 Yanjing Wu
Yanjing Wu Yongle Wu
Yongle Wu Kun Liu
Kun Liu Hui Liu
Hui Liu Shanshan Wang
Shanshan Wang Jian Huang
Jian Huang Huiguo Ding
Huiguo Ding- 1Department of Gastroenterology and Hepatology, Beijing You’an Hospital, Affiliated with Capital Medical University, Beijing, China
- 2Department of General Surgery, Beijing Friendship Hospital, Affiliated with Capital Medical University, Beijing, China
- 3Department of Pathology, Beijing You’an Hospital, Affiliated with Capital Medical University, Beijing, China
- 4Beijing Institute of Hepatology, Beijing You’an Hospital, Affiliated with Capital Medical University, Beijing, China
- 5Experimental Center, Beijing Friendship Hospital, Affiliated with Capital Medical University, Beijing, China
Background and Aims: The multiple renal cysts (MRC) occur in some patients with noncirrhotic portal hypertension (NCPH) could be a subset of ciliopathy. However, the potential genetic influencers and/or determinants in NCPH with MRC are largely unknown. The aim of this study was to explore the potential candidate variants/genes associated with those patients.
Methods: 8,295 cirrhotic patients with portal hypertension were enrolled in cohort 1 and 267 patients affected with NCPH were included in cohort 2. MRC was defined as at least two cysts in both kidneys within a patient detected by ultrasonography or computed tomography. Whole-genome sequencing (WGS) was performed in nine patients (four from cohort 1 and five from cohort 2). Then we integrated WGS and publicly available single-cell RNA sequencing (scRNA-seq) to prioritize potential candidate genes. Genes co-expressed with known pathogenic genes within same cell types were likely associated NCPH with MRC.
Results: The prevalence of MRC in NCPH patients (19.5%, 52/267) was significantly higher than cirrhotic patients (6.2%, 513/8,295). Further, the clinical characteristics of NCPH patients with MRC were distinguishable from cirrhotic patients, including late-onset, more prominent portal hypertension however having preserved liver functions. In the nine whole genome sequenced patients, we identified three patients with early onset harboring compound rare putative pathogenic variants in the known disease gene PKHD1. For the remaining patients, by assessing cilia genes profile in kidney and liver scRNA-seq data, we identified CRB3 was the most co-expressed gene with PKHD1 that highly expressed in ureteric bud cell, kidney stromal cell and hepatoblasts. Moreover, we found a homozygous variant, CRB3 p.P114L, that caused conformational changes in the evolutional conserved domain, which may associate with NCPH with MRC.
Conclusion: ScRNA-seq enables unravelling cell heterogeneity with cell specific gene expression across multiple tissues. With the boosting public accessible scRNA-seq data, we believe our proposed analytical strategy would effectively help disease risk gene identification.
Introduction
Cirrhotic portal hypertension complicated esophageal-gastric variceal bleeding (EGVB), ascites, hepatic encephalopathy (HE), acute kidney injury (AKI) or hepatorenal syndrome (HRS) and splenomegaly accompanied by severe liver disfunctions, is almost accounted for 80–85%. In clinical practice, a small number of patients present with portal hypertension, such as splenomegaly and or EGVB, HE, their clinical manifestations are similar to the liver cirrhosis,but in fact, these patients do not have liver cirrhosis, that is non-cirrhotic portal hypertension (NCPH) (Khanna and Sarin, 2019; Gao et al., 2020). Currently, chronic infections, autoimmune disorders, and genetic determinants have been reported to be associated with pathogenesis of NCPH (Vilarinho et al., 2016; Wu et al., 2019). Interestingly, the NCPH is common in cystic fibrosis-associated liver disease (Boëlle et al., 2019). Therefore, we speculate that liver disease in multiple renal cysts (MRC) present as NCPH, except for a subset of patients with ciliopathy affected by hepatorenal fibrocystic diseases (HFDs), such as autosomal recessive polycystic kidney disease (ARPKD) or Caroli syndrome (Abdul Majeed et al., 2020; McConnachie et al., 2021). HFDs are a group of ciliopathies and genetic disorders that involve developmental abnormalities in the portobiliary system in association with fibrocystic degeneration of the kidney (Lasagni et al., 2021). HFDs can cause enlarged kidneys, cyst formation, biliary duct dilation, and congenital hepatic fibrosis (CHF), resulting in portal hypertension (Myram et al., 2021). Therefore, patients with NCPH with MRC may be a non-classical genetic mutations or HFD phenotype.
Currently, single-cell RNA sequencing (scRNA-seq) has revolutionized developmental biology and genomics. ScRNA-seq is a powerful tool that can be used to elucidate the cellular composition in the interest tissue, to define undescribed rare cell subsets, to dissect regulators controlling cell fate transition, to pinpoint cell-type specific responses to stress or stimulation, and to identify mechanisms of cell-cell crosstalk (Han et al., 2020). In this study, we applied a novel analytic approach that integrated whole-genome sequencing (WGS) and scRNA-seq data to survey potential genetic modifiers or candidate disease genes in NCPH patients with MRC.
Methods
Study Design and Patients
Total 8,295 cirrhotic portal hypertensive patients, diagnosed by medical history, signs and imaging of portal hypertension according to chinese guidelines on the management of liver cirrhosis (Xu et al., 2020), were enrolled in cohort 1. The etiologies of liver cirrhosis included hepatitis B (65.04%), hepatitis C (15.79%), alcoholic cirrhosis (12.35%), and autoimmune liver disease (6.82%). Two hundred sixty-seven patients with NCPH in cohort 2, confirmed by radiologists, hepatologists and pathologists based on enhanced computed tomography (CT) or nuclear magnetic resonance imaging (MRI) and/or pathology according to guidelines on the management of NCPH of EASL (Khanna and Sarin, 2014). The signs and symptoms, laboratory and endoscopy data were obtained from Electronic Medical Record Management System (EMRS). The portal and splenic vein diameter and splenic thickness were measured using abdominal ultrasonography. Model for end-stage liver disease (MELD) and Child-Pugh score were assessed for the severity of cirrhosis. Transient elastography liver stiffness measurement (LSM) was performed using FibroScan™ (Echosens, Paris, France). The MRC were defined as more than two cysts in both kidneys detected by ultrasonography or CT.
The study protocol was performed in compliance with the Declaration of Helsinki and approved by the Ethics Committees of Beijing You’an Hospital Capital Medical University. Signed informed consent was obtained from each participant for using samples, materials and publication.
Whole-Genome DNA Sequencing
The peripheral blood of nine patients with MRC (four from cohort 1 and five from cohort 2 separately) were collected for WGS to explore potential genetic modifiers or candidate disease genes. First, the genomic DNA was extracted from the peripheral blood of those patients. The whole-genome DNA Sequencing libraries were prepared according to the manufacturer’s instructions. The raw reads were produced by a BGISEQ-500 sequencer at an average depth of 40×. The rare putatively pathogenic variants were validated by Sanger sequencing.
Genomic DNA Analysis
Sequencing data were quality controlled with adapter and aligned to human reference genome build hg19 (http://www.gencodegenes.org/releases/19.html) with BWA aligner (Li and Durbin, 2009). The GATK best practice Haplotype Caller pipeline was implemented for SNV and indel calling (Li et al., 2009; McKenna et al., 2010). SV was called with lumpy software, and CNV was detected with FreeC software (Layer et al., 2014). All SNV variants were annotated using ANNOVAR for bioinformatics analysis (Wang et al., 2010). Several genomic databases, including the 1,000 Genomes (1000G), ExAC (Exome Aggregation Consortium), Exome Sequencing Project (ESP), gnom AD (both WES and WGS databases), and CG46, were used to assess the variant frequency in the population. MCAP, SIFT, Polyphen2-HDIV, Polyphen2-HVAR, MutationTaster, MutationAssessor, and Clinvar were implemented to annotate the effect of missense variants. GERPs were used to evaluate the conservation of the variant locus. Rare putative pathogenic variants were filtered as follows (Gao et al., 2020): the allele frequency of the candidate variant should be lower than 0.01 among 1000G, ExAC, ESP 6500, Genome Aggregation (GA) and Complete Genomics 46 (CG46) Databases (Khanna and Sarin, 2019); amino acid changing variants were kept, and GERP scores should be higher than 2.0 (Vilarinho et al., 2016); truncating variants were kept, and for missense variants, MCAP scores higher than 0.6 were automatically kept, whereas for other variants, the effect should be annotated as “Deleterious” or “Highly pathogenic” by at least two software programs for MCAP scores between 0.025 and 0.6. PKHD1 protein domain prediction was obtained using SMART (Letunic et al., 2021). Cilia genes were obtained from Syscilia.org for downstream analysis.
Single-Cell RNA Sequencing Analysis
Five sets of scRNA-seq data were used in this study. Summarized gene expression matrices derived from multiple organs from human fetuses were obtained from the GEO database via accession number GSE156793 (Cao et al., 2020). Single-cell expression data from kidneys were generated from mice under accession number GSE140023 (Conway et al., 2020). The adult human kidney and human liver single cell transcriptome was achieved by accession GSE114530 (Hochane et al., 2019), GSE131685 (He et al., 2020) and GSE159929 (Liao et al., 2020), respectively. The single cell RNA sequencing analysis was implemented with Seurat package (Version 3.9.9). Cells were discarded according to the following criteria (Gao et al., 2020): cells that had fewer than 400 genes (UMI >0) (Khanna and Sarin, 2019); cells that had fewer than 600 UMI or over 10,000 UMI; and (Vilarinho et al., 2016) cells that had more than 15% mitochondrial UMI counts. After the above quality control, for mouse kidney, human fetal kidney, human adult kidney and human liver scRNA-seq analysis, we performed log-normalization with the “vst” method and identified 2000, 3,000, 3,000 and 3,000 variable features, respectively. We then scaled by setting the parameter “vars.to.regress” to “percent.mito” and “nCount_RNA”. Principal component analysis (PCA) was performed using the “RunPCA” function. The number of PCs was chosen by visualization plot with the “ElbowPlot” function. A shared nearest neighbor (SNN) graph was constructed using the “FindNeighbors” function with the top 40 PCs, then cells were clustered by the “FindClusters” function with the “resolution” parameter set to 0.5. The “RunUMAP” function was used for the visualization plot with the “umap-learn” method, setting “n.neighbors” to 40L, “dims” to 1:40, and “min.dist” to 0.3. Marker genes for each cluster were detected using the “FindAllMarkers” function, setting the parameter “min.pct” to 0.3 and “logfc.threshold” to 0.6. Subsequently, cell clusters were annotated manually to the major cell types according to known markers. Any cluster with multiple markers of two types of cells was manually discarded as a doublet.
Protein Conformation Modeling
The full-length human CRB3 was not annotated in the PDB Database; thus, the intact wild-type and mutated CRB3 protein sequences were annotated by Phyre2 (Kelley et al., 2015). Then, protein conformational alteration was predicted using Chimera software (Pettersen et al., 2004).
Data Visualization and Statistics
Microsoft R Open (version 3.6.1, https://mran.microsoft.com/) was used. The R packages ggplot 2 (version 3.1.0) and pheatmap (version 1.0.12) were used to generate graphs of the data. Continuous variables were compared using independent T test if data were normally distributed or Mann Whitney U test. The categorical variables were compared using X2 tests performed with SPSS 22.0 (IBM, United States).
Results
Prevalence and Clinical Characteristics of NCPH Patients With MRC
The prevalence of MRC in NCPH patients accounted for 19.5% (52/267). It was significantly higher than that in cirrhotic patients with portal hypertension (6.2%, 513/8,295), p < 0.05. The clinical characteristics of NCPH patients with MRC were compared with 92 hepatitis B virous (HBV) related cirrhosis without MRC, which randomly selected (1:2) in cohort 1 (Table 1). The NCPH patients with MRC had a relatively older onset complications of portal hypertension (p < 0.05). In terms of the manifestations of portal hypertension, the proportion of EGVB was prominent (p < 0.05), while ascites was less (p < 0.001). The platelet counts were higher than that of hepatitis B cirrhosis (p < 0.001). Although there was no significant difference in LSM between the two groups, the ratio of platelet counts/LSM in NCPH patients with MRC was significantly higher than it in hepatitis B cirrhosis without MRC patients (p < 0.001). The clinical characteristics of nine patients with MRC enrolled for WGS were summarized in Table 2. Three patients (P1-3) showed CHF by liver biopsy (Figure 1A), in which two patients exhibit early-onset in childhood or adolescence and underwent splenectomy procedure before their visit to our hospital. The endoscopy and CT imaging confirmed EGV, dilated bile ducts and multiple renal cysts in those patients (Figures 1B,C).
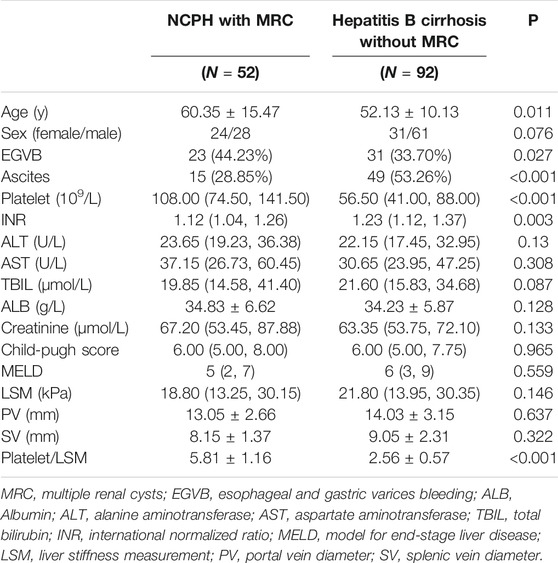
TABLE 1. Comparison of clinical characteristics between NCPH patients with MRC and Hepatitis B cirrhosis without MRC.
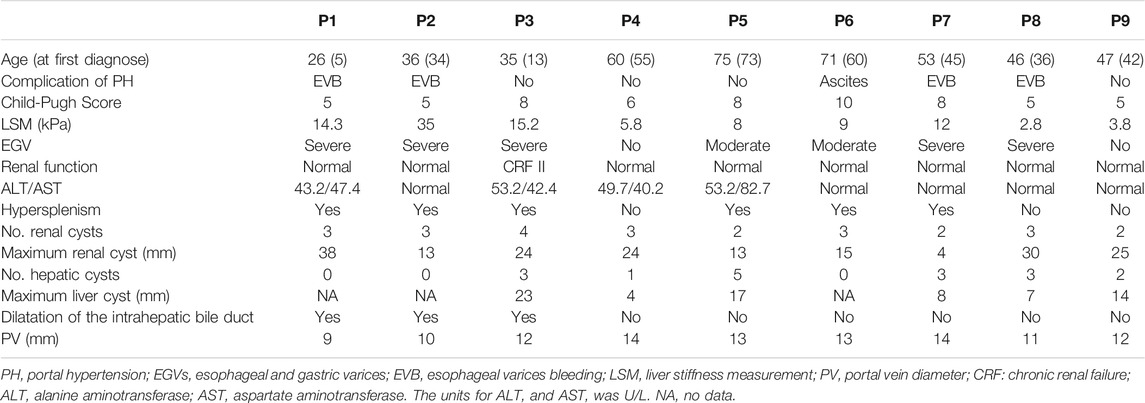
TABLE 2. The clinical data for nine patients with MRC enrolled for whole-genome sequencing.
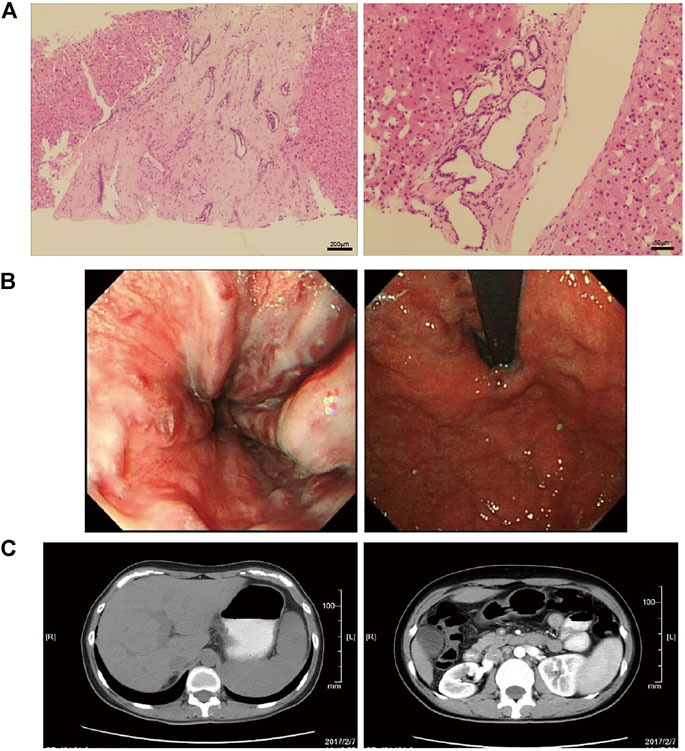
FIGURE 1. The pathological and clinical presentation of one CHF patient with NCPH. (A) The left panel shows diffuse fibrosis in the liver. The right panel shows a small bile duct hamartoma. (B) Endoscopy showed esophageal varices (left) and gastric varices (right). (C) The abdomen CT images show slight dilatation of the intrahepatic bile duct and splenomegaly (left), and multiple renal cysts in the bilateral kidneys (right).
The others NCPH patients (P4 and P5) showed late-onset of complication, their clinical manifestations, including large hepatorenal cysts and normal LSM, implied underlying HFDs although there is no pathological evidence. Moreover, four HBV-positive patients (P6-P9) were diagnosed at middle age with normal liver function may also pinpoint to the need of dissecting genetic factors for HFD phenotypic expression.
Early Onset Harbored Compound Rare Pathogenic Variants in PKHD1
The copy number variations or structural variants spanning known HFD-related genes were not observed. Subsequently, we identified all gene mutations in the known Caroli syndrome-causing gene PKHD1 in 3 CHF patients with NCPH (Supplementary Table S1). All missense mutations in PKHD1 had GERP scores higher than 5.4, indicating that these mutations were in evolutionarily highly conserved regions. Moreover, all three patients harbored an additional truncation mutation, which were not reported in any public databases. Therefore, we assumed that the patients carried recessive mutations in PKHD1, and validated the mutations by Sanger sequencing (Figure 2A). In addition, analysis of PKHD1 mutation distributions in different protein domains showed that all mutations were located in the extracellular domain (Figure 2B). Further, by using scRNA-seq data to explore PKHD1 expression, we found that PKHD1 was highly expressed in ureteric bud cells and stromal cells and moderately expressed in metanephric cells in the kidney. In contrast, PKHD1 was highly expressed on hepatoblasts in the liver (Figure 2C). Moreover, we also detected PKD1 compound mutations, which may explain that the patient P1 had early disease onset at the age of five and large renal cysts (38 mm).

FIGURE 2. Gene mutations analysis of PKHD1. (A) The rare putative pathogenic variants in PKHD1 were validated by Sanger sequencing. (B) Schematic representation of variant distributions in the PKHD1 domain. (C) Violin plot of PKHD1 expression in mouse kidney and human liver tissues by scRNA-seq analysis. The Y axis was normalized expression level for PKHD1 in different cells.
Potential Candidate Genes Associated With HFDs Phenotype
To narrow the potential HFD phenotype-associated genes, we retrieved all known cilia genes from the literature and the European project SYSCILIA (Boldt et al., 2016). After prioritizing candidate disease-associated cilia genes by scRNA-seq analysis, the cilia-related mutations derived from the enrolled patients were listed in Supplementary Table S1, such as CRB3 p.P114L. We first plotted the expression for candidate cilia genes in multiple human fetal organs, and annotated different cell types, particularly in the kidneys and liver (Figures 3A,B). We further assessed the cilia genes of cell clustering and annotated different cell type in adult mice kidney and adult human liver using uniform manifold approximation and projection (UMAP). We found that CRB3, TUBA4A, PTCH1 and CEP290 co-expressed with PKHD1 (Figures 3C,D). Similar co-expression pattern were also seen in human fetal kidney (Figures 3E,F) and adult kidney (Figures 3G,H). Followed by cell clustering and annotation in adult human liver, the CRB3 and PKHD1 co-expression were spotted in hepatoblasts (Figures 3I,J). To further investigate the functional effect of the CRB3 p.P114L variants, we performed protein structure remodeling and predicted that the mutation would lead to protein conformational alterations in the PDZ-domain (Figure 4A). The bona fide tight junction assembly acquired the capacity for PRKC1/PARD6A complex translocation to the apical surface by interacting with the CRB3 C-terminus. Hence, we speculated that this mutation might ultimately blocking CRB3 function. With additional scRNA-seq data, we believe that this newly proposed analytical strategy may help clinicians and geneticists to map disease-related genes (Figure 4B).
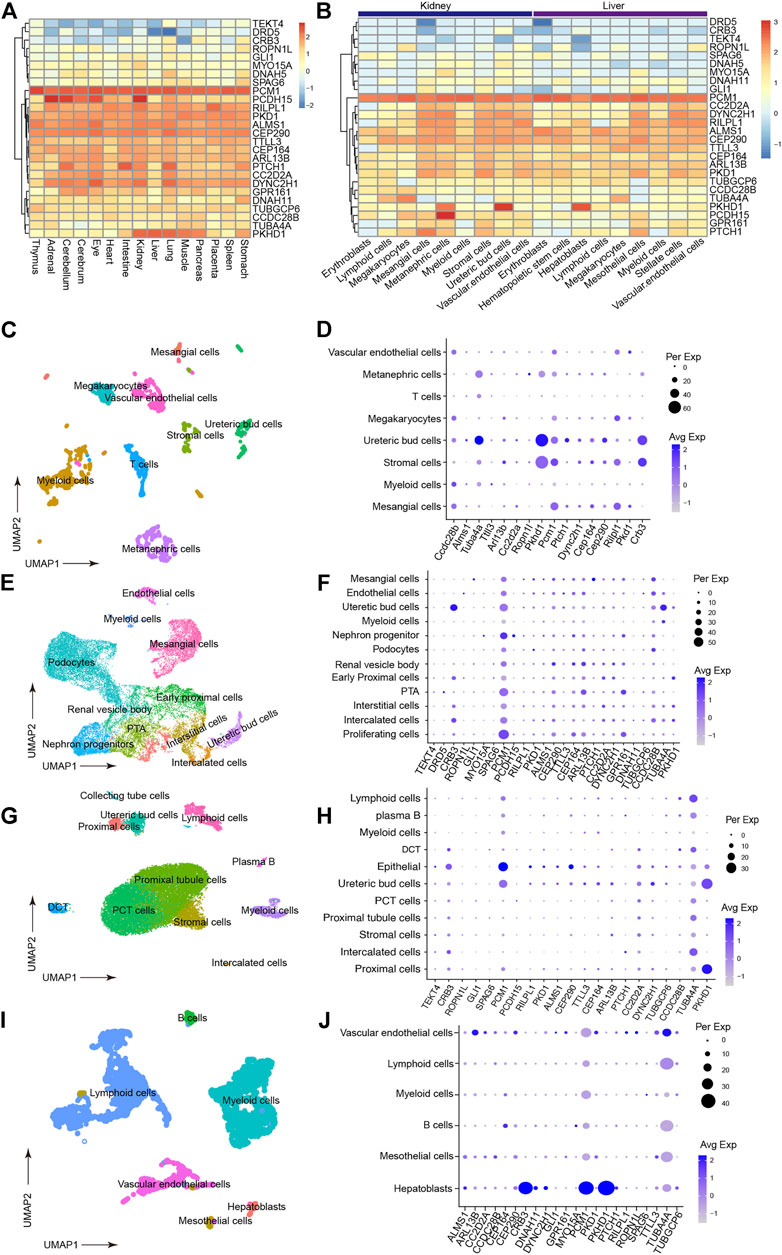
FIGURE 3. Prioritization of detected cilia genes with scRNA-seq. (A) Heatmap of detected cilia gene expression in multiple human fetal organs using data under accession GSE156793. Gene expression have been scaled with normalized read counts for each gene. (B) Heatmap of detected cilia gene expression in different cell types from human fetal kidney and liver tissues (from GSE156793). Gene expression have been scaled with normalized read counts for each gene. (C) UMAP plot showing the clustering of different cell types in mouse kidneys (from GSE140023). (D) Dot plot of cilia genes in mouse kidney (from GSE140023). The expression inferred Pkhd1, Crb3 and Tuba4a had similar expression pattern. Dot size indicated percentage of cell which expressed gene of interest and color indicated expression level. (E) UMAP plot showing the clustering of different cell types in human fetal kidney (from GSE114530). (F) Dot plot reflecting the cell type expression patterns of the detected cilia genes (from GSE114530). This result supported that PKHD1 and CRB3 had similar expression pattern. (G) UMAP plot showing the clustering of different cell types in human fetal kidney (from GSE131685). (H) Dot plot reflecting the cell type expression patterns of the detected cilia genes (from GSE131685). (I) UMAP plot showing the clustering of different cell types in the adult human liver (from GSE159929). (J) Dot plot reflecting the cell type expression patterns of the detected cilia genes (from GSE159929).
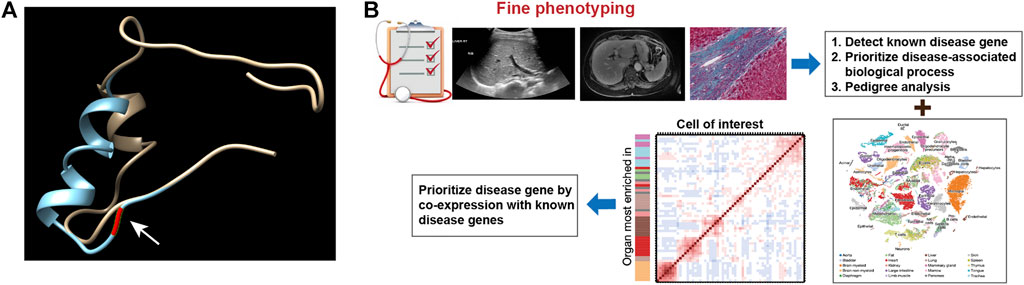
FIGURE 4. (A) Prediction of the conformation of CRB3 protein. The yellow chain shows the wild-type protein, and the blue chain shows the mutated protein. Residue 114 is pointed by arrow and highlighted in red on the protein backbone. (B) Schematic illustration of the novel analytic strategy to identify disease-related genes.
Discussions
Currently, the etiologies and genetic pathogenesis of NCPH with CHF, especially idiopathic non-cirrhotic portal hypertension (INCPH), have not been fully elucidated (Lanktree et al., 2021). The MRC is more common in CHF and NCPH (Vilarinho et al., 2016; Boëlle et al., 2019; McConnachie et al., 2021). We have reasons to believe that MRC, as a cilia, may have genetic variation disorders, especially non-classical genetic mutations of HFD phenotype in INCPH with MRC patients. In this study, the prevalence of MRC in NCPH patients was accounted almost for 19.5%, which was significantly higher than the cirrhotic portal hypertension patients with known etiologies. The clinical characteristics of NCPH with MRC from these data were older-onset of the complications of portal hypertension, obvious manifestations of portal hypertension, such as EGVB, and having preserved liver functions.
Genome-wide single-cell analysis represents the ultimate frontier of genomics research (Boldt et al., 2016). In particular, scRNA-seq studies have been boosted in the last few years of new technologies enabling the study of the transcriptomic landscape of thousands of single cells in complex pathogenesis of diseases (Ying et al., 2021). Owing to the dramatic improvement in scRNA-seq technology, especially integrating WGS and scRNA-seq, tissue-specific expression at the single-cell level has improved our understanding of biological processes (Zeggini et al., 2019). In this study, WGS and scRNA-seq were performed to survey potential genetic modifiers or candidate disease genes in NCPH patients with MRC. The results showed that genes also expressed in ureteric bud cells, stromal cells, and hepatoblasts may have additive effects on NCPH with MRC. We also found that CRB3, TUBA4A, PTCH1 and CEP290 co-expressed with PKHD1 at hepatoblasts in liver using UMAP. Interestingly, we discovered that patient P5 carried a homozygous candidate mutation in CRB3 without family history of MRC. The CRB3 encodes an apical transmembrane protein that regulates the morphogenesis of tight junctions in mammalian epithelial cells (Lemmers et al., 2004). CRB3 protein plays an important role in apicobasal polarity formation, such as cyst formation (Hurd et al., 2003). Furthermore, CRB3 participates in interactions with TAZ/YAP, thereby affecting transforming growth factor (TGF)-β signaling. Disruption of CRB3 function enhances TGF-β signaling and predisposes cells to TGF-β-mediated epithelial-to-mesenchymal transition (Varelas et al., 2010). Therefore, loss of function of CRB3 could potentially be linked to cyst formation and/or fibrosis. Importantly, further narrowing of the candidate gene selection showed that CRB3 could be a novel disease risk gene for HFDs. Although patients carrying a homozygous mutation in CRB3 showed late disease onset, this mutation affects PDZ domain conformation and might alleviate protein function rather than cause complete loss of function. However, further studies are needed for functional validation of the pathogenicity of this gene.
One of the limitations of this study is the lack of parental genomic materials and family pedigree of MRC, making it challenging to further prioritize selected candidates. In addition, the present study lacks WGS analysis in MRC patients with mutations in known pathogenic genes, such as ARPKD phenotypes associated genes and PKHD1 or PKD genes because of small sample size of NCPH with MRC and hepatitis B cirrhosis patients without MRC. Furthermore, the novel identified CRB3 p.P114L variant has not been undergone biological function study, and we will conduct the research in the future.
Conclusion
CRB3 gene is commonly co-expressed with PKHD1 in NCPH with MRC. The homozygous variant in CRB3 may be associated with genetic pathogenesis of NCPH with MRC. Therefore, we speculate that there may be non-classical genetic mutations in NCPH patients with MRC. CRB3 may be a novel homozygous candidate gene mutation.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee Review Board of You’an Hospital, Capital Medical University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YaW, YoW, and HD designed the study; JH and HD supervised the study; YoW, HL, and SW performed experiments and clinical examinations; KL and JH performed data analysis; YaW performed data interpretation; YoW and KL wrote the manuscript. JH and HD made revisions. HD providing funding. All authors read the manuscript and approved it before submission.
Funding
This work was supported by the National Natural Science Foundation (grant no. 81970525), and Digestive Medical Coordinated Development Center of Beijing Hospitals Authority (grant no. XXZ0801).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We sincerely appreciated the enrolled patients for their support and understanding. Additionally, we also would like to thank Editage for language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.775470/full#supplementary-material
References
Abdul Majeed, N., Font-Montgomery, E., Lukose, L., Bryant, J., Veppumthara, P., Choyke, P. L., et al. (2020). Prospective Evaluation of Kidney and Liver Disease in Autosomal Recessive Polycystic Kidney Disease-Congenital Hepatic Fibrosis. Mol. Genet. Metab. 131, 267–276. doi:10.1016/j.ymgme.2020.08.006
Boëlle, P. Y., Debray, D., Guillot, L., Clement, A., and Corvol, H. (2019). Cystic Fibrosis Liver Disease: Outcomes and Risk Factors in a Large Cohort of French Patients. Hepatology 69, 1648–1656. doi:10.1002/hep.30148
Boldt, K., van Reeuwijk, J., van Reeuwijk, J., Lu, Q., Koutroumpas, K., Nguyen, T.-M. T., et al. (2016). An Organelle-Specific Protein Landscape Identifies Novel Diseases and Molecular Mechanisms. Nat. Commun. 7, 11491. doi:10.1038/ncomms11491
Cao, J., O’Day, D. R., Pliner, H. A., Kingsley, P. D., Deng, M., Daza, R. M., et al. (2020). A Human Cell Atlas of Fetal Gene Expression. Science 370, eaba7721. doi:10.1126/science.aba7721
Conway, B. R., O’Sullivan, E. D., Cairns, C., O’Sullivan, J., Simpson, D. J., Salzano, A., et al. (2020). Kidney Single-Cell Atlas Reveals Myeloid Heterogeneity in Progression and Regression of Kidney Disease. J. Am. Soc. Nephrol. 31, 2833–2854. doi:10.1681/ASN.2020060806
Gao, Z.-Q., Han, Y., Li, L., and Ding, H.-G. (2020). Pharmacological Management of Portal Hypertension: Current Status and Future. Chin. Med. J. (Engl) 133, 2362–2364. doi:10.1097/CM9.0000000000001004
Han, X., Zhou, Z., Fei, L., Sun, H., Wang, R., Chen, Y., et al. (2020). Construction of a Human Cell Landscape at Single-Cell Level. Nature 581, 303–309. doi:10.1038/s41586-020-2157-4
He, S., Wang, L.-H., Liu, Y., Li, Y.-Q., Chen, H.-T., Xu, J.-H., et al. (2020). Single-Cell Transcriptome Profiling of an Adult Human Cell Atlas of 15 Major Organs. Genome Biol. 21, 294. doi:10.1186/s13059-020-02210-0
Hochane, M., van den Berg, P. R., Fan, X., Bérenger-Currias, N., Adegeest, E., Bialecka, M., et al. (2019). Single-Cell Transcriptomics Reveals Gene Expression Dynamics of Human Fetal Kidney Development. Plos Biol. 17, e3000152. doi:10.1371/journal.pbio.3000152
Hurd, T. W., Gao, L., Roh, M. H., Macara, I. G., and Margolis, B. (2003). Direct Interaction of Two Polarity Complexes Implicated in Epithelial Tight junction Assembly. Nat. Cel Biol. 5, 137–142. doi:10.1038/ncb923
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 10, 845–858. doi:10.1038/nprot.2015.053
Khanna, R., and Sarin, S. K. (2014). Non-Cirrhotic Portal Hypertension - Diagnosis and Management. J. Hepatol. 60, 421–441. doi:10.1016/j.jhep.2013.08.013
Khanna, R., and Sarin, S. K. (2019). Noncirrhotic Portal Hypertension. Clin. Liver Dis. 23, 781–807. doi:10.1016/j.cld.2019.07.006
Lanktree, M. B., Haghighi, A., di Bari, I., Song, X., and Pei, Y. (2021). Insights into Autosomal Dominant Polycystic Kidney Disease from Genetic Studies. Clin. J. Am. Soc. Nephrol. 16, 790–799. doi:10.2215/CJN.02320220
Lasagni, A., Cadamuro, M., Morana, G., Fabris, L., and Strazzabosco, M. (2021). Fibrocystic Liver Disease: Novel Concepts and Translational Perspectives. Transl Gastroenterol. Hepatol. 6, 26. doi:10.21037/tgh-2020-04
Layer, R. M., Chiang, C., Quinlan, A. R., and Hall, I. M. (2014). LUMPY: A Probabilistic Framework for Structural Variant Discovery. Genome Biol. 15, R84. doi:10.1186/gb-2014-15-6-r84
Lemmers, C., Michel, D., Lane-Guermonprez, L., Delgrossi, M.-H., Médina, E., Arsanto, J.-P., et al. (2004). CRB3 Binds Directly to Par6 and Regulates the Morphogenesis of the Tight Junctions in Mammalian Epithelial Cells. Mol. Biol. Cel. Mar. 15, 1324–1333. doi:10.1091/mbc.e03-04-0235
Letunic, I., Khedkar, S., and Bork, P. (2021). SMART: Recent Updates, New Developments and Status in 2020. Nucleic Acids Res. 49, D458–D460. doi:10.1093/nar/gkaa937
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map Format and SAMtools. Bioinformatics 25, 2078–2079. doi:10.1093/bioinformatics/btp352
Liao, J., Yu, Z., Chen, Y., Bao, M., Zou, C., Zhang, H., et al. (2020). Single-Cell RNA Sequencing of Human Kidney. Sci. Data 7, 4. doi:10.1038/s41597-019-0351-8
McConnachie, D. J., Stow, J. L., and Mallett, A. J. (2021). Ciliopathies and the Kidney: A Review. Am. J. Kidney Dis. 77, 410–419. doi:10.1053/j.ajkd.2020.08.012
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 20, 1297–1303. doi:10.1101/gr.107524.110
Myram, S., Venzac, B., Lapin, B., Battistella, A., Cayrac, F., Cinquin, B., et al. (2021). A Multitubular Kidney-On-Chip to Decipher Pathophysiological Mechanisms in Renal Cystic Diseases. Front. Bioeng. Biotechnol. 9, 624553. doi:10.3389/fbioe.2021.624553
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera? A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 25, 1605–1612. doi:10.1002/jcc.20084
Varelas, X., Samavarchi-Tehrani, P., Narimatsu, M., Weiss, A., Cockburn, K., Larsen, B. G., et al. (2010). The Crumbs Complex Couples Cell Density Sensing to Hippo-Dependent Control of the TGF-β-SMAD Pathway. Develop. Cel. 19, 831–844. doi:10.1016/j.devcel.2010.11.012
Vilarinho, S., Sari, S., Yilmaz, G., Stiegler, A. L., Boggon, T. J., Jain, D., et al. (2016). Recurrent Recessive Mutation in Deoxyguanosine Kinase Causes Idiopathic Noncirrhotic Portal Hypertension. Hepatology 63, 1977–1986. doi:10.1002/hep.28499
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 38, e164. doi:10.1093/nar/gkq603
Wu, H., Vu, M., Dhingra, S., Ackah, R., Goss, J. A., Rana, A., et al. (2019). Obliterative Portal Venopathy without Cirrhosis Is Prevalent in Pediatric Cystic Fibrosis Liver Disease with Portal Hypertension. Clin. Gastroenterol. Hepatol. 17, 2134–2136. doi:10.1016/j.cgh.2018.10.046
Xu, X.-Y., Ding, H.-G., Li, W.-G., Xu, J.-H., Han, Y., Jia, J.-D., et al. (2020). Chinese Guidelines on the Management of Liver Cirrhosis (Abbreviated Version). World J. Gastroenterol. 26, 7088–7103. doi:10.3748/wjg.v26.i45.7088
Ying, P., Huang, C., Wang, Y., Guo, X., Cao, Y., Zhang, Y., et al. (2021). Single-cell RNA Sequencing of Retina:new Looks for Gene Marker and Old Diseases. Front. Mol. Biosci. 8, 699906. doi:10.3389/fmolb.2021.699906
Keywords: whole-genome sequencing, single-cell RNA sequencing, gene mutations, multiple renal cysts, noncirrhotic portal hypertension
Citation: Wu Y, Wu Y, Liu K, Liu H, Wang S, Huang J and Ding H (2021) Identification of Genetic Predisposition in Noncirrhotic Portal Hypertension Patients With Multiple Renal Cysts by Integrated Analysis of Whole-Genome and Single-Cell RNA Sequencing. Front. Genet. 12:775470. doi: 10.3389/fgene.2021.775470
Received: 14 September 2021; Accepted: 26 October 2021;
Published: 12 November 2021.
Edited by:
Peng Wang, Harbin Medical University, ChinaReviewed by:
Chungang Feng, Nanjing Agricultural University, ChinaYi Ding, Allen Institute for Brain Science, United States
Copyright © 2021 Wu, Wu, Liu, Liu, Wang, Huang and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Huang, aHVhbmdqMTk2NkBob3RtYWlsLmNvbQ==; Huiguo Ding, ZGluZ2h1aWd1b0BjY211LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship