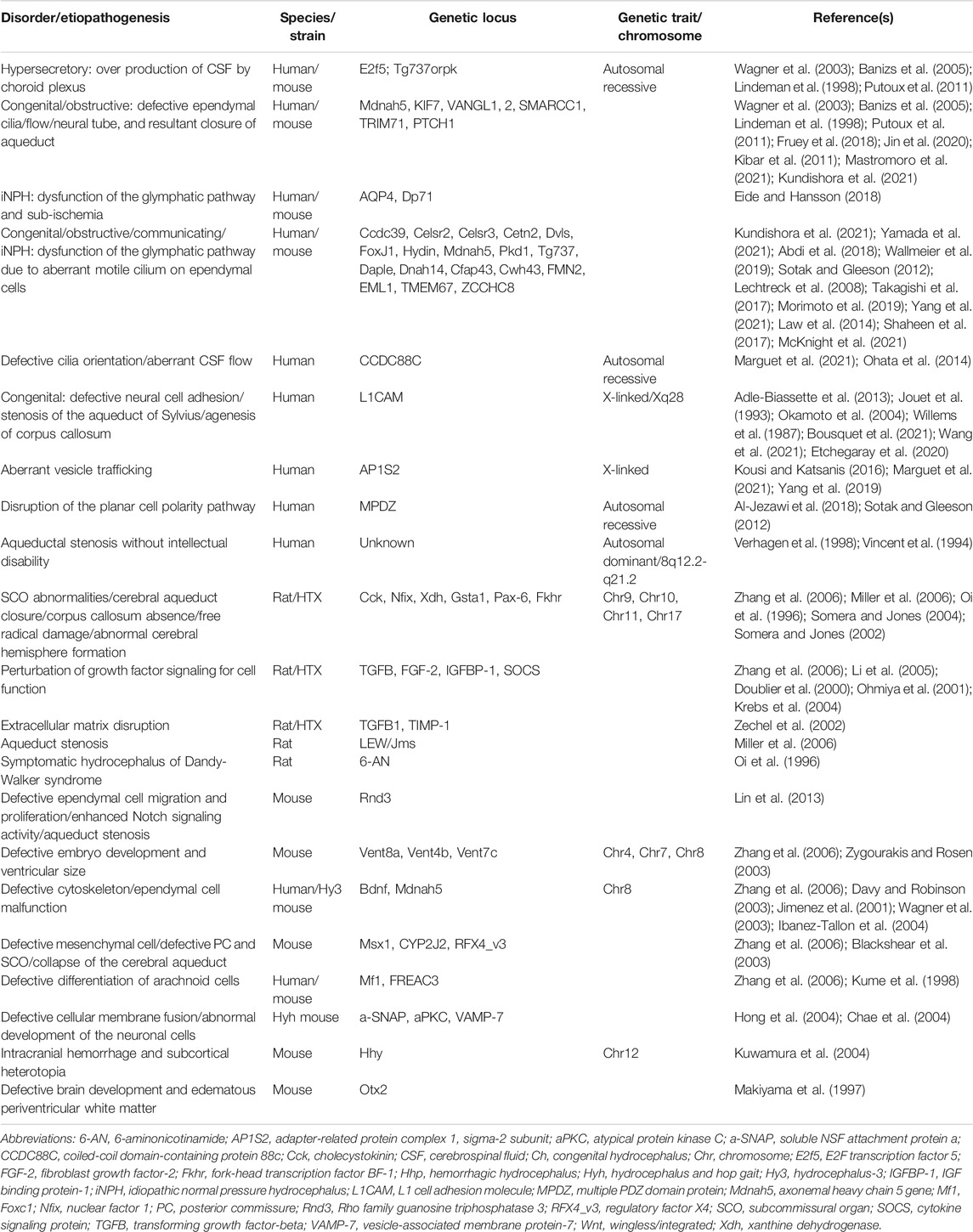
TABLE 2. Summary of genetic mechanism in the pathogenesis of hydrocephalus.
Three major congenital hydrocephalus models are Texas strain (HTX), LEW/Jms, and 6-aminonicotinamide (6-AN)-related hydrocephalus in rats. HTX rats proceed to cerebral aqueduct closure and consequently develop hydrocephalus. LEW/Jms rats demonstrate aqueduct stenosis and hydrocephalus in early prenatal life before pulmonary maturation. 6-AN–related hydrocephalus is similar to Dandy-Walker syndrome (Oi et al., 1996; Miller et al., 2006; Zhang et al., 2006).
The HTX rat is a well-studied congenital hydrocephalus model with ventricular enlargement in late gestation resulting from the cerebral aqueduct and subcommissural organ (SCO) abnormalities. SCO is a key structure for cerebral aqueduct patency and CSF flow. The reduced SCO glycoprotein level had been demonstrated to correlate to both aqueduct stenosis and enlarged lateral ventricle size in the HTX rats (Somera and Jones, 2002; Somera and Jones, 2004). Consistently, a human study of hydrocephalic fetuses also demonstrated a smaller SCO (Castaneyra-Perdomo et al., 1994). Following the development of cerebral aqueduct stenosis, the clinical feature manifests as early communicating to final obstructive hydrocephalus in fetuses (Oi et al., 1996; Miller et al., 2006).
The aqueduct closure in HTX rats may result from retrograde degeneration of apoptotic cells (Mori et al., 2002) and failure in cell proliferation (Draper et al., 2001; Mashayekhi et al., 2001) in the thalamus. The loci of correspondent genes have been indicated on chromosomes (Chr) 9 (peak markers D9Rat2), 10 (between markers D10Rat136 and D10Rat135), 11 (peak markers D11Arb2 and D11Rat46), and 17 (peak markers D17mit4 and D17Rat154), respectively (Zhang et al., 2006). Further genetic studies in the midbrain region of HTX rats showed that the related genes may include cholecystokinin (Cck), Nfix, Xanthine dehydrogenase (Xdh), Gsta1, Pax-6, and fork-head transcription factor BF-1 (Fkhr). Most of these genes have been investigated in clinical studies. For example, Cck levels decreased in hydrocephalus patients (Galard et al., 1997). Cck is known to be involved in learning and memory processes, and most hydrocephalic patients exhibit learning and memory deficits (Ding et al., 2001). Nfix is a member of the nuclear factor 1 (Nf1) family. Nfix deficiency causes corpus callosum absence and ventricular enlargement (Shu et al., 2003). Xanthine dehydrogenase catalyzes xanthine oxidation. The conversion of xanthine dehydrogenase to xanthine oxidase could lead to the concomitant generation of free radical products (Battelli et al., 1998). Gsta1 is an enzyme for free radical scavenging. The accumulation of free radicals is associated with the pathogenesis of hydrocephalus (Socci et al., 1999). Both Pax-6 and Fkhr have been shown to be associated with hydrocephalus (Kume et al., 1998; Kume et al., 2000). Animals with Pax-6 deficiency failed to develop the SCO, and Fkhr is known to be involved in the cerebral hemisphere formation (Hatini et al., 1999; Huh et al., 1999).
The hydrocephalic HXT rats expressed significantly higher transforming growth factor-beta (Tgfb) levels than their normal siblings, suggesting that the perturbation of growth factor signaling may also be involved in the pathogenesis of hydrocephalus (Li et al., 2005; Zhang et al., 2006). Together with other growth factors, such as fibroblast growth factor-2 (FGF-2) and IGF binding protein-1 (IGFBP-1), they are important cytokines and growth molecules in the brain, and play an essential role in cell proliferation, differentiation, survival, and motility. Hydrocephalus has been observed in animal models that overexpress these growth factors (Johanson et al., 1999; Doublier et al., 2000), possibly due to aberrant neuronal differentiation in the postnatal cerebral cortex. The proposed mechanisms may refer to the skeletal and predominant cranial changes which obstruct CSF flow and reduce CSF absorption into the systemic circulation due to increased venous pressure and overgrowth of aberrant brain compounds (Ohmiya et al., 2001; Li et al., 2005; Khonsari et al., 2012). In contrast, the loss of the suppressor of cytokine signaling (SOCS) protein function will lead to increased expression of cytokines and the development of hydrocephalus due to its inhibitory role (Krebs et al., 2004). Furthermore, extracellular matrix (ECM) disruption may also be involved in the pathogenesis of hydrocephalus. In the Tgfb1 overexpression model, the changes of matrix metalloproteinase-9 (Mmp-9) and its inhibitor Timp-1 were found to be important factors for developing hydrocephalus due to ECM environment alterations (Zechel et al., 2002).
Another congenital hydrocephalus strain, LEW/Jms rats, manifests as a similar strain with the HTX rats. The inheritance of hydrocephalus in these rats is possibly autosomal recessive or semidominant, but none of the loci has been identified (Jones et al., 2003).
Hydrocephalus could result from aqueductal stenosis induced through genetic deletion of Rho family guanosine triphosphatase 3 (Rnd3) and regulating the Notch signaling activity (Lin et al., 2013). Rnd3 regulates cell migration and proliferation, and cell actin cytoskeleton dynamics. Deletion of the Rnd3 gene results in severe obstructive hydrocephalus with enlargements of the lateral and third ventricles, but normal fourth ventricle size, indicating the blocking of the cerebral aqueduct. The molecular mechanism was reported to be Rnd3 deficiency and consequent enhancement of Notch signaling activity, resulting in the overgrowth of aqueduct ependymal cells and obstructive hydrocephalus. Accordingly, this type of hydrocephalus could be resolved by the inhibition of Notch activity.
Quantitative trait locus (QTL) genetic mapping had been performed in mouse models, and three QTL loci were identified on Chr 8, Chr 4, and Chr 7, and labeled as Vent8a, Vent4b, and Vent7c, respectively. They are associated with congenital hydrocephalus by affecting ventricular size in the developing embryo (Zygourakis and Rosen, 2003; Zhang et al., 2006).
Although the autosomal recessive congenital hydrocephalus-1 (hy1) and hydrocephalus-2 (hy2) mice have been reported, the genetic loci have not been identified (Zhang et al., 2006). In contrast, the autosomal recessive mutation hydrocephalus-3 (hy3) mouse has been investigated comprehensively. A hy3 mouse manifests severe communicating hydrocephalus in the perinatal stage. Another transgenic mouse line OVE459 is caused by a Bdnf transgene–induced insertional mutation on a single locus on chromosome 8, and the insertion locus is overlapped with that of hy3. The transgene insertion resulted in a rearrangement of the hy3 gene exons in OVE459 mice (Zhang et al., 2006). Hy3 protein (Hydin) is homologous to Caldesmon, an actin-binding protein, suggesting that Hydin interacts with the cytoskeleton (Davy and Robinson, 2003). In the ventricular system of hy3 mice, Hydin is confined to the ciliated ependymal cell layer. Studies demonstrate that ependymal cell malfunction may contribute to hydrocephalus (Jimenez et al., 2001; Wagner et al., 2003). The protein Dynein, coded by axonemal heavy chain 5 gene (Mdnah5), is also specifically expressed in ependymal cells and is essential for the structure and function of ependymal cilia. In Mdnah5-mutant mice, the ependymal flow disturbance and consequent aqueduct closure finally result in the development of hydrocephalus. In hydrocephalus patients, the correlation of ciliary defect and aqueductal stenosis also proves the relevance of this pathogenesis in humans (Ibanez-Tallon et al., 2004).
Hydrocephalus may also result from the abnormality of mesenchymal cells. In mice, the Msx1 gene is expressed along the dorsal midline and involved in epithelial–mesenchymal interactions in organogenesis. The mouse with homozygous Msx1 mutants showed malformation or absence of posterior commissure (PC) and SCO, and subsequently developed the collapse of the cerebral aqueduct and hydrocephalus. Failure of SCO development can also be caused by inserting the Cyp2j2 transgene to interfere with the regulatory factor X4 (Rfx4) (Blackshear et al., 2003). Disruption of Rfx4 can reduce Msx2 expression and subsequently prevent the formation of SCO, and finally lead to hydrocephalus development.
Spontaneous congenital hydrocephalus (ch) can also be caused by mutation of gene Mf1 (Foxc1) (Kume et al., 1998; Zhang et al., 2006). Mf1-encoded protein is expressed in many embryonic tissues, including mesenchyme, endothelial cells, and meninges. Mice with Mf1 mutation were found to show multiple developmental disorders, including abnormal differentiation of arachnoid cells in meninges. In addition, patients with deletion of FREAC3, a homolog of Mf1, were also found to develop hydrocephalus (Kume et al., 1998).
A mouse with autosomal recessive hydrocephalus and hop gait (Hyh) manifests ventricle enlargement and hop gait at birth. The Hyh gene was identified as soluble NSF-attachment protein α (α-Snap) (Hong et al., 2004). α-Snap plays an important role in cellular membrane fusion (Chae et al., 2004). The membrane fusion is essential for inter- and intracellular molecular transportation and communication (Hong et al., 2004). In the Hyh mouse, abnormal localization of many apical proteins was identified; these proteins including SNAP receptor (Snare), beta-catenin, E-cadherin, vesicle-associated membrane protein-7 (Vamp-7), and atypical protein kinase C (aPkc) (Chae et al., 2004). The Hyh mouse demonstrated a small cerebral cortex and died postnatally from deteriorative hydrocephalus. The small cortex exhibited an abnormal development of the neuronal cells.
Another autosomal recessive mouse model is hemorrhagic hydrocephalus (Hhy). The Hhy locus has been localized on Chr 12, and the mouse is characterized by intracranial hemorrhage, hydrocephalus, and subcortical heterotopia (Kuwamura et al., 2004). The mouse manifested as communicating hydrocephalus with patent aqueduct and no histological abnormalities in choroid plexus and subarachnoid space. Following the development of hydrocephalus, numerous hemorrhages throughout the brain parenchyma and meninges could be observed.
Autosomal dominant hydrocephalus can be caused by Otx2 mutation. The mouse was characterized by a lateral ventricle dilation and a ballooned cerebrum. The histological examination showed edematous changes in periventricular white matter, suggesting that the function of Otx2 may have been a brain developmental organizer (Makiyama et al., 1997).
Congenital hydrocephalus could be related to abnormal brain development and cellular dysfunction, suggesting genetic involvement in the pathogenesis of hydrocephalus (Stoll et al., 1992). It is estimated that up to 40% of cases of hydrocephalus refer to a possible genetic etiology (Haverkamp et al., 1999), and over 100 genes have been described to be mutated in syndromic hydrocephalus cases (Kousi and Katsanis, 2016). Thereinto, X-linked hydrocephalus comprises about 5–15% of genetic-related congenital hydrocephalus (Haverkamp et al., 1999; Zhang et al., 2006; Adle-Biassette et al., 2013), and bona fide mutations have been described in four genes, that is, L1 cell adhesion molecule (L1CAM), sigma 2 subunit of the adapter protein 1 complex (AP1S2), multiple PDZ domain proteins (MPDZ), and coiled-coil domain-containing protein 88c (CCDC88C) (Tarpey et al., 2006; Al-Dosari et al., 2013; Shaheen et al., 2017; Yang et al., 2019; Etchegaray et al., 2020; Marguet et al., 2021; Wang et al., 2021). Since the phenotypes are heterogenic, there may be more genetic loci in congenital hydrocephalus. Although the loci or genes for congenital hydrocephalus have not yet been identified fully, the mutations of the respondent genes may be expressed as X-linked and/or autosomal dominant in fashion.
The genes responsible for X-linked human congenital hydrocephalus are L1CAM (L1 protein), located at Xq28 (Jouet et al., 1993; Adle-Biassette et al., 2013), and AP1S2 (Shaheen et al., 2017; Marguet et al., 2021). The exact mechanisms of the gene mutation and consequent L1 protein dysfunction are still not clear. However, a possible correlation between L1 protein dysfunction and the pathogenesis of hydrocephalus is the disruption of neural cell membrane proteins that play an important role during brain development. The L1 protein is expressed in neurons and Schwann cells and is a member of the immunoglobulin family for neural cell adhesion (Okamoto et al., 2004). As a neuronal adhesion molecule, L1CAM mediates functions of cell–cell adhesion; growth-cone morphology; the guidance of neurite outgrowth, myelination, axon bundling, and pathfinding; long-term potentiation, neuronal cell survival and migration, and synaptogenesis (Adle-Biassette et al., 2013). Mutations in L1CAM cause hydrocephalus with stenosis of the aqueduct of Sylvius and agenesis of the corpus callosum, hypoplasia of corticospinal tracts and the anterior cerebellar vermis, fusion of the thalami, and spastic paraplegia (Willems et al., 1987; Bousquet et al., 2021). The precise mechanism of L1CAM defect–related ventricular dilation remains unclear, and the hypotheses are possibly due to L1CAM-mediated decrease in white matter elasticity, increased CSF pressure and ventricular vulnerability, abnormal development of the midline structure, and narrowing of the CSF pathway (Kousi and Katsanis, 2016). AP1S2 encodes the sigma 2 subunit of the AP1 Adaptin protein for regulating lysosomal protein sorting, and the mutation of AP1S2 was reported to relate to X-linked congenital hydrocephalus through aberrant vesicle trafficking (Kousi and Katsanis, 2016; Shaheen et al., 2017; Marguet et al., 2021).
CCDC88C encodes DAPLE protein and acts as a negative regulator of the non-canonical wingless/integrated (WNT) signaling pathway; its mutation results in hydrocephalus through defective cilia orientation and aberrant CSF flow (Ohata et al., 2014; Marguet et al., 2021). MPDZ was reported to cause autosomal recessive non-syndromic communicating hydrocephalus through the disruption of the planar cell polarity pathway (Al-Jezawi et al., 2018).
Autosomal dominant congenital hydrocephalus was reported in one kindred. In contrast to X-linked or recessive congenital hydrocephalus with stenosis of the aqueduct of Sylvius, these patients were also associated with aqueductal stenosis but not related to intellectual disability and pyramidal tract dysfunction (Verhagen et al., 1998). Another hydrocephalus kindred with an identified microdeletion of 8q12.2-q21.2 was also transmitted in an autosomal dominant fashion (Vincent et al., 1994). In dystroglycanopathies, hydrocephalus is driven by aqueduct obstruction due to aberrational integrity of basement membranes in the brain, caused by mutant genes encoding for glycosyltransferases, that is, protein O-mannosyltransferase 1 (POMT1), Fukutin-related protein (FKRP), acetylglucosaminyltransferase-like protein (LARGE), and protein kinase B (AKT) (Langenbach and Rando, 2002; Akasaka-Manya et al., 2004; Satz et al., 2008). For syndromic hydrocephalus, the pathogenic mechanisms may relate to neural tube defects with resultant ependymal cilium dysfunction and aqueduct stenosis, and the mutant genes may include vang-like1, 2 (VANGL1, 2), SMARCC1, TRIM71, and PTCH1 (Kibar et al., 2011; Furey et al., 2018; Jin et al., 2020; Kundishora et al., 2021; Mastromoro et al., 2021). Both PIK3CA and PTEN genes connected with the PI3K–AKT–mTOR pathway are correlated with the pathogenesis of hydrocephalus through the mechanisms of cerebellar overgrowth and obstruction of CSF flow (Jin et al., 2020; Mastromoro et al., 2021).
In contrast to congenital hydrocephalus, acquired or adult-onset idiopathic normal pressure hydrocephalus (iNPH) is characterized by gait disturbance, urinary incontinence, and cognitive impairment. Adult-onset hydrocephalus may develop either due to decompensation of congenital hydrocephalus or sequel to an acquired CSF circulation disturbance. The acquired CSF circulation disorder may be due to late-onset aqueductal stenosis or disruption of CSF absorption (Chahlavi et al., 2001; Edwards et al., 2004). Although an X-linked adult-onset iNPH (Katsuragi et al., 2000) and an autosomal dominant fashion of familial NPH (Portenoy et al., 1984) have been reported, the related genetic studies have not been carried out yet.
Challenging to traditional CSF physiology, increasing investigations raise the new concept of CSF dynamics that, rather than choroid plexus, CSF production also comes from the interstitial fluid efflux, and the major CSF outflow pathway may go through the lymphatic drainages rather than dural venous sinuses through arachnoid villi (Louveau et al., 2016; Da Mesquita et al., 2018; Louveau et al., 2018; Mestre et al., 2018; Ringstad and Eide, 2020; Yamada et al., 2021). The CSF outflow through lymphatic drainages mainly goes through the subarachnoid spaces to the meningeal lymphatics and cervical lymphatics via the cribriform plate and nasal lymphatics (Proulx, 2021). This new CSF dynamic concept has been termed as the glymphatic system (Figure 2).
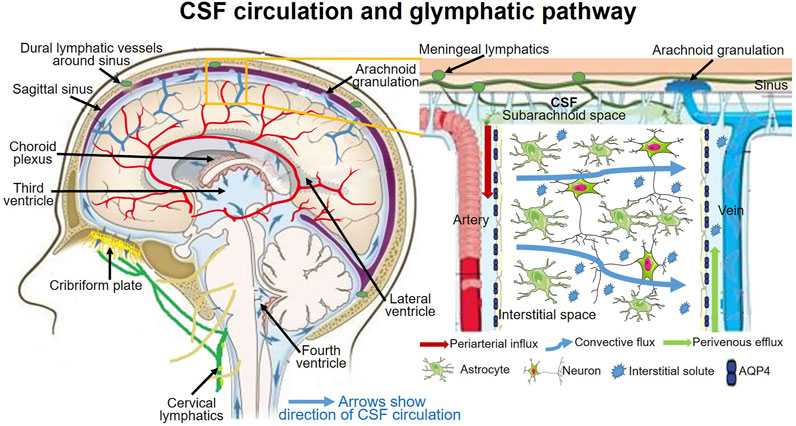
FIGURE 2. CSF circulation and glymphagitc pathway.
The glial-lymphatic or glymphatic system is a recently identified fluid clearance system with a highly-organized transport pathway. This pathway subserves the flow of CSF from the subarachnoid space into the brain through perivascular spaces of the large leptomeningeal and penetrating arteries (Iliff et al., 2012; Ma et al., 2017), also known as Virchow-Robins spaces (Iliff et al., 2012; Pizzo et al., 2018); and thence into the brain interstitium. In the interstitium, the fluid disperses via a polarized net fluid movement toward the venous perivascular and perineuronal spaces (Iliff et al., 2012). Meanwhile, the CSF flows across the glial basement membrane and astroglial end-feet fringing the brain parenchyma (Iliff et al., 2012). In the astrocytic end-feet, the water channel aquaporin-4 (AQP4) is highly expressed and facilitates the flow of CSF into the brain parenchyma, where it mixes with the interstitial fluid (ISF) (Iliff et al., 2012; Kress et al., 2014). The pathway finally directs the flow toward the venous perivascular space, perineuronal sheaths of cranial and spinal nerves, meningeal lymphatic vessels, and arachnoid granulations, ultimately clearing solutes from the neuropil into meningeal and cervical lymphatic drainage vessels (Figure 2) (Louveau et al., 2015; Absinta et al., 2017; Ma et al., 2017).
In rodents, the glymphatic pathway is primarily active during sleep, when the clearance of harmful metabolites such as amyloid β (Aβ) increases two-fold relative to the waking state (Rasmussen et al., 2018; Hablitz and Nedergaard, 2021). However, this action decreases in most iNPH (Ringstad et al., 2017; Eide and Ringstad, 2019) due to the high incidence of sleep disorders, such as obstructive sleep apnea, in iNPH patients (Roman et al., 2018). The glymphatic dysfunction in iNPH patients was supposed to be caused by inefficient arterial pulsation due to excessive breathing during obstructive sleep apnea (Roman et al., 2019), resulting in decreased brain fluid and cerebral metabolite clearance through paravascular exchange of CSF and ISF. This paravascular CSF-ISF exchange increases by 60% during sleep compared with being awake (Xie et al., 2013). Moreover, increased awakenings during obstructive sleep apnea disturb sleep-associated circulation of interstitial CSF into the glymphatic flux, further contributing to hydrocephalus. In addition, glymphatic dysfunction resulted from the blockage of the airway during obstructive sleep apnea was reported to be associated with the aggregation of Aβ and other toxic solutes, which is closely linked with cognitive impairment as manifested in iNPH and other dementia diseases (Ju et al., 2019). Aβ is degraded and cleared via multiple pathways (Tarasoff-Conway et al., 2015), and the glymphatic pathway is a significant factor in the net clearance of Aβ (Xu et al., 2015). Impaired glymphatic function in AQP4 knockout mice resulted in a 55% reduction in the parenchymal clearance of radio-labeled Aβ (Iliff et al., 2012) due to reduced efflux of ISF to the CSF (Xu et al., 2015). Furthermore, some iNPH patients responding to CSF diversion were presented with persistent disorders of cognitive function and abnormal pulsatile ICP (Villani et al., 1995; Silverberg et al., 2002; Zhang et al., 2020; Kundishora et al., 2021; Thavarajasingam et al., 2021), indicating impaired intracranial compliance and underlying pathological changes. A main histopathological finding was astrogliosis and reduction of AQP4 and of Dp71 in astrocytic perivascular end-feet. The altered AQP4 and Dp71 complex possibly contributed to the sub-ischemia and related pathological changes prevalent in the brain tissue of iNPH (Eide and Hansson, 2018). Therefore, the dysfunction of the glymphatic system may play an important role in the pathogenesis of iNPH patients.
As an essential constituent of the glymphatic system, ependymal cilia line the ventricles and interventricular connections. One of the critical functions of ependymal cilia is to effectively drive directional CSF ependymal flow through a regular synchronous beating in the glymphatic pathway (Ibanez-Tallon et al., 2004). In addition, during brain development, the patency of the aqueduct also requires an ependymal flow to maintain, and any abnormality of this flow can lead to aqueduct stenosis and hydrocephalus subsequently. Moreover, cilia also play a role in the regulation of CSF production. Therefore, aberrant molecular changes of ependymal cilia have closely related to the pathogenesis of hydrocephalus through the glymphatic mechanism. For example, mutations of Mdnah5 and Kif7 will lead to lack of ependymal flow and closure of aqueduct, and dysfunctions of Tg737orpk and E2F transcription factor 5 (E2f5) will result in CSF overproduction through the increased secretory activity of the choroid plexus; both abnormalities will result in hydrocephalus (Lindeman et al., 1998; Ibanez-Tallon et al., 2004; Banizs et al., 2005; Putoux et al., 2011).
The tight interplay between primary cilia and centrosomes makes it difficult to allocate independent composition and role to either organelle in the processes of neocortical development (Wilsch-Brauninger and Huttner, 2021). The centrosome plays a major role in the primary microtubule organization. Its dysfunction has a major impact on brain size and functionality, including ciliogenesis, suggesting a possible involvement in the glymphatic system (Marthiens and Basto, 2020; Yang et al., 2021b; Narita and Takeda, 2021). The centrosome is a complex organelle composed of two perpendicular cylindrical microtubule-based structures termed as the proximal and distal ends or centrioles. The proximal end of the centrosome is critical for pericentriolar material recruitment and microtubule-organizing activity, whereas the distal end is essential for appendage formation, membrane docking, and primary ciliogenesis (Paintrand et al., 1992). Based on its property of specialized structures and a large collection of proteins and other molecules, centrosomes exhibit a highly orchestrated biogenesis cycle. Any error in their regulation may lead to structurally and functionally compromised centrosomes and subsequent disorders. For example, the formation of the functional primary cilia depends on the intraflagellar transport (IFT) system (Pedersen et al., 2008). Ift88 is a crucial member of the IFT-B complex, involved in anterograde transport. Kinesin family member 3a (Kif3a) is a subunit of the heterotrimeric kinesin complex and is responsible for anterograde transport too. Arl13b encodes a ciliary localized small GTPase of the ARF/ARL family, being essential for ciliary axoneme structure and signaling (Caspary et al., 2007). The genetic ablations of Kif3a, Ift88, and Arl13b at early stages of cortical development lead to a generally dysmorphic telencephalon including reduced cortical neurogenesis, lack of olfactory bulbs, and ventriculomegaly (Willaredt et al., 2008; Higginbotham et al., 2013; Insolera et al., 2014; Snedeker et al., 2017; Yang et al., 2021b). Notably, the genetic ablations of Ift88 or Kif3a in the neural crest and midbrain also lead to a significant cortical malformation and ventriculomegaly (Snedeker et al., 2017), suggesting the requirement of primary cilia in non-forebrain tissue to regulate cortical development and ventricle size. In addition, a previous study showed that deletion of Kif3a using Nestin-Cre led to cortical disorganization and robust ventriculomegaly (Wilson et al., 2012). Remarkably, the Nes-Cre8 transgene is widely expressed in both forebrain and surrounding non-forebrain tissues, indicating a contribution of cortical and non-cortical ciliary defects in this mutant (Petersen et al., 2002). Moreover, mutation of Polo-like kinase 4 (Plk4) gene and resultant centrosome dysfunction and cilium defect have been identified as causes in the pathophysiology of autosomal recessive developmental disorders, Seckel syndrome (SCKL) (Martin et al., 2014). SCKL patients show short stature and brain structural anomalies (Griffith et al., 2008; Martin et al., 2014; Gambarotto et al., 2019). These findings suggest that centrosome membrane anchorage, in the process of ciliogenesis, plays a vital role in regulating cortical development and ventricle size.
Based on the new CSF dynamic concept, the pathogenic mechanisms of iNPH or communicating hydrocephalus that correlate with the alterations in motile cilia and cerebrospinal fluid dynamics have been extensively investigated, and the related mutant genes have also been reported (Table 2). The mutant genes related to motile cilium dysfunction on ependymal cells, and consequent hydrocephalus development may include Ccdc39, Celsr2, Celsr3, Cetn2, Dvl, Foxj1, Hydin, Mdnah5, Pkd1, Tg737, Daple, Dnah14, Cfap43, Cwh43, Eml1, Fmn2, Tmem67, and Zcchc8 (Law et al., 2014; Shaheen et al., 2017; Kundishora et al., 2021; McKnight et al., 2021; Yamada et al., 2021). For example, Foxj1 mutation results in ciliopathy and hydrocephalus due to dysfunction of motile cilia (Abdi et al., 2018; Wallmeier et al., 2019). Celsr2 is a planar cell polarity gene, and mutation results in hydrocephalus through ependymal cilia dysfunction (Sotak and Gleeson, 2012). Mutation of gene Hydin, which encodes a key protein within motile cilia in the ventricles, may also cause hydrocephalus (Lechtreck et al., 2008). Daple regulates the direction of CSF flow through motile cilium beating in the ependymal cells, and mutation of Daple results in communication hydrocephalus (Takagishi et al., 2017). Mutation of Cfap43 causes a morphologic abnormality of ependymal cilia and consequent hydrocephalus (Morimoto et al., 2019). Cwh43 modifies the glycosylphosphatidylinositol-anchored proteins on the ependymal cells, and the mutant Cwh43 is related to iNPH in both humans and mice. The clinical features manifest as late-onset communicating hydrocephalus with symptoms of gait and balance dysfunction (Yang et al., 2021a).
The clinical manifestation and progression, as well as experimental investigations, indicate that hydrocephalus is a complex disease with polygenic involvement, rather than a simple CSF accumulation disorder. Although the current studies have revealed that some genetic mutations are involved in the pathogenesis of hydrocephalus, how these mutations are associated with the disorder of CSF circulation and their pathogenic roles in the pathological progression of hydrocephalus still remain largely unknown. Previous studies indicated that a lot of genetic mutations were relevant to the disorders of ciliary and/or centrosome, resulting in the dysfunction of the glymphatic system. However, how these mutations and their interactions contribute to the pathogenesis of hydrocephalus needs to be further elucidated. Moreover, there is still a lack of basic knowledge on the mechanisms underlying the cognitive functional impairment of hydrocephalus. Therefore, further extensive studies should be conducted to explore the underlying molecular mechanisms of identified and/or unidentified genes in the pathophysiology of hydrocephalus. Based on our knowledge, we propose that the genetic mutations relevant to ciliary and centrosomal proteins and the interaction between glymphatic system and ciliary/centrosomal structures/functions may be a critical molecular mechanism in the pathophysiology of hydrocephalus. In addition, based on these fundamental molecular mechanisms, it is noteworthy that environmental and other acquired risks or etiological factors are also involved in the facilitation of ventricular enlargement.
With regard to the environment and acquired elements, alcohol consumption, diabetes, hypertension, inflammation, aging, and sleep apnea are remarkable risk factors for iNPH development (Sakata-Haga et al., 2004; Kahle et al., 2016; Omran et al., 2017; Holth et al., 2019; Ghaffari-Rafi et al., 2020; Zhang et al., 2020; Yamada et al., 2021). Alcohol consumption demonstrated a correlation with iNPH development possibly due to ethanol-induced decrease in beating frequency of motile cilia and consequent dysfunction on the ependymal cells of ventricles (Omran et al., 2017; Ghaffari-Rafi et al., 2020). Although iNPH patients present with increased morbidity of diabetes, the causal relationship has not been concluded (Hudson et al., 2019; Rasanen et al., 2020). The speculative pathogenesis may be attributed to high concentration of sugar and increased fluid viscosity in CSF. The studies have indicated that increased fluid viscosity increases the shear stress on the ventricular wall and accelerates the dilatation of the vasculature (Roux et al., 2020). Hypertension and aging were also reported to be potential risk factors for ventriculomegaly. The underlining mechanism was considered to be related to the decline in the glymphatic function (Mestre et al., 2018; Ringstad and Eide, 2020). As previously discussed, obstructive sleep apnea was frequently associated with iNPH (Roman et al., 2018), and up to 90% of iNPH patients suffered from obstructive sleep apnea (Roman et al., 2019). In addition to the previously discussed pathogenic mechanism, obstructive sleep apnea reduces oxygen intake and decreases venous return to the heart, resulting in further retrograde intracranial venous hypertension and ventricular enlargement. It is worth noting that sleep and glymphatic system have become critical elements in the pathogenesis of iNPH, and they may be possibly manipulated through the changes of cardiac-induced arterial pulsation and intracranial pressure pulsatility with the cardiac-gated device used in our previous studies (Iliff et al., 2013; Luciano et al., 2017).
In summary, it is essential to recognize that the current studies of genetic foundation and predisposing risk factors are promising approaches toward addressing the usual concern about whether an observed phenomenon is a consequence or a cause of hydrocephalus. Although the genetic study of hydrocephalus in humans is limited, many genetic loci of hydrocephalus have been identified in animal models. This approach offers researchers significant insights into the molecular etiology of impaired brains during the pathogenesis of hydrocephalus. In addition, genetic conditions affecting motile cilia lay the foundation, and acquired risk factors facilitate the development of hydrocephalus. Thereinto, the glymphatic system plays a key role in the activities of brain’s physiological metabolism and pathological process of hydrocephalus. Moreover, dysfunction of the glymphatic system and sleep disorders are emerging as popular topics in the studies of pathogenesis and possible therapeutic management of hydrocephalus, especially for iNPH. Therefore, these types of studies will facilitate a greater understanding of the molecular mechanism in hydrocephalus and provide valuable insights into the pathogenesis of iNPH and other neurological diseases. Possible new mechanisms other than altered CSF circulation and resorption, if uncovered via genetic research, may also help to explain why patients with hydrocephalus may still experience symptomatic progression despite functioning shunts. Ultimately, such knowledge will be useful in the improvement of patient care in different ways and also guide the optimal treatment decisions for patients as early as possible.
JY conceived the presented idea. JY, JL, XZ, JG, and CY undertook the literature review and synthesized the draft. JY contributed ideas throughout the process and approved the final draft. JY, JL, XZ, JG, and CY looked over and edited the final draft.
This work was supported by the Xiamen Municipal Bureau of Science and Technology (grant 3502Z20209117 to JY) and Scientific Research Fund of Xiang’an Hospital of Xiamen University (grant PM202109150001 to JL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdi, K., Lai, C.-H., Paez-Gonzalez, P., Lay, M., Pyun, J., and Kuo, C. T. (2018). Uncovering Inherent Cellular Plasticity of Multiciliated Ependyma Leading to Ventricular wall Transformation and Hydrocephalus. Nat. Commun. 9 (1), 1655. doi:10.1038/s41467-018-03812-w
Absinta, M., Ha, S.-K., Nair, G., Sati, P., Luciano, N. J., Palisoc, M., et al. (2017). Human and Nonhuman Primate Meninges Harbor Lymphatic Vessels that Can Be Visualized Noninvasively by MRI. Elife 6. doi:10.7554/eLife.29738
Adle-Biassette, H., Saugier-Veber, P., Fallet-Bianco, C., Delezoide, A.-L., Razavi, F., Drouot, N., et al. (2013). Neuropathological Review of 138 Cases Genetically Tested for X-Linked Hydrocephalus: Evidence for Closely Related Clinical Entities of Unknown Molecular Bases. Acta Neuropathol. 126 (3), 427–442. doi:10.1007/s00401-013-1146-1
Akasaka-Manya, K., Manya, H., Kobayashi, K., Toda, T., and Endo, T. (2004). Structure-function Analysis of Human Protein O-Linked Mannose β1,2-N-acetylglucosaminyltransferase 1, POMGnT1. Biochem. Biophysical Res. Commun. 320 (1), 39–44. doi:10.1016/j.bbrc.2004.05.129
Al-Dosari, M. S., Al-Owain, M., Tulbah, M., Kurdi, W., Adly, N., Al-Hemidan, A., et al. (2013). Mutation inMPDZcauses Severe Congenital Hydrocephalus. J. Med. Genet. 50 (1), 54–58. doi:10.1136/jmedgenet-2012-101294
Al-Jezawi, N. K., Al-Shamsi, A. M., Suleiman, J., Ben-Salem, S., John, A., Vijayan, R., et al. (2018). Compound Heterozygous Variants in the Multiple PDZ Domain Protein (MPDZ) Cause a Case of Mild Non-progressive Communicating Hydrocephalus. BMC Med. Genet. 19 (1), 34. doi:10.1186/s12881-018-0540-x
Banizs, B., Pike, M. M., Millican, C. L., Ferguson, W. B., Komlosi, P., Sheetz, J., et al. (2005). Dysfunctional Cilia lead to Altered Ependyma and Choroid Plexus Function,and Result in the Formation of Hydrocephalus. Development 132 (23), 5329–5339. doi:10.1242/dev.02153
Battelli, M., Buonamici, L., Virgili, M., Abbondanza, A., and Contestabile, A. (1998). Simulated Ischaemia-Reperfusion Conditions Increase Xanthine Dehydrogenase and Oxidase Activities in Rat Brain Slices. Neurochem. Int. 32 (1), 17–21. doi:10.1016/s0197-0186(97)00052-1
Blackshear, P. J., Graves, J. P., Stumpo, D. J., Cobos, I., Rubenstein, J. L. R., and Zeldin, D. C. (2003). Graded Phenotypic Response to Partial and Complete Deficiency of a Brain-specific Transcript Variant of the Winged helix Transcription Factor RFX4. Development 130 (19), 4539–4552. doi:10.1242/dev.00661
Bousquet, I., Bozon, M., Castellani, V., Touraine, R., Piton, A., Gérard, B., et al. (2021). X-linked Partial Corpus Callosum Agenesis with Mild Intellectual Disability: Identification of a Novel L1CAM Pathogenic Variant. Neurogenetics 22 (1), 43–51. doi:10.1007/s10048-020-00629-y
Caspary, T., Larkins, C. E., and Anderson, K. V. (2007). The Graded Response to Sonic Hedgehog Depends on Cilia Architecture. Develop. Cel 12 (5), 767–778. doi:10.1016/j.devcel.2007.03.004
Castañeyra-Perdomo, A., Meyer, G., Carmona-Calero, E., Bañuelos-Pineda, J., Méndez-Medina, R., Ormazabal-Ramos, C., et al. (1994). Alterations of the Subcommissural Organ in the Hydrocephalic Human Fetal Brain. Brain Res. Dev. Brain Res. 79 (2), 316–320. doi:10.1016/0165-3806(94)90138-4
Chae, T. H., Kim, S., Marz, K. E., Hanson, P. I., and Walsh, C. A. (2004). The Hyh Mutation Uncovers Roles for αSnap in Apical Protein Localization and Control of Neural Cell Fate. Nat. Genet. 36 (3), 264–270. doi:10.1038/ng1302
Chahlavi, A., El-Babaa, S. K., and Luciano, M. G. (2001). Adult-onset Hydrocephalus. Neurosurg. Clin. North America 12 (4), 753–760. ix. doi:10.1016/s1042-3680(18)30032-9
Da Mesquita, S., Louveau, A., Vaccari, A., Smirnov, I., Cornelison, R. C., Kingsmore, K. M., et al. (2018). Publisher Correction: Functional Aspects of Meningeal Lymphatics in Ageing and Alzheimer's Disease. Nature 564 (7734), E7. doi:10.1038/s41586-018-0689-7
Davy, B. E., and Robinson, M. L. (2003). Congenital Hydrocephalus in Hy3 Mice Is Caused by a Frameshift Mutation in Hydin, a Large Novel Gene. Hum. Mol. Genet. 12 (10), 1163–1170. doi:10.1093/hmg/ddg122
Ding, Y., Lai, Q., McAllister II, J. P., and Canady, A. I. (2001). Impaired Motor Learning in Children with Hydrocephalus. Pediatr. Neurosurg. 34 (4), 182–189. doi:10.1159/000056017
Doublier, S., Duyckaerts, C., Seurin, D., and Binoux, M. (2000). Impaired Brain Development and Hydrocephalus in a Line of Transgenic Mice with Liver-specific Expression of Human Insulin-like Growth Factor Binding Protein-1. Growth Horm. IGF Res. 10 (5), 267–274. doi:10.1054/ghir.2000.0168
Draper, C. E., Owen-Lynch, P. J., Bannister, C. M., and Miyan, J. (2001). Proliferation of Cerebral Cortical Cells from the Hydrocephalic HTx Rat: an In Vitro Study. Eur. J. Pediatr. Surg. 11 Suppl 1 (Suppl. 1), S51–S52.
Edwards, R. J., Dombrowski, S. M., Luciano, M. G., and Pople, I. K. (2004). Chronic Hydrocephalus in Adults. Brain Pathol. 14 (3), 325–336. doi:10.1111/j.1750-3639.2004.tb00072.x
Eide, P. K., and Hansson, H.-A. (2018). Astrogliosis and Impaired Aquaporin-4 and Dystrophin Systems in Idiopathic normal Pressure Hydrocephalus. Neuropathol. Appl. Neurobiol. 44 (5), 474–490. doi:10.1111/nan.12420
Eide, P. K., and Ringstad, G. (2019). Delayed Clearance of Cerebrospinal Fluid Tracer from Entorhinal Cortex in Idiopathic normal Pressure Hydrocephalus: A Glymphatic Magnetic Resonance Imaging Study. J. Cereb. Blood Flow Metab. 39 (7), 1355–1368. doi:10.1177/0271678X18760974
Etchegaray, A., Juarez-Peñalva, S., Petracchi, F., and Igarzabal, L. (2020). Prenatal Genetic Considerations in Congenital Ventriculomegaly and Hydrocephalus. Childs Nerv Syst. 36 (8), 1645–1660. doi:10.1007/s00381-020-04526-5
Furey, C. G., Choi, J., Jin, S. C., Zeng, X., Timberlake, A. T., Nelson-Williams, C., et al. (2018). De Novo Mutation in Genes Regulating Neural Stem Cell Fate in Human Congenital Hydrocephalus. Neuron 99 (2), 302–314. e304. doi:10.1016/j.neuron.2018.06.019
Galard, R., Poca, M. A., Catalán, R., Tintoré, M., Castellanos, J. M., and Sahuquillo, J. (1997). Decreased Cholecystokinin Levels in Cerebrospinal Fluid of Patients with Adult Chronic Hydrocephalus Syndrome. Biol. Psychiatry 41 (7), 804–809. doi:10.1016/s0006-3223(96)00098-4
Gambarotto, D., Pennetier, C., Ryniawec, J. M., Buster, D. W., Gogendeau, D., Goupil, A., et al. (2019). Plk4 Regulates Centriole Asymmetry and Spindle Orientation in Neural Stem Cells. Develop. Cel 50 (1), 11–24. e10. doi:10.1016/j.devcel.2019.04.036
Ghaffari-Rafi, A., Gorenflo, R., Hu, H., Viereck, J., and Liow, K. (2020). Role of Psychiatric, Cardiovascular, Socioeconomic, and Demographic Risk Factors on Idiopathic normal Pressure Hydrocephalus: A Retrospective Case-Control Study. Clin. Neurol. Neurosurg. 193, 105836. doi:10.1016/j.clineuro.2020.105836
Griffith, E., Walker, S., Martin, C.-A., Vagnarelli, P., Stiff, T., Vernay, B., et al. (2008). Mutations in Pericentrin Cause Seckel Syndrome with Defective ATR-dependent DNA Damage Signaling. Nat. Genet. 40 (2), 232–236. doi:10.1038/ng.2007.80
Hablitz, L. M., and Nedergaard, M. (2021). The Glymphatic System: A Novel Component of Fundamental Neurobiology. J. Neurosci. 41 (37), 7698–7711. doi:10.1523/JNEUROSCI.0619-21.2021
Hatini, V., Ye, X., Balas, G., and Lai, E. (1999). Dynamics of Placodal Lineage Development Revealed by Targeted Transgene Expression. Dev. Dyn. 215 (4), 332–343. doi:10.1002/(sici)1097-0177(199908)215:4<332:aid-aja5>3.0.co;2-r
Haverkamp, F., Wölfle, J., Aretz, M., Krämer, A., Höhmann, B., Fahnenstich, H., et al. (1999). Congenital Hydrocephalus Internus and Aqueduct Stenosis: Aetiology and Implications for Genetic Counselling. Eur. J. Pediatr. 158 (6), 474–478. doi:10.1007/s004310051123
Higginbotham, H., Guo, J., Yokota, Y., Umberger, N. L., Su, C.-Y., Li, J., et al. (2013). Arl13b-regulated Cilia Activities Are Essential for Polarized Radial Glial Scaffold Formation. Nat. Neurosci. 16 (8), 1000–1007. doi:10.1038/nn.3451
Holth, J. K., Fritschi, S. K., Wang, C., Pedersen, N. P., Cirrito, J. R., Mahan, T. E., et al. (2019). The Sleep-Wake Cycle Regulates Brain Interstitial Fluid Tau in Mice and CSF Tau in Humans. Science 363 (6429), 880–884. doi:10.1126/science.aav2546
Hong, H.-K., Chakravarti, A., and Takahashi, J. S. (2004). From the Cover: The Gene for Soluble N-Ethylmaleimide Sensitive Factor Attachment Protein Is Mutated in Hydrocephaly with Hop Gait (Hyh) Mice. Proc. Natl. Acad. Sci. 101 (6), 1748–1753. doi:10.1073/pnas.0308268100
Hudson, M., Nowak, C., Garling, R. J., and Harris, C. (2019). Comorbidity of Diabetes Mellitus in Idiopathic normal Pressure Hydrocephalus: a Systematic Literature Review. Fluids Barriers CNS 16 (1), 5. doi:10.1186/s12987-019-0125-x
Huh, S., Hatini, V., Marcus, R. C., Li, S. C., and Lai, E. (1999). Dorsal-ventral Patterning Defects in the Eye of BF-1-Deficient Mice Associated with a Restricted Loss of Shh Expression. Develop. Biol. 211 (1), 53–63. doi:10.1006/dbio.1999.9303
Ibañez-Tallon, I., Pagenstecher, A., Fliegauf, M., Olbrich, H., Kispert, A., Ketelsen, U.-P., et al. (2004). Dysfunction of Axonemal Dynein Heavy Chain Mdnah5 Inhibits Ependymal Flow and Reveals a Novel Mechanism for Hydrocephalus Formation. Hum. Mol. Genet. 13 (18), 2133–2141. doi:10.1093/hmg/ddh219
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 4 (147), 147ra111. doi:10.1126/scitranslmed.3003748
Iliff, J. J., Wang, M., Zeppenfeld, D. M., Venkataraman, A., Plog, B. A., Liao, Y., et al. (2013). Cerebral Arterial Pulsation Drives Paravascular CSF-Interstitial Fluid Exchange in the Murine Brain. J. Neurosci. 33 (46), 18190–18199. doi:10.1523/Jneurosci.1592-13.2013
Insolera, R., Bazzi, H., Shao, W., Anderson, K. V., and Shi, S.-H. (2014). Cortical Neurogenesis in the Absence of Centrioles. Nat. Neurosci. 17 (11), 1528–1535. doi:10.1038/nn.3831
Jiménez, A. J., Tomé, M., Páez, P., Wagner, C., Rodríguez, S., Fernández-Llebrez, P., et al. (2001). A Programmed Ependymal Denudation Precedes Congenital Hydrocephalus in thehyhMutant Mouse. J. Neuropathol. Exp. Neurol. 60 (11), 1105–1119. doi:10.1093/jnen/60.11.1105
Jin, S. C., Dong, W., Kundishora, A. J., Panchagnula, S., Moreno-De-Luca, A., Furey, C. G., et al. (2020). Exome Sequencing Implicates Genetic Disruption of Prenatal Neuro-Gliogenesis in Sporadic Congenital Hydrocephalus. Nat. Med. 26 (11), 1754–1765. doi:10.1038/s41591-020-1090-2
Johanson, C. E., Szmydynger-Chodobska, J., Chodobski, A., Baird, A., McMillan, P., and Stopa, E. G. (1999). Altered Formation and Bulk Absorption of Cerebrospinal Fluid in FGF-2-Induced Hydrocephalus. Am. J. Physiology-Regulatory, Integr. Comp. Physiol. 277 (1), R263–R271. doi:10.1152/ajpregu.1999.277.1.R263
Jones, H. C., Carter, B. J., and Morel, L. (2003). Characteristics of Hydrocephalus Expression in the LEW/Jms Rat Strain with Inherited Disease. Childs Nerv Syst. 19 (1), 11–18. doi:10.1007/s00381-002-0671-3
Jouet, M., Rosenthal, A., MacFarlane, J., Kenwrick, S., and Donnai, D. (1993). A Missense Mutation Confirms the L1 Defect in X-Linked Hydrocephalus (HSAS). Nat. Genet. 4 (4), 331. doi:10.1038/ng0893-331
Ju, Y.-E. S., Zangrilli, M. A., Finn, M. B., Fagan, A. M., and Holtzman, D. M. (2019). Obstructive Sleep Apnea Treatment, Slow Wave Activity, and Amyloid-β. Ann. Neurol. 85 (2), 291–295. doi:10.1002/ana.25408
Kahle, K. T., Kulkarni, A. V., Limbrick, D. D., and Warf, B. C. (2016). Hydrocephalus in Children. The Lancet 387 (10020), 788–799. doi:10.1016/S0140-6736(15)60694-8
Katsuragi, S., Teraoka, K., Ikegami, K., Amano, K., Yamashita, K., Ishizuka, K., et al. (2000). Late Onset X-Linked Hydrocephalus with normal Cerebrospinal Fluid Pressure. Psychiatry Clin. Neurosci. 54 (4), 487–492. doi:10.1046/j.1440-1819.2000.00740.x
Khonsari, R. H., Delezoide, A.-L., Kang, W., Hébert, J. M., Bessières, B., Bodiguel, V., et al. (2012). Central Nervous System Malformations and Deformations inFGFR2-Related Craniosynostosis. Am. J. Med. Genet. 158A (11), 2797–2806. doi:10.1002/ajmg.a.35598
Kibar, Z., Salem, S., Bosoi, C., Pauwels, E., De Marco, P., Merello, E., et al. (2011). Contribution of VANGL2 Mutations to Isolated Neural Tube Defects. Clin. Genet. 80 (1), 76–82. doi:10.1111/j.1399-0004.2010.01515.x
Koleva, M., and De Jesus, O. (2021). Hydrocephalus. National Center for Biotechnology Information. Bethesda, Maryland, U.S. NBK560875 [bookaccession].
Kousi, M., and Katsanis, N. (2016). The Genetic Basis of Hydrocephalus. Annu. Rev. Neurosci. 39, 409–435. doi:10.1146/annurev-neuro-070815-014023
Krebs, D. L., Metcalf, D., Merson, T. D., Voss, A. K., Thomas, T., Zhang, J.-G., et al. (2004). Development of Hydrocephalus in Mice Lacking SOCS7. Proc. Natl. Acad. Sci. 101 (43), 15446–15451. doi:10.1073/pnas.0406870101
Kress, B. T., Iliff, J. J., Xia, M., Wang, M., Wei, H. S., Zeppenfeld, D., et al. (2014). Impairment of Paravascular Clearance Pathways in the Aging Brain. Ann. Neurol. 76 (6), 845–861. doi:10.1002/ana.24271
Kume, T., Deng, K.-Y., Winfrey, V., Gould, D. B., Walter, M. A., and Hogan, B. L. M. (1998). The Forkhead/winged helix Gene Mf1 Is Disrupted in the Pleiotropic Mouse Mutation Congenital Hydrocephalus. Cell 93 (6), 985–996. doi:10.1016/s0092-8674(00)81204-0
Kume, T., Deng, K., and Hogan, B. L. (2000). Murine Forkhead/winged helix Genes Foxc1 (Mf1) and Foxc2 (Mfh1) Are Required for the Early Organogenesis of the Kidney and Urinary Tract. Development 127 (7), 1387–1395. doi:10.1242/dev.127.7.1387
Kundishora, A. J., Singh, A. K., Allington, G., Duy, P. Q., Ryou, J., Alper, S. L., et al. (2021). Genomics of Human Congenital Hydrocephalus. Childs Nerv Syst. 37, 3325–3340. doi:10.1007/s00381-021-05230-8
Kuwamura, M., Kinoshita, A., Okumoto, M., Yamate, J., and Mori, N. (2004). Hemorrhagic Hydrocephalus (Hhy): a Novel Mutation on Mouse Chromosome 12. Develop. Brain Res. 152 (1), 69–72. doi:10.1016/j.devbrainres.2004.05.006
Langenbach, K. J., and Rando, T. A. (2002). Inhibition of Dystroglycan Binding to Laminin Disrupts the PI3K/AKT Pathway and Survival Signaling in Muscle Cells. Muscle Nerve 26 (5), 644–653. doi:10.1002/mus.10258
Law, R., Dixon-Salazar, T., Jerber, J., Cai, N., Abbasi, A. A., Zaki, M. S., et al. (2014). Biallelic Truncating Mutations in FMN2, Encoding the Actin-Regulatory Protein Formin 2, Cause Nonsyndromic Autosomal-Recessive Intellectual Disability. Am. J. Hum. Genet. 95 (6), 721–728. doi:10.1016/j.ajhg.2014.10.016
Lechtreck, K.-F., Delmotte, P., Robinson, M. L., Sanderson, M. J., and Witman, G. B. (2008). Mutations in Hydin Impair Ciliary Motility in Mice. J. Cel Biol 180 (3), 633–643. doi:10.1083/jcb.200710162
Li, X., Miyajima, M., and Arai, H. (2005). Analysis of TGF-Beta2 and TGF-Beta3 Expression in the Hydrocephalic H-Tx Rat Brain. Childs Nerv Syst. 21 (1), 32–38. doi:10.1007/s00381-004-1034-z
Lin, X., Liu, B., Yang, X., Yue, X., Diao, L., Wang, J., et al. (2013). Genetic Deletion of Rnd3 Results in Aqueductal Stenosis Leading to Hydrocephalus through Up-Regulation of Notch Signaling. Proc. Natl. Acad. Sci. 110 (20), 8236–8241. doi:10.1073/pnas.1219995110
Lindeman, G. J., Dagnino, L., Gaubatz, S., Xu, Y., Bronson, R. T., Warren, H. B., et al. (1998). A Specific, Nonproliferative Role for E2F-5 in Choroid Plexus Function Revealed by Gene Targeting. Genes Develop. 12 (8), 1092–1098. doi:10.1101/gad.12.8.1092
Louveau, A., Herz, J., Alme, M. N., Salvador, A. F., Dong, M. Q., Viar, K. E., et al. (2018). CNS Lymphatic Drainage and Neuroinflammation Are Regulated by Meningeal Lymphatic Vasculature. Nat. Neurosci. 21 (10), 1380–1391. doi:10.1038/s41593-018-0227-9
Louveau, A., Smirnov, I., Keyes, T. J., Eccles, J. D., Rouhani, S. J., Peske, J. D., et al. (2016). Corrigendum: Structural and Functional Features of central Nervous System Lymphatic Vessels. Nature 533 (7602), 278. doi:10.1038/nature16999
Louveau, A., Smirnov, I., Keyes, T. J., Eccles, J. D., Rouhani, S. J., Peske, J. D., et al. (2015). Structural and Functional Features of central Nervous System Lymphatic Vessels. Nature 523 (7560), 337–341. doi:10.1038/nature14432
Luciano, M. G., Dombrowski, S. M., Qvarlander, S., El-Khoury, S., Yang, J., Thyagaraj, S., et al. (2017). Novel Method for Dynamic Control of Intracranial Pressure. J. Neurosurg. 126 (5), 1629–1640. doi:10.3171/2016.4.JNS152457
Ma, Q., Ineichen, B. V., Detmar, M., and Proulx, S. T. (2017). Outflow of Cerebrospinal Fluid Is Predominantly through Lymphatic Vessels and Is Reduced in Aged Mice. Nat. Commun. 8 (1), 1434. doi:10.1038/s41467-017-01484-6
Makiyama, Y., Shoji, S. i., and Mizusawa, H. (1997). Hydrocephalus in the Otx2+/−Mutant Mouse. Exp. Neurol. 148 (1), 215–221. doi:10.1006/exnr.1997.6638
Marguet, F., Vezain, M., Marcorelles, P., Audebert-Bellanger, S., Cassinari, K., Drouot, N., et al. (2021). Neuropathological Hallmarks of Fetal Hydrocephalus Linked to CCDC88C Pathogenic Variants. Acta Neuropathol. Commun. 9 (1), 104. doi:10.1186/s40478-021-01207-5
Marthiens, V., and Basto, R. (2020). Centrosomes: The Good and the Bad for Brain Development. Biol. Cel 112 (6), 153–172. doi:10.1111/boc.201900090
Martin, C.-A., Ahmad, I., Klingseisen, A., Hussain, M. S., Bicknell, L. S., Leitch, A., et al. (2014). Mutations in PLK4, Encoding a Master Regulator of Centriole Biogenesis, Cause Microcephaly, Growth Failure and Retinopathy. Nat. Genet. 46 (12), 1283–1292. doi:10.1038/ng.3122
Mashayekhi, F., Bannister, C. M., and Miyan, J. A. (2001). Failure in Cell Proliferation in the Germinal Epithelium of the HTx Rats. Eur. J. Pediatr. Surg. 11 (Suppl. 1), S57–S59.
Mastromoro, G., De Luca, A., Marchionni, E., Spagnuolo, A., Ventriglia, F., Manganaro, L., et al. (2021). External Hydrocephalus as a Prenatal Feature of noonan Syndrome. Ann. Hum. Genet. 85 (6), 249–252. doi:10.1111/ahg.12436
McKnight, I., Hart, C., Park, I.-H., and Shim, J. W. (2021). Genes Causing Congenital Hydrocephalus: Their Chromosomal Characteristics of Telomere Proximity and DNA Compositions. Exp. Neurol. 335, 113523. doi:10.1016/j.expneurol.2020.113523
Mestre, H., Tithof, J., Du, T., Song, W., Peng, W., Sweeney, A. M., et al. (2018). Flow of Cerebrospinal Fluid Is Driven by Arterial Pulsations and Is Reduced in Hypertension. Nat. Commun. 9 (1), 4878. doi:10.1038/s41467-018-07318-3
Miller, J. M., Kumar, R., McAllister, J. P., Krause, G. S., and Krause, G. S. (2006). Gene Expression Analysis of the Development of Congenital Hydrocephalus in the H-Tx Rat. Brain Res. 1075 (1), 36–47. doi:10.1016/j.brainres.2005.12.094
Mori, F., Tanji, K., Yoshida, Y., and Wakabayashi, K. (2002). Thalamic Retrograde Degeneration in the Congenitally Hydrocephalic Rat Is Attributable to Apoptotic Cell Death. Neuropathology 22 (3), 186–193. doi:10.1046/j.1440-1789.2002.00445.x
Morimoto, Y., Yoshida, S., Kinoshita, A., Satoh, C., Mishima, H., Yamaguchi, N., et al. (2019). Nonsense Mutation in CFAP43 Causes normal-pressure Hydrocephalus with Ciliary Abnormalities. Neurology 92 (20), e2364–e2374. doi:10.1212/WNL.0000000000007505
Narita, K., and Takeda, S. (2021). Ultrastructural Evidence for an Unusual Mode of Ciliogenesis in Mouse Multiciliated Epithelia. Microscopy (Oxf) 70 (3), 308–315. doi:10.1093/jmicro/dfaa074
Ohata, S., Nakatani, J., Herranz-Pérez, V., Cheng, J., Belinson, H., Inubushi, T., et al. (2014). Loss of Dishevelleds Disrupts Planar Polarity in Ependymal Motile Cilia and Results in Hydrocephalus. Neuron 83 (3), 558–571. doi:10.1016/j.neuron.2014.06.022
Ohmiya, M., Fukumitsu, H., Nitta, A., Nomoto, H., Furukawa, Y., and Furukawa, S. (2001). Administration of FGF-2 to Embryonic Mouse Brain Induces Hydrocephalic Brain Morphology and Aberrant Differentiation of Neurons in the Postnatal Cerebral Cortex. J. Neurosci. Res. 65 (3), 228–235. doi:10.1002/jnr.1146
Oi, S., Sato, O., Yamada, H., and Matsumoto, S. (1996). Experimental Models of Congenital Hydrocephalus and Comparable Clinical Problems in the Fetal and Neonatal Periods. Child's Nerv Syst. 12 (6), 292–302. doi:10.1007/bf00301016
Okamoto, N., Del Maestro, R., Valero, R., Monros, E., Poo, P., Kanemura, Y., et al. (2004). Hydrocephalus and Hirschsprung's Disease with a Mutation of L1CAM. J. Hum. Genet. 49 (6), 334–337. doi:10.1007/s10038-004-0153-4
Omran, A. J. A., Saternos, H. C., Althobaiti, Y. S., Wisner, A., Sari, Y., Nauli, S. M., et al. (2017). Alcohol Consumption Impairs the Ependymal Cilia Motility in the Brain Ventricles. Sci. Rep. 7 (1), 13652. doi:10.1038/s41598-017-13947-3
Paintrand, M., Moudjou, M., Delacroix, H., and Bornens, M. (1992). Centrosome Organization and Centriole Architecture: Their Sensitivity to Divalent Cations. J. Struct. Biol. 108 (2), 107–128. doi:10.1016/1047-8477(92)90011-x
Pedersen, L. B., Veland, I. R., Schrøder, J. M., and Christensen, S. T. (2008). Assembly of Primary Cilia. Dev. Dyn. 237 (8), 1993–2006. doi:10.1002/dvdy.21521
Petersen, P. H., Zou, K., Hwang, J. K., Jan, Y. N., and Zhong, W. (2002). Progenitor Cell Maintenance Requires Numb and Numblike during Mouse Neurogenesis. Nature 419 (6910), 929–934. doi:10.1038/nature01124
Pizzo, M. E., Wolak, D. J., Kumar, N. N., Brunette, E., Brunnquell, C. L., Hannocks, M. J., et al. (2018). Intrathecal Antibody Distribution in the Rat Brain: Surface Diffusion, Perivascular Transport and Osmotic Enhancement of Delivery. J. Physiol. 596 (3), 445–475. doi:10.1113/JP275105
Portenoy, R. K., Berger, A., and Gross, E. (1984). Familial Occurrence of Idiopathic normal-pressure Hydrocephalus. Arch. Neurol. 41 (3), 335–337. doi:10.1001/archneur.1984.04050150117029
Proulx, S. T. (2021). Cerebrospinal Fluid Outflow: a Review of the Historical and Contemporary Evidence for Arachnoid Villi, Perineural Routes, and Dural Lymphatics. Cell. Mol. Life Sci. 78 (6), 2429–2457. doi:10.1007/s00018-020-03706-5
Putoux, A., Thomas, S., Coene, K. L. M., Davis, E. E., Alanay, Y., Ogur, G., et al. (2011). KIF7 Mutations Cause Fetal Hydrolethalus and Acrocallosal Syndromes. Nat. Genet. 43 (6), 601–606. doi:10.1038/ng.826
Räsänen, J., Huovinen, J., Korhonen, V. E., Junkkari, A., Kastinen, S., Komulainen, S., et al. (2020). Diabetes Is Associated with Familial Idiopathic normal Pressure Hydrocephalus: a Case-Control Comparison with Family Members. Fluids Barriers CNS 17 (1), 57. doi:10.1186/s12987-020-00217-0
Rasmussen, M. K., Mestre, H., and Nedergaard, M. (2018). The Glymphatic Pathway in Neurological Disorders. Lancet Neurol. 17 (11), 1016–1024. doi:10.1016/S1474-4422(18)30318-1
Rekate, H. L. (2008). The Definition and Classification of Hydrocephalus: a Personal Recommendation to Stimulate Debate. Fluids Barriers CNS 5, 2. doi:10.1186/1743-8454-5-2
Ringstad, G., and Eide, P. K. (2020). Cerebrospinal Fluid Tracer Efflux to Parasagittal Dura in Humans. Nat. Commun. 11 (1), 354. doi:10.1038/s41467-019-14195-x
Ringstad, G., Vatnehol, S. A. S., and Eide, P. K. (2017). Glymphatic MRI in Idiopathic normal Pressure Hydrocephalus. Brain 140 (10), 2691–2705. doi:10.1093/brain/awx191
Román, G. C., Jackson, R. E., Fung, S. H., Zhang, Y. J., and Verma, A. K. (2019). Sleep-Disordered Breathing and Idiopathic Normal-Pressure Hydrocephalus: Recent Pathophysiological Advances. Curr. Neurol. Neurosci. Rep. 19 (7). doi:10.1007/s11910-019-0952-9
Román, G. C., Verma, A. K., Zhang, Y. J., and Fung, S. H. (2018). Idiopathic normal-pressure Hydrocephalus and Obstructive Sleep Apnea Are Frequently Associated: A Prospective Cohort Study. J. Neurol. Sci. 395, 164–168. doi:10.1016/j.jns.2018.10.005
Roux, E., Bougaran, P., Dufourcq, P., and Couffinhal, T. (2020). Fluid Shear Stress Sensing by the Endothelial Layer. Front. Physiol. 11, 861. doi:10.3389/fphys.2020.00861
Sakata-Haga, H., Sawada, K., Ohnishi, T., and Fukui, Y. (2004). Hydrocephalus Following Prenatal Exposure to Ethanol. Acta Neuropathol. 108 (5), 393–398. doi:10.1007/s00401-004-0901-8
Satz, J. S., Barresi, R., Durbeej, M., Willer, T., Turner, A., Moore, S. A., et al. (2008). Brain and Eye Malformations Resembling Walker-Warburg Syndrome Are Recapitulated in Mice by Dystroglycan Deletion in the Epiblast. J. Neurosci. 28 (42), 10567–10575. doi:10.1523/JNEUROSCI.2457-08.2008
Shaheen, R., Sebai, M. A., Patel, N., Ewida, N., Kurdi, W., Altweijri, I., et al. (2017). The Genetic Landscape of Familial Congenital Hydrocephalus. Ann. Neurol. 81 (6), 890–897. doi:10.1002/ana.24964
Shu, T., Butz, K. G., Plachez, C., Gronostajski, R. M., and Richards, L. J. (2003). Abnormal Development of Forebrain Midline Glia and Commissural Projections inNfiaKnock-Out Mice. J. Neurosci. 23 (1), 203–212. doi:10.1523/jneurosci.23-01-00203.2003
Silverberg, G. D., Huhn, S., Jaffe, R. A., Chang, S. D., Saul, T., Heit, G., et al. (2002). Downregulation of Cerebrospinal Fluid Production in Patients with Chronic Hydrocephalus. J. Neurosurg. 97 (6), 1271–1275. doi:10.3171/jns.2002.97.6.1271
Snedeker, J., Schock, E. N., Struve, J. N., Chang, C.-F., Cionni, M., Tran, P. V., et al. (2017). Unique Spatiotemporal Requirements for Intraflagellar Transport Genes during Forebrain Development. PLoS One 12 (3), e0173258. doi:10.1371/journal.pone.0173258
Socci, D. J., Bjugstad, K. B., Jones, H. C., Pattisapu, J. V., and Arendash, G. W. (1999). Evidence that Oxidative Stress Is Associated with the Pathophysiology of Inherited Hydrocephalus in the H-Tx Rat Model. Exp. Neurol. 155 (1), 109–117. doi:10.1006/exnr.1998.6969
Somera, K. C., and Jones, H. (2002). Subcommissural Organ Dysfunction in H-Tx Rats with Early-Onset Hydrocephalus. Eur. J. Pediatr. Surg. 12 (Suppl. 1), S45–S47.
Somera, K. C., and Jones, H. C. (2004). Reduced Subcommissural Organ Glycoprotein Immunoreactivity Precedes Aqueduct Closure and Ventricular Dilatation in H-Tx Rat Hydrocephalus. Cel Tissue Res. 315 (3), 361–373. doi:10.1007/s00441-003-0843-9
Sotak, B. N., and Gleeson, J. G. (2012). Can't Get There from Here: Cilia and Hydrocephalus. Nat. Med. 18 (12), 1742–1743. doi:10.1038/nm.3011
Stoll, C., Alembik, Y., Dott, B., and Roth, M. P. (1992). An Epidemiologic Study of Environmental and Genetic Factors in Congenital Hydrocephalus. Eur. J. Epidemiol. 8 (6), 797–803. doi:10.1007/bf00145322
Takagishi, M., Sawada, M., Ohata, S., Asai, N., Enomoto, A., Takahashi, K., et al. (2017). Daple Coordinates Planar Polarized Microtubule Dynamics in Ependymal Cells and Contributes to Hydrocephalus. Cel Rep. 20 (4), 960–972. doi:10.1016/j.celrep.2017.06.089
Tarasoff-Conway, J. M., Carare, R. O., Osorio, R. S., Glodzik, L., Butler, T., Fieremans, E., et al. (2015). Clearance Systems in the Brain-Implications for Alzheimer Disease. Nat. Rev. Neurol. 11 (8), 457–470. doi:10.1038/nrneurol.2015.119
Tarpey, P. S., Stevens, C., Teague, J., Edkins, S., O’Meara, S., Avis, T., et al. (2006). Mutations in the Gene Encoding the Sigma 2 Subunit of the Adaptor Protein 1 Complex, AP1S2, Cause X-Linked Mental Retardation. Am. J. Hum. Genet. 79 (6), 1119–1124. doi:10.1086/510137
Thavarajasingam, S. G., El-Khatib, M., Rea, M., Russo, S., Lemcke, J., Al-Nusair, L., et al. (2021). Clinical Predictors of Shunt Response in the Diagnosis and Treatment of Idiopathic normal Pressure Hydrocephalus: a Systematic Review and Meta-Analysis. Acta Neurochir 163, 2641–2672. doi:10.1007/s00701-021-04922-z
Verhagen, W. I. M., Bartels, R. H. A. M., Fransen, E., van Camp, G., Renier, W. O., and Grotenhuis, J. A. (1998). Familial Congenital Hydrocephalus and Aqueduct Stenosis with Probably Autosomal Dominant Inheritance and Variable Expression. J. Neurol. Sci. 158 (1), 101–105. doi:10.1016/s0022-510x(98)00097-5
Villani, R., Tomei, G., Gaini, S. M., Grimoldi, N., Spagnoli, D., and Bello, L. (1995). Long-term Outcome in Aqueductal Stenosis. Child's Nerv Syst. 11 (3), 180–185. doi:10.1007/bf00570262
Vincent, C., Kalatzis, V., Compain, S., Levilliers, J., Slim, R., Graia, F., et al. (1994). A Proposed New Contiguous Gene Syndrome on 8q Consists of Branchio-Oto-Renal (BOR) Syndrome, Duane Syndrome, a Dominant Form of Hydrocephalus and Trapeze Aplasia; Implications for the Mapping of the BOR Gene. Hum. Mol. Genet. 3 (10), 1859–1866. doi:10.1093/hmg/3.10.1859
Wagner, C., Batiz, L. F., Rodríguez, S., Jiménez, A. J., Páez, P., Tomé, M., et al. (2003). Cellular Mechanisms Involved in the Stenosis and Obliteration of the Cerebral Aqueduct ofhyhMutant Mice Developing Congenital Hydrocephalus. J. Neuropathol. Exp. Neurol. 62 (10), 1019–1040. doi:10.1093/jnen/62.10.1019
Wallmeier, J., Frank, D., Shoemark, A., Nöthe-Menchen, T., Cindric, S., Olbrich, H., et al. (2019). De Novo Mutations in FOXJ1 Result in a Motile Ciliopathy with Hydrocephalus and Randomization of Left/Right Body Asymmetry. Am. J. Hum. Genet. 105 (5), 1030–1039. doi:10.1016/j.ajhg.2019.09.022
Wang, R., Chen, H., Wang, X., Huang, S., Xie, A., and Wu, X. (2021). Prenatal Diagnosis of a Nonsense Mutation in the L1CAM Gene Resulting in Congenital Hydrocephalus: A Case Report and Literature Review. Exp. Ther. Med. 22 (6), 1416. doi:10.3892/etm.2021.10807
Willaredt, M. A., Hasenpusch-Theil, K., Gardner, H. A. R., Kitanovic, I., Hirschfeld-Warneken, V. C., Gojak, C. P., et al. (2008). A Crucial Role for Primary Cilia in Cortical Morphogenesis. J. Neurosci. 28 (48), 12887–12900. doi:10.1523/JNEUROSCI.2084-08.2008
Willems, P. J., Brouwer, O. F., Dijkstra, I., Wilmink, J., Opitz, J. M., and Reynolds, J. F. (1987). X-linked Hydrocephalus. Am. J. Med. Genet. 27 (4), 921–928. doi:10.1002/ajmg.1320270419
Wilsch-Bräuninger, M., and Huttner, W. B. (2021). Primary Cilia and Centrosomes in Neocortex Development. Front. Neurosci. 15, 755867. doi:10.3389/fnins.2021.755867
Wilson, S. L., Wilson, J. P., Wang, C., Wang, B., and McConnell, S. K. (2012). Primary Cilia and Gli3 Activity Regulate Cerebral Cortical Size. Devel Neurobio 72 (9), 1196–1212. doi:10.1002/dneu.20985
Xie, L., Kang, H., Xu, Q., Chen, M. J., Liao, Y., Thiyagarajan, M., et al. (2013). Sleep Drives Metabolite Clearance from the Adult Brain. Science 342 (6156), 373–377. doi:10.1126/science.1241224
Xu, Z., Xiao, N., Chen, Y., Huang, H., Marshall, C., Gao, J., et al. (2015). Deletion of Aquaporin-4 in APP/PS1 Mice Exacerbates Brain Aβ Accumulation and Memory Deficits. Mol. Neurodegeneration 10, 58. doi:10.1186/s13024-015-0056-1
Yamada, S., Ishikawa, M., and Nozaki, K. (2021). Exploring Mechanisms of Ventricular Enlargement in Idiopathic normal Pressure Hydrocephalus: a Role of Cerebrospinal Fluid Dynamics and Motile Cilia. Fluids Barriers CNS 18 (1), 20. doi:10.1186/s12987-021-00243-6
Yang, H. W., Lee, S., Yang, D., Dai, H., Zhang, Y., Han, L., et al. (2021a). Deletions in CWH43 Cause Idiopathic normal Pressure Hydrocephalus. EMBO Mol. Med. 13 (3), e13249. doi:10.15252/emmm.202013249
Yang, J., Hu, X., Ma, J., and Shi, S.-H. (2021b). Centrosome Regulation and Function in Mammalian Cortical Neurogenesis. Curr. Opin. Neurobiol. 69, 256–266. doi:10.1016/j.conb.2021.06.003
Yang, J., Simonneau, C., Kilker, R., Oakley, L., Byrne, M. D., Nichtova, Z., et al. (2019). Murine MPDZ ‐linked Hydrocephalus Is Caused by Hyperpermeability of the Choroid Plexus. EMBO Mol. Med. 11 (1). doi:10.15252/emmm.201809540
Zechel, J., Gohil, H., Lust, W. D., and Cohen, A. (2002). Alterations in Matrix Metalloproteinase-9 Levels and Tissue Inhibitor of Matrix Metalloproteinases-1 Expression in a Transforming Growth Factor-? Transgenic Model of Hydrocephalus. J. Neurosci. Res. 69 (5), 662–668. doi:10.1002/jnr.10326
Zhang, J., Williams, M. A., and Rigamonti, D. (2006). Genetics of Human Hydrocephalus. J. Neurol. 253 (10), 1255–1266. doi:10.1007/s00415-006-0245-5
Zhang, X. J., Guo, J., and Yang, J. (20202020). Cerebrospinal Fluid Biomarkers in Idiopathic normal Pressure Hydrocephalus. Neuroimmunology and Neuroinflammation 7, 109–119. doi:10.20517/2347-8659.2019.018
Keywords: etiological study of hydrocephalus, molecular mechanism, risk factors, pathogenesis, CSF
Citation: Li J, Zhang X, Guo J, Yu C and Yang J (2022) Molecular Mechanisms and Risk Factors for the Pathogenesis of Hydrocephalus. Front. Genet. 12:777926. doi: 10.3389/fgene.2021.777926
Received: 18 September 2021; Accepted: 03 December 2021;
Published: 03 January 2022.
Edited by:
Babak Behnam, National Sanitation Foundation International, United StatesReviewed by:
Xuanye Cao, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2022 Li, Zhang, Guo, Yu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Yang, anVueWFuZ3lhbmdAaG90bWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.