 Venceslas Douillard1
Venceslas Douillard1 Erick C. Castelli2
Erick C. Castelli2 Steven J. Mack3
Steven J. Mack3 Jill A. Hollenbach4,5
Jill A. Hollenbach4,5 Pierre-Antoine Gourraud1
Pierre-Antoine Gourraud1 Nicolas Vince1†*
Nicolas Vince1†* Sophie Limou1,6*†on behalf of the Covid-19|HLA & Immunogenetics Consortium and the SNP-HLA Reference Consortium
Sophie Limou1,6*†on behalf of the Covid-19|HLA & Immunogenetics Consortium and the SNP-HLA Reference Consortium- 1Centre de Recherche en Transplantation et Immunologie, CHU Nantes, Inserm, Centre de Recherche en Transplantation et Immunologie, Université de Nantes, Nantes, France
- 2Unesp—Universidade Estadual Paulista, Botucatu, Brazil
- 3Division of Allergy, Immunology and Bone Marrow Transplantation, Department of Pediatrics, School of Medicine, University of California, San Francisco, San Francisco, CA, United States
- 4Department of Neurology, University of California, San Francisco, San Francisco, CA, United States
- 5Department of Epidemiology and Biostatistics, University of California, San Francisco, San Francisco, CA, United States
- 6Ecole Centrale de Nantes, Department of Computer Sciences and Mathematics in Biology, Nantes, France
The current SARS-CoV-2 pandemic era launched an immediate and broad response of the research community with studies both about the virus and host genetics. Research in genetics investigated HLA association with COVID-19 based on in silico, population, and individual data. However, they were conducted with variable scale and success; convincing results were mostly obtained with broader whole-genome association studies. Here, we propose a technical review of HLA analysis, including basic HLA knowledge as well as available tools and advice. We notably describe recent algorithms to infer and call HLA genotypes from GWAS SNPs and NGS data, respectively, which opens the possibility to investigate HLA from large datasets without a specific initial focus on this region. We thus hope this overview will empower geneticists who were unfamiliar with HLA to run MHC-focused analyses following the footsteps of the Covid-19|HLA & Immunogenetics Consortium.
Introduction to Human Leukocyte Antigens: Creating Immunity From Diversity
The classical HLA proteins are expressed on the surface of human cells. Although their primary role is to present exogenous and endogenous peptides, they were first described as “antigens” due to their interaction with T-cells in transplant rejection (Dausset, 1958). Along with other genes in the MHC region, the products of the HLA genes are essential in the adaptive immune response. By presenting peptides to both CD8+ (HLA class I molecules) and CD4+ T cells (HLA class II molecules), HLA proteins initiate an immune response against foreign (non-self) peptides which may be defective products of translation, neo-antigens generated by mutated genes from tumor cells, or pathogenic in origin. In addition, class I HLA proteins interact with the KIR ligands of NK cells, including KIR and LILRB, which are important in innate immunity (Carrington et al., 2008; Kulkarni et al., 2008; Trowsdale and Moffett, 2008). Thus, HLA molecules are key features of both innate and adaptive immune responses. HLA genes central role in immunity against infectious diseases and their importance for transplantation have made them the subject of much study.
HLA proteins are coded by multiple genes on the short arm of chromosome 6 at the 6p21 locus; this region containing HLA genes is referred to as the Major Histocompatibility Complex (MHC) for its seminal role in transplantation (Dausset, 1981; Montgomery et al., 2018). Although there is a common confusion between the two terms HLA and MHC, HLA specifically refers to the genes involved in antigen processing and presentation whereas the MHC corresponds to a whole locus, with HLA and other immune-related genes such as the complement system. The MHC region is the most gene-dense region of the human genome, with 1% of the human coding genes (>200) found in 0.1% of the genome length (Shiina et al., 2009). The MHC region is commonly defined as a 4 Mb segment on chromosome 6 (MOG 29657002–33192499 COL11A2, GRCh38. p13 assembly) (Beck et al., 1999). However, due to extended patterns of linkage disequilibrium (LD), an extended MHC (xMHC) is often referred to in immunogenomics (25726063–33400556, GRCh38. p13 assembly) (Horton et al., 2004). The MHC region is divided into three regions based on gene sequence similarities and functions, class I, II, and III in which approximately 40% of the genes are immune-related. HLA genes are found in the class I and class II regions and are commonly divided in two categories: classical HLA proteins present peptides to T-cells, whereas non-classical HLA are mostly involved either in peptide presentation with other receptors, with immune modulation, or with various steps of classical HLA formation and loading.
The MHC class I region contains 12 HLA pseudogenes and 6 HLA genes (HLA-A, -B, -C, -E, -F, and -G), including three classical (HLA-A, -B, and -C) that are ubiquitously expressed as a heterodimer with beta-2 microglobulin at the cells’ surface. Class I HLA molecules and their bound peptides are specifically recognized by CD8+ T cells receptors. The non-classical HLA class I molecules (HLA-G, -E, and -F) present different expression patterns. HLA-E and HLA-F are usually ubiquitously expressed in low levels, and they interact with ligands in T and NK cells (such as HLA-E with NKG2A). HLA-G is predominantly expressed at the maternal-fetal interface and has primarily been associated with maternal–fetal tolerance by interacting CD8 from T cells and LILRB1, LILRB2, and KIR2DL4 from NK cells (Donadi et al., 2011).
The Class II region comprises four non-classical genes (HLA-DMA, -DMB, -DOA, -DOB), mostly related to peptide loading, and 17 classical HLA genes (e.g., HLA-DRA, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DPA1, and others) that are expressed in Antigen Presenting Cells (APC) such as B cells, monocytes, macrophages, dendritic cells as well as epithelial cells under inflammatory signals (Rock et al., 2016). Unlike class I HLA molecules, class II molecules are heterodimers, consisting of α and β chains, encoded by the corresponding HLA genes (e.g., HLA-DPA1 and HLA-DPB1 produce the HLA-DP molecule), which facilitates molecular diversity. The HLA-DR beta chain can be encoded by nine different genes (DRB1-9) with complex patterns of expression, and gene content adding additional layers of complexity (Faner et al., 2009). Finally, the class III region, located between the class I and II regions, is the most gene-dense region of the MHC; this region contains genes encoding elements of the complement system, chaperone genes, cytokines such as TNF and LTA, but no HLA genes.
Finally, there are other important non-HLA genes in the MHC, such as TAP1 and TAP2, both related to peptide pumping from the cytoplasm to the endoplasmic reticulum (Praest et al., 2018), MICA and MICB, both induced in viral infections and tumors and activate NK-mediate killing (Ghadially et al., 2017), the tripartite motif (TRIM) family, related to cell cycle progression, autophagy, and viral replication restriction (van Tol et al., 2017), PSORS1C1, conferring susceptibility to psoriasis and systemic sclerosis (Allanore et al., 2011), and others.
In addition to their large number and potential for many combinations, the HLA genes display unparalleled genetic diversity, with more than 27,000 alleles and almost 17,000 unique proteins (June 03, 2021, https://www.ebi.ac.uk/ipd/imgt/hla/stats.html) identified for the five most polymorphic loci (HLA-A, -C, -B, -DRB and -DQB1). This diversity of HLA molecules is concentrated in the peptide-binding groove, which allows the presentation of peptides of various shapes and sizes, hence conferring broad protection against pathogens at the population level. At the same time, the polymorphic nature of HLA is also found on non-coding parts of the genes, such as the promoter and have an impact on expression (Kulkarni et al., 2011; Vince et al., 2016; Lima et al., 2019). Over evolutive time, together with founder effects, multiple pathogen-challenges have exerted selective pressures on HLA alleles in human populations across the globe (Meyer and Thomson, 2001; Spurgin and Richardson, 2010), shaping allele frequency differences and selecting very specific or even private HLA alleles in some populations (Brandt et al., 2018; Meyer et al., 2018). The progress of genomics, and immunogenomics over the last decade, had deepened our understanding of HLA role in human diseases though the use of genome-wide association studies (GWAS) (Kennedy et al., 2017; Dendrou et al., 2018).
The COVID-19 HLA and Immunogenetics Consortium (CHIC) has been created during the pandemic to coordinate efforts on HLA analysis. The CHIC provided a website with information on HLA data and current projects (The COVID-19 HLA and Immunogenetics Consortium, 2020a). It is supported by a database (The COVID-19 HLA and Immunogenetics Consortium, 2020b) and its role is the centralization of relevant HLA and clinical data for COVID-19 study. It contains HLA data of 2,892 individuals from nine projects. These data are freely available and new data can be easily uploaded upon account creation. In addition, the website allows HLA allele frequencies visualization, and use of HLA data management and analysis tools. An HLA Imputation Portal (HIP) is set up to allow geneticists to infer individuals HLA alleles using SNP genotyping data, relying on multi-ethnic models from Zheng et al. (Zheng et al., 2014). This tool may help leverage SNP data to gain power in HLA association studies. The CHIC also produced a broad review on immunogenetic parameters (e.g., HLA, KIR, complement, cytokines and chemokines receptors) and their role in COVID-19 (Aguiar et al., 2021). A more specific review of COVID-19 and HLA associations (Douillard et al., 2021) highlights links between the pathology and HLA at different levels, from allele frequency correlation to HLA associations and haplotypes. The consortium will gradually improve its portal by providing access to additional and more diverse imputation reference panels, and by recruiting more individuals. Results from GWASs showed no association between HLA SNPs and COVID-19 infection (COVID-19 Host Genetics Initiative, 2021) but demonstrated an association with COVID-19 severity; dedicated HLA allele association studies identified potential signals of interest (Castelli et al., 2021). The spread of HLA tools, allowing HLA allele inference from whole-exome or whole-genome sequencing as well as from GWAS SNP data will significantly increase the sample size from available cohorts to maximize the statistical discovery power of HLA-centric studies. In this report, we pursue this effort to provide an overview of methods for generating HLA data along with several analytical strategies to capitalize on this genetic information. We will also cover additional immunogenomic parameters, as MHC-related associations still have much to reveal (Trowsdale and Knight, 2013). We hope this work will empower researchers to include HLA-focused investigations in their palette and will contribute to promote efforts for in-depth explorations of the relationship between HLA and immune-related outcomes in this pandemic era.
Generating and Working With HLA Data
Performing immunogenetic studies can be a challenge for those unfamiliar with the specifics of HLA nomenclature. An individual HLA genotype can be obtained through multiple molecular techniques, the complexity of its nomenclature allows the alleles in a genotype to be described in different styles, and these data can be stored in a variety of file standards. Overall, HLA information can take multiple forms, requiring a comprehensive understanding of the nomenclature in order to run proper statistical analyses and find relevant associations.
Generating HLA Data
Originally, immunologists conducted microlymphocytotoxicity assays, testing patients T/B cells (for HLA class I) or B cells (for HLA class II) against different anti-sera or monoclonal antibodies in the presence of complement. Sera or antibodies recognizing the HLA antigens on cells would activate the complement and lyse the cell; this serology staining would reveal the patient HLA serotype (Park and Terasaki, 2000). Serology was however limited by the underlying complexity of HLA and it resulted in poor performances in transplantation (Hurley, 2021). The need to improve this performance and technique evolution, with the advent of PCR, conducted HLA specialists to switch to molecular typing. Molecular techniques were adopted for HLA typing; these methods allowed systematic identification of HLA alleles, based on sequence polymorphisms, providing a ‘higher resolution’ result that distinguishes many more allele categories than serological methods. This molecular typing consistently improved in resolution throughout the years driving nomenclature evolution along the way. Sequence-specific (PCR-SSO) methods rely on the hybridization of hundreds of labeled SSO probes targeting unique sequences in polymorphic regions. Sequence-specific priming (PCR-SSP) methods directly amplify elements of the HLA genes with PCR primers containing sequence-specific 3′ end polymorphisms, resulting in less ambiguity (inability to distinguish alleles with similar nucleotide sequences), than SSO methods (Meral and Beksaç, 2007).
Sanger sequencing-based typing (PCR-SBT) methods initially provided sequences of the exons that encoded the peptide-binding groove, and later overlapping sets of sequences for entire genes. PCR-SBT was the gold standard for HLA genotyping until the development of next-generation sequencing (NGS) methods (Meral and Beksaç, 2007; De Santis et al., 2013). Except for the last one, PCR-SBT, previous methodologies were not suitable to detect new variants, and their goals were detecting known polymorphisms.
The application of NGS was explored in 2012, as part of the 16th International HLA and Immunogenetics Workshop (IHIW), but, given issues with mapping of short reads, allelic imbalance, phasing, and high costs, PCR-SBT remained the gold standard. More recently, the integration of NGS technologies with bioinformatic solutions for immunogenetics has improved the speed and accuracy of NGS HLA genotyping with lower error rates and fewer ambiguities than PCR-SBT (Baier et al., 2019; Jekarl et al., 2021), and the application of NGS was the focus of the 17th IHIW in 2017 (Vayntrub et al., 2020). Moreover, NGS is ideal to detect new HLA variants. Researchers now routinely identify novel HLA alleles (Nilsson et al., 2018; Ralazamahaleo et al., 2019; Loginova et al., 2020; Ananeva et al., 2021a; Ananeva et al., 2021b; Cheranev et al., 2021; Loginova et al., 2021) using NGS and confirm them using SBT with PCR-SBT error often responsible for non-concordance between the two. NGS-based sequencing of multiple exons and introns has led to increases in the growth of the IPD-IMGT-HLA Database collection (Robinson et al., 2015; Robinson et al., 2019). Unfortunately, the total number of new alleles may be underestimated as it is not uncommon for new alleles to be NGS-typed without Sanger validation.
So-called third-generation NGS generates unambiguous, phased HLA genotypes, using instruments like the PacBio SMRT (Mayor et al., 2015) or Oxford Nanopore Technology MinION (De Santis et al., 2020) to avoid multiple molecular techniques. This approach is faster than SBT and generates phased polymorphism with longer reads. Researchers using Oxford Nanopore Technology systems have successfully sequenced 11 HLA loci with low ambiguities in under 6 h (Mosbruger et al., 2020).
HLA Nomenclature
Soon after cell-surface antigens were identified as polymorphic between individuals, the WHO Nomenclature Committee for Factors of the HLA System was formed to develop a specific nomenclature for HLA genes, proteins and allelic variants (Allen et al., 1968). The original “HL-A″ factor serologically typed with multiple antibodies with an individual type (e.g., HL-A (1,2/7,8) identifying them as positive for factors 1,2,7,8, and confirmed two distinct haplotypes from parental typing. As dozens of HLA genes and thousands of alleles were identified, the nomenclature was expanded to accommodate new complexity while building on the historical serological vocabulary. In 1987, the nomenclature was updated to accommodate newly available protein and nucleotide sequences (Antigens, 1987). The modern locus names were adopted at this time, and four-digits names were assigned to alleles, which were only defines as protein variants. In 2010, the current field-delimited nomenclature was adopted to account for the growing number of silent and non-coding nucleotide variants (Marsh et al., 2010).
A modern HLA allele name consists of up to four “fields”, each of which includes a two- or more digit number, each separated by a colon (Figure 1).
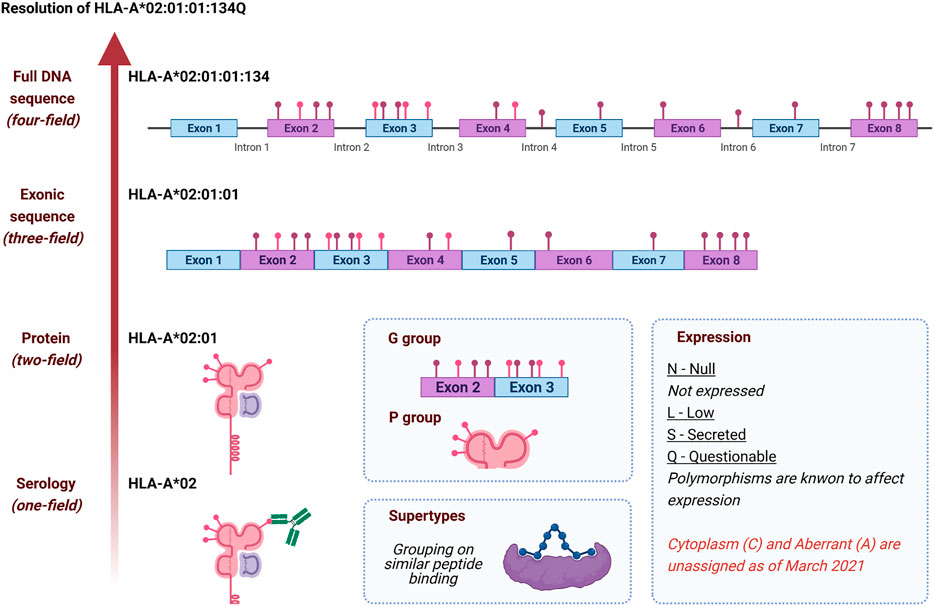
FIGURE 1. History and development of HLA nomenclature as illustrated by HLA-A*02:01:01:134Q. Each level of resolution corresponds to a group of HLA alleles fitting the description, except for the full DNA sequence, a unique HLA allele. Colored pins represent non-synonymous polymorphism (pink) and synonymous or intronic polymorphisms (purple); the displayed polymorphism is only indicative and does not reflect HLA-A*02:01:01:134Q sequence. P and G groups are named with the lowest numbered two-field (HLA-A*02:01) and three-field (HLA-A*02:01:01) HLA allele name, respectively. Class II P and G groups are based on exon 2 only, while class I P and G groups are based on exons 2 and 3. Supertypes are not defined as part of the official nomenclature (Wang and Claesson, 2014; del Guercio et al., 1995; Sidney et al., 1995). Created with biorender.com.
The first and second fields represent a historical serological group, and a unique protein sequence, respectively. All allele names have at least two fields. Alleles sharing the 1st, and 2nd fields with a different 3rd field encode the same protein but have unique silent-substitution in the exonic sequence, whereas sequence differences contained in the introns are written in the 4th field. The four fields of an allele name can also be suffixed with a single-letter “expression variant”, identifying alleles that are either not expressed, expressed at a low or questionable level, or secreted. For example, HLA-A*02:01:01 represents an exonic sequence shared by e.g., HLA-A*02:01:01:01 and HLA-A*02:01:01:134Q. In the latter case, the expression of HLA-A*02:01:01:134Q is Questionable, due to a potential alternate splicing nucleotide variant in intron 2. Allele names can be truncated to fewer fields for different applications, with each truncation described as a level of “resolution” (e.g., HLA-A*02 is a one-field resolution allele).
In addition to this allele nomenclature, specific groups of alleles have been defined. P and G groups refer to multiple alleles sharing either the same peptide or nucleotide sequence for the peptide-binding groove, respectively. For instance, HLA-A*02:01:01:134Q and HLA-A*02:252 both belong to the A*02:01P P group; the two proteins are globally different but share the same peptide-binding groove. HLA-A*02:01:01:134Q and HLA-A*02:89:01 belong to the A*02:01:01G G group as they share identical peptide-binding groove encoding exon sequences.
HLA supertypes are groups of alleles sharing similar peptide-binding repertoires. Supertypes are defined by “structural similarities, shared peptide-binding motifs, and identification of cross-reacting peptides” (Wang and Claesson, 2014). Using this classification, HLA-A*02:01:01:134Q potentially belongs with HLA-A*02:02, A*02:05, A*69:01 in the A2 supertype. (del Guercio et al., 1995). Some studies of the HLA molecules’ evolution have interpreted HLA diversity differently. Kaufman et al. (Kaufman, 2018; Di et al., 2021) have proposed promiscuous and generalist HLA categories when Di et al. have challenged the concepts of supertypes and function peptide-binding groove groups.
HLA Data Formats
The modern and legacy nomenclature systems are still in use, which often makes data comparison and meta-analysis difficult. In addition, HLA alleles are stored in multiple formats which impact their use with bioinformatic tools. TSV or CSVs have been used to store HLA genotypes, usually organizing individuals in rows and HLA genes in columns (with two columns for each gene). Such files are often generated manually, but are used by multiple population genetic and disease-association applications (Lancaster et al., 2007; Excoffier and Lischer, 2010; Pappas et al., 2016). More strictly-defined bioinformatic-oriented formats include HLA PED (or HPED) (Choi et al., 2021), an HLA-focused extension of the PED format (Purcell et al., 2007); Variant Call Format (VCF), as used by BEAGLE (Browning et al., 2018), in which HLA allele names are recoded as multiple binary identifiers, and Histoimmunogenetic Markup Language (HML), an XML format developed specifically for exchanging HLA and Killer-cell Immunoglobulin-like Receptor (KIR) genotype data (Milius et al., 2015).
The IPD-IMGT/HLA Database releases new and updated reference sequences and allele names every 3 months. Individuals datasets may have been generated under any release version, which is why tools like the Allele Name Translation Tool (ANTT) have been developed to standardize datasets to a common release version. (Mack and Hollenbach, 2010). Development of a standardized means of storing and sharing data is still underway. In 2015, the MIRING reporting guideline (Mack et al., 2015) introduced standardized data elements and a controlled vocabulary for HLA genotype data and meta-data, which were implemented in HML (Milius et al., 2015). An HML message includes information on the IPD-IMGT/HLA Database version, the entity and how they generated the data, as well as references to external sources (e.g., reference sequences and aligned read). HML is used to transmit HLA genotyping data to the National Marrow Donor Program (and other similar registries and donor centers), but has yet to be adopted for genetic–analysis applications. Most of the existent HLA analysis applications require fewer data elements than are included in an HML message.
Given the number of different applications of HLA data, new informatics tools can influence the interpretation of this information. Multiple ancillary tools have been developed for HLA research. Whether they allow researchers to run rapid association analyses, extract new information from data, or link HLA genotypes to novel fields of translational research, all contribute to the advances in the HLA research (Table 1).

TABLE 1. Tools for HLA analyses.
Inferring and Imputing HLA Alleles: From Complex Read-Mapping to the Study of Linkage Disequilibrium
HLA inference is an umbrella term comprising multiple bioinformatic tools and statistical methods to obtain individuals’ HLA genotypes. Inference implies using missing information to obtain HLA genotypes, this can generally refer to using untargeted sequencing data, which have insufficient sequence read depth, to thoroughly recover the HLA alleles polymorphisms (Klasberg et al., 2019).
Inference From Whole-Genome Sequencing and Whole-Exome Sequencing
Unlike NGS typing techniques which targets HLA genes (as many commercial kits apply), untargeted sequencing does not focus on HLA. Whole-genome sequencing (WGS) methods aim to identify all genetic variations of an individual genome, while whole-exome sequencing (WES) is designed to target all exons. Initially, these methods did not support the calling of HLA alleles; low coverage and short read-lengths led to poor HLA typing accuracy (Bauer et al., 2016). Low coverage does allow identification of HLA alleles, due to their high levels of polymorphism and extensive conserved sequences among genes, and improvements were needed (Hosomichi et al., 2015). Moreover, general pipelines for analyzing NGS data from WGS do not work for HLA genes; because they present high sequence similarity, it is very common that a short read (a sequence generated in NGS procedures) from one gene aligns to another gene (cross-mapping), leading to genotyping errors (e.g., HLA-A and HLA-H, or HLA-C and HLA-B) (Castelli et al., 2018). The intense polymorphism observed in HLA genes may bias read alignment when using a single genome reference, especially when one individual presents too many modifications compared to the reference genome. This issue overestimates reference allele frequencies and causes genotyping errors (Brandt et al., 2015). Therefore, it is mandatory to use methods tailored for HLA genes to get reliable genotypes and haplotypes at the SNP level from NGS data.
Multiple algorithms have been developed and refined (Klasberg et al., 2019). These include: 1) classic read-mapping with HLA-specific quality control steps or different scores, hla-mapper (http://www.castelli-lab.net/apps/hla-mapper) (Castelli et al., 2018) which also works on KIR genes and provide genotyping and haplotyping at the SNP level, seq2HLA (Boegel et al., 2012) and HLAforest (Kim and Pourmand, 2013), among other tools; 2) population graph reference methods (e.g., HLA*PRG:LA), which identify probability edges between polymorphisms nodes and project read data onto these to evaluate the most likely alleles.
Recent reviews and tool comparisons on the optimal methods for non-HLA targeted sequencing data are already available (see (Klasberg et al., 2019; Chen et al., 2021)). In 2020, Chen et al. found that HLA-HD was the most accurate tool for producing HLA genotypes from WGS and WES. However, the study focused on the performance of five tools only. Notably, most of the tools they studied achieved much higher accuracies than previously reported by Bauer et al., 2016, which emphasizes a drastic improvement in read coverage and processing in the MHC region (Bauer et al., 2016). Finally, researchers successfully implemented these tools in association studies, promoting their importance for HLA-centric epidemiological studies (Juhos et al., 2015; Xie et al., 2017; Mimori et al., 2019; Vince et al., 2020a).
HLA Allele Imputation
HLA genotyping data can also be generated using HLA imputation tools, which generate genotypes for individuals on the basis of LD between GWAS-derived SNP data for the MHC region and specific HLA alleles. These methods ultimately rely on reference datasets of HLA and SNP genotypes for the same individuals, and have become increasingly accurate in their predictions as new algorithms are developed.
Following the opportunity brought by SNP to SNP imputation, SNP to HLA imputation algorithms offered a quick and easy way to obtain HLA genotypes from widely available GWAS SNP genotyping data (McCarthy et al., 2016). SNP to HLA imputation relies on reference panels of individuals with known SNPs and HLA genotypes, to generate links between SNPs, haplotypes, and HLA alleles using machine learning algorithms (Figure 2).
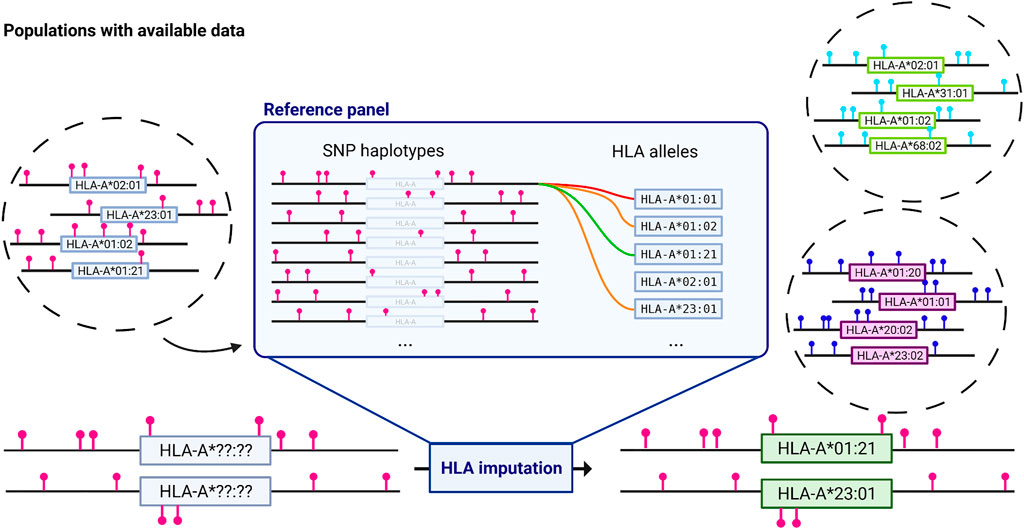
FIGURE 2. HLA imputation from GWAS data. Reference panels are created from individuals with known SNP and HLA data. Depending on the method, an algorithm will deduce the probability of a specific HLA allele in the population given a SNP haplotype. These new found links are stored for that reference panel and applied to new SNP data to infer HLA genotypes. HLA-A is given as an example with a truncated list of alleles; other MHC genes are imputed using the same method. Different populations are represented in different circles and imply different allele frequencies. Pinpoints represent SNPs and are only indicative. HLA imputation results are highly dependent on the population chosen for the reference panel. Created with biorender.com.
The first published algorithms, SNP2HLA (Jia et al., 2013) and HLA*IMP (Dilthey et al., 2011), were based on different implementations of hidden Markov models; SNP2HLA used BEAGLE (Browning and Browning, 2009), a haplotyping and SNP genotype imputation tool. In 2014, Zheng et al. proposed HIBAG, an attribute bagging method tailored for HLA data (Zheng et al., 2014), which showed better performance than pre-existing tools, and at the time was the only method to provide population-specific reference panels for hundreds of individuals while enabling construction of personalized reference panels building. Initial independent reviews suggested that SNP2HLA performed better on 3,265 samples from BioVU, a de-identified electronic health record database coupled to a DNA biorepository (Karnes et al., 2017). However, later reviews (Kuniholm et al., 2016; Pappas et al., 2018) and studies (Ritari et al., 2020) have favored HIBAG for HLA imputation, notably on more complex HLA data.
In practice, both SNP2HLA and HIBAG are commonly used to conduct HLA imputation or creation of new reference panels. Overall accuracy differences are low for European panels that had been extensively assessed. An important point still under investigation is the impact of population diversity in reference panels. While some researchers advocate for the creation of exhaustive multi-ethnic reference panels (Degenhardt et al., 2019), others have shown that specific populations (e.g., insular or admixed require more restrained reference panels (Khor et al., 2015; Ritari et al., 2020).
The difficulty in determining if a reference panel is suitable for HLA imputation is related to how well it matches to target data, on the frequency of common alleles and the presence of rare HLA alleles, specific to some population (especially in underrepresented populations). This has led to the creation of reference panels with limited HLA diversity. While accuracy values are often reported as the ultimate answer to a model viability, these values can be misleading. For a rare HLA allele in a validation dataset, a 90% accuracy value can be achieved if that allele should be imputed 20 times out of 2,000 alleles (i.e., 1,000 individuals) but is never predicted. Therefore, other metrics (e.g., sensitivity, specificity, or F1 score (Cook et al., 2021)), must not be overlooked. Admixed populations are formed by individuals from different genetic backgrounds in variable proportions, and HLA imputation can be sub-optimal if the reference panel is only drawn from one of the ancestral populations. Conversely, a reference panel from an admixed population with a different overall genome proportion from the individuals being imputed may also provide inaccurate results.
To effect worldwide improvement in HLA imputation efforts, we led the creation of an international consortium, the SNP-HLA Reference Consortium (SHLARC), whose aim is to gather data to represent the extreme diversity of HLA alleles, fostering accurate imputation (Vince et al., 2020b). We further advocate for improvements to current HLA imputation tools and for the development of a platform promoting easy access to HLA imputation for immunogeneticists. Though HLA imputation is not yet suited for clinical settings, generalization of HLA association studies offers a new way to investigate immune pathologies (Meyer and Nunes, 2017).
New versions HLA*IMP (Motyer et al., 2016) and SNP2HLA have been released (e.g., MHC*IMP (Squire et al., 2020), CookHLA (Cook et al., 2021), and Deep-HLA (Naito et al., 2021)) that apply new algorithms. These highlight the community intense interest in HLA imputation. CookHLA is an updated version of SNP2HLA (based on the BEAGLE algorithm) that better accounts for LD in the HLA region and makes use of the genetic map option to better impute individuals who are not well represented in the reference panels. For its part, Deep-HLA seems especially promising as deep learning may lead to better imputation of rare alleles.
Bioinformatic Analyses of HLA Information
The pressing challenge of understanding the COVID-19 pandemic, given previous associations with infectious diseases, has led researchers to scrutinize HLA using any available resource. In addition to issues of nomenclature and on-going technological evolution of typing methods, the complexity of HLA analyses is also derived from the multiple forms these analyses can take. On the one hand, HLA allele frequencies and predicted binding affinity of pathogen peptides to HLA alleles allow for a first step in the HLA world, as they are easily available, but are limited to investigate its actual role. On the other hand, the in-depth implication of HLA is revealed when looking at SNP association in the MHC region, and specifically when looking at allele associations, but their realization is hindered by high costs and technical difficulties. The study of HLA is multi-layered, with a continuum of methods peaking with analysis of individual data and multi-locus haplotypes, all of which contributing to a comprehensive understanding of the role of HLA in a given analysis.
HLA Allele Frequencies
The diversity of HLA alleles across geographically separated populations is thought to be the result of balancing selection due to local pathogens (Meyer and Thomson, 2001). The allelefrequencies.net database has the most extensive collection of HLA allele frequencies in diverse populations (Middleton et al., 2003). In addition, HLA typing conducted by bone marrow registries may constitute a local estimation of HLA allele distribution in a population (Sacchi et al., 2019; Schmidt et al., 2020). It is possible to statistically analyze the correlation (e.g., via linear regression or Pearson coefficient) between a quantitative value, such as the number of COVID-19 cases, and the HLA allele frequencies obtained from a different sample in every studied population (e.g., in a database or registry).
However, while these correlations are faster and easier to obtain than new HLA genotypes, they may result in spurious correlations because: 1) most of the HLA alleles (and observed haplotypes) have a low frequency. For example, according to allelefrequencies.net, in the 416,581 individuals from the African-American NMDP population in the United States, two-thirds of the 321 HLA-B alleles at two-field resolution have frequencies below 0.003% (24 or less occurrences). Assuming that reference population samples are representative is not always accurate. A possible solution is to focus on common HLA alleles; 2) statistical tests are often applied without multiple-testing correction, regardless of the number of tests; 3) the confounding variables, both genetic (e.g., ancestry) and environmental (e.g., comorbidities), are often overlooked.
In any case, correlation is not causation. Therefore, the high number of HLA alleles and biased frequencies are bound to create spurious links between their presence and any phenotype. Therefore, to thoroughly investigate the relationship between HLA and phenotype, it is of the utmost importance to conduct studies and control for other genetic factors such as population stratification, linkage disequilibrium, or comorbidities (some linked to HLA polymorphism itself such as diabetes). Statistical bias could also be reduced by working on a higher number of samples and correcting for multiple testing. It is also worth considering different resolution levels of information, from in silico studies to full haplotype information.
In silico Peptide Binding
HLA molecules present endogenous and exogenous peptides, however, affinities for these peptides vary greatly depending on the peptide conformation and the peptide cleft topology and chemistry. Whether an HLA allele presents several or few peptides derived from one specific pathogen is one mechanism potentially explaining the strong immune response or tolerance towards it. Researchers can use prediction tools, such as NetMHCpan (Nielsen and Andreatta, 2016; Jurtz et al., 2017), trained on binding affinity and elution assays, to evaluate the number of potentially bound peptides for any HLA class I allele. The “pan” methods, contrary to the “allele-specific” methods, use similarities in sequence data to predict the peptide binding capacity of HLA alleles for which no information is available. Other tools exist and have been reviewed by Mei et al., in 2020 (Mei et al., 2019). Such predictions, coupled with HLA genotype data of individuals, give a theoretical insight into the possible adaptive immune response of a person. In these tools, the peptidome of the studied pathogen is informatically divided into peptide sequences of limited size (8–12 residues to account for the size of peptides presented by class I molecules), and the number of alleles predicted to bind a large number of peptides is inferred to represent better presentation to T cells, and a protective role against the pathogen. However, the only way to definitely determine peptide binding affinity is through laboratory experiments.
Genome-wide Association Studies
Genotyping data obtained with SNP arrays has proven to be fast and inexpensive for investigating the genetic component of complex traits and diseases (Claussnitzer et al., 2020), compared to more thorough and exhaustive sequencing technologies. Without assumptions regarding the region potentially involved in the studied trait, GWAS helped discover protective and risk alleles, particularly in the HLA region (Kennedy et al., 2017). Contrary to the use of independent HLA allele frequencies for studying a pathology, association studies assess the difference between affected individuals and unaffected individuals or the distribution of a particular quantitative trait. Both genetic and phenotypic data are individual and not population-based, reducing biases. The statistically significant SNP (aka, top hits) are linked to genes by proximity, and investigation by pathway analysis can reveal additional biological information on their effect. More recently, transcriptome-wide association studies have allowed more accurate investigation of the impact of a SNP on the expression of genes (Wainberg et al., 2019). In addition, some SNPs can be highly correlated to an HLA allele (e.g., rs2395029 and HLA-B*57:01 have been described multiple times as in complete linkage disequilibrium (de Bakker et al., 2006)), and therefore provide additional functional information for biological interpretation. Finally, statistical regression models can take into account potential confounding factors (e.g., genetic ancestry and population stratification, sex, age, comorbidities) to control for limiting biases.
Given the complex LD patterns across the MHC region, SNP association analyses are not usually precise enough to identify specific disease-associated HLA alleles. LD patterns may differ between populations. For example, the rs2395029 tags HLA-B*57:01 in Europeans but displays reduced LD in African-Americans (Colombo et al., 2008). The complex LD patterns and the high number of genes in the MHC region, make it difficult to pinpoint an SNP to a specific HLA allele in most cases.
HLA Allele Association Studies
Association studies of HLA alleles offer a more relevant biological explanation, based on peptide presentation. HLA allele data can come from different sources, including various epochs of HLA typing and HLA imputation from SNPs (see above). These data can be analyzed as is, or low resolution HLA data can be “upgraded” using the HLA-Upgrade tool from the Easy-HLA website, which statistically impute the most probable two-field genotype based on a haplotype database (Geffard et al., 2020). Once HLA data from multiple sources have been standardized for allele content and resolution, a frequency cut-off value is usually applied to test only those alleles with sufficient occurrences in the dataset to guarantee statistical power in the analysis. HLA alleles being highly polymorphic, they often display lower frequencies, and a larger sample size is usually required to obtain significant results compared to SNP analyses.
Regression models, which are commonly used for SNP association, are the most versatile and common statistical models implemented to test associations between individual HLA alleles and phenotypes of interest (linear models for continuous and logistic for discrete phenotypes, respectively). Regression models can work with multiple covariables, allowing the disentanglement of the HLA effect and confounding factors such as population stratification, sex, gender, and others. Similar to GWAS SNP analyses, HLA alleles are tested individually as biallelic markers for each HLA gene, as each individual can exhibit 0, 1, or 2 occurrences of a given allele. As HLA molecules are expressed co-dominantly (Hughes and Nei, 1988), the dominant genetic model is commonly preferred to allelic or recessive models to assess HLA allele associations. However, it should be mentioned that different alleles might present different expression levels due to promoter and 3′UTR variations and final protein stability. Indeed, this is another HLA world: the effect of variants in the expression levels, which sometimes are directly linked with disease susceptibility (Kulkarni et al., 2011).
As in GWAS analysis, the overall performance of a statistical model can be evaluated with a Quantile-Quantile (QQ) plot, representing the observed p-value distribution for each HLA allele compared to the expected distribution under the null hypothesis. Any deviation from this distribution is highlighted by a deviation from a straight line. (Murdoch et al., 2008). Different scenarios can be described: 1) observed p-values mostly follow the null hypothesis, indicating that the statistical model accurately fits the data; 2) observed p-values deviate below the null hypothesis line, indicating that the statistical model is probably underpowered; 3) observed p-values deviate above the null hypothesis line, indicating that the statistical model may not be well parameterized and some confounding factors are not enough considered. Once the robustness of the analysis is confirmed, it is important to obtain a comprehensive visualization of the results with Manhattan plots, for instance, displaying–log10 (p-value) along with the list of test HLA alleles ordered numerically (as seen in Vince et al. (Vince et al., 2020a)). Volcano plots can also display the significativity of alleles along with their effect size, allowing a global view of their impact. Finally, the significance threshold accounting for multiple testing can be determined with the Bonferroni correction (5% α threshold divided by the number of tests) or other corrections such as the FDR, or permutations.
Easy-HLA: Going Beyond HLA Alleles to HLA Genes Haplotypes, HLA Expression Levels, Specific HLA Amino Acids, KIR Ligand Groups
New tools have been developed to facilitate the analysis of additional immunogenetic parameters (e.g. KIR ligands, see Figure 3).
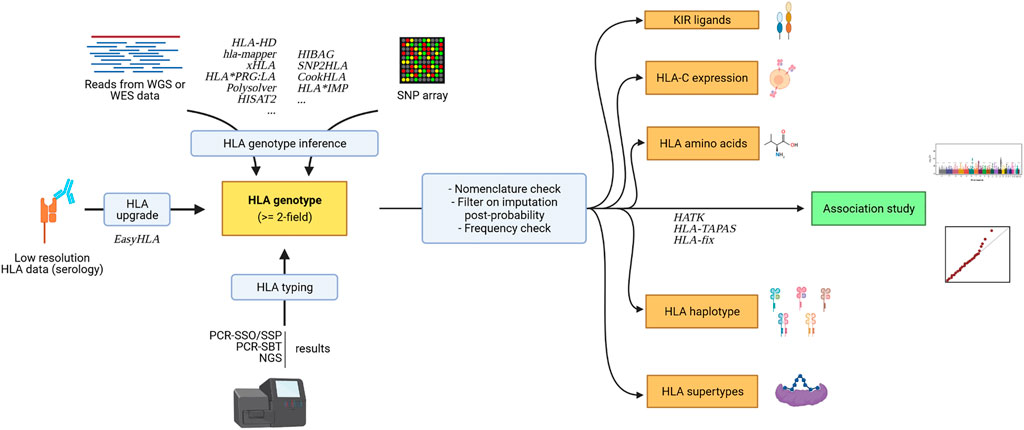
FIGURE 3. Association study pipeline for HLA data and surrounding immunogenetic factors. Created with biorender.com.
HLA genotypes can be used to infer additional immunogenetic parameters that can further be analyzed (see Figure 3.) to get a clearer understanding of the relationship between immunity and pathologies. While one HLA allele already represents a haplotype of SNPs within a gene, as it is a collection of polymorphisms in the gene of interest, researchers have demonstrated the importance of looking at multiple HLA alleles on the same chromosome, which is referred to as an HLA haplotype. Association studies can be done on haplotypes, but many haplotype frequencies can be even lower than constituent allele frequencies. In a clinical setting, the collection of haplotype information is also useful, notably in HSCT transplants, for identifying haploidentical individuals. These haplotypes can be inferred using the HLA-2-Haplo tool from Easy-HLA website (Geffard et al., 2020), for instance. A straightforward, reliable, but expansive strategy to get HLA gene haplotypes is the analysis of trios (mother, father, and offspring) or third-generation long-read sequencing such as PacBio SMRT.
Easy-HLA also infers HLA-C expression levels, HLA alleles amino acids, and KIR ligand groups. Recently, high HLA-C expression levels were associated with better control of HIV (Apps et al., 2013; Vince et al., 2016). Class I HLA alleles have also been grouped according to their dependence on tapasin, a major actor in peptide loading, which proved to be an interesting subdivision for studying HIV-1 control (Bashirova et al., 2020). Moreover, testing HLA allele amino acids may indicate a specific function of a given residue across several alleles, as with this study by McLaren et al., again in HIV control (McLaren et al., 2012). Finally, studying KIR ligand groups along with KIR typing as previously described (Martin and Carrington, 2013; Vince et al., 2014) can reveal the binding patterns of specific HLA alleles. For example, HLA-A and HLA-B molecules bearing the Bw4+ motif bind specifically to KIR3DL1. Similarly, HLA-C group 1 (C1) allele-encoded molecules carry an asparagine at position 80 and specifically bind KIR2DL2/3, as opposed to group 2 (C2) allele-encoded molecules, which carry a lysine and specifically bind KIR2DL1 (Parham et al., 2012). Grouping HLA alleles according to different functional parameters can increase the power of detecting a true positive signal and represent an opportunity to come closer to the biological cause behind HLA genetic association with diseases.
Conclusion
However intricate it may be, the MHC region, and HLA in particular, is the perfect candidate to investigate infectious or auto-immune diseases, as its primary biological role is to present antigen to the immune system. HLA research was able to grow in different directions from in silico studies on peptide binding to association studies of HLA alleles, giving leads on HLA involvement in pathologies. That said, HLA-focused analysis requires special care because its immense diversity and low-frequency distribution may potentially result in spurious associations when tested incorrectly or in a small cohort. Fortunately, many tools have been and are still developed to obtain high-quality HLA information for a low cost with statistical inference, through HLA inference from NGS data or HLA imputation from SNP GWAS data or HLA resolution upgrading from HLA genotypes. Researchers considering to explore HLA should take advantage of existing resources and mobilize them when taking on new challenges, such as with the SARS-CoV-2 research.
Author Contributions
VD contributed in writing the review and produced figures. EC, SM, JH, P-AG, NV, and SL contributed in writing and editing various sections of the review.
Funding
NV has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 846520. This work is supported by the ATIP-Avenir Inserm program, the Region Pays de Loire ConnectTalent. This work was also supported by United States, National Institutes of Health (NIH) National Institute of Allergy and Infectious Disease (NIAID) grants R01AI128775 (JH, SJM), and R01AI158861 (JH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aguiar, V. R. C., Augusto, D. G., Castelli, E. C., Hollenbach, J. A., Meyer, D., Nunes, K., et al. (2021). An Immunogenetic View of COVID-19. Genet. Mol. Biol. 44 (1), 1–24. doi:10.1590/1678-4685-gmb-2021-0036
Aguiar, V. R. C., Masotti, C., Camargo, A. A., and Meyer, D. (2020). “HLApers: HLA Typing and Quantification of Expression with Personalized Index,” in Methods in Molecular Biology. Editor S. Boegel (New York, NY: Springer US), Vol. 2120, 101–112. doi:10.1007/978-1-0716-0327-7_7
Allanore, Y., Saad, M., Dieudé, P., Avouac, J., Distler, J. H. W., Amouyel, P., et al. (2011). Genome-Wide Scan Identifies TNIP1, PSORS1C1, and RHOB as Novel Risk Loci for Systemic Sclerosis. Plos Genet. 7 (7), e1002091. doi:10.1371/journal.pgen.1002091
Allen, F. H., Amos, D. B., Batchelor, J. R., Bodmer, W. F., Ceppellini, R., Dausset, J., et al. (1968). Nomenclature for Factors of the HL-A System. Bull. World Health Organ. 39 (3), 483–486. Available at: http://www.ncbi.nlm.nih.gov/pubmed/5303912.
Ananeva, A., Sergeeva, I., Gusev, O., and Shagimardanova, E. (2021). Three Novel HLA‐C Alleles Identified in Russian Individuals: C*04:01:124 , C*12:02:38, and C*12:03:64. HLA 97 (3), 237. doi:10.1111/tan.14178
Ananeva, A., Leksina, Y., Andryushkina, A., and Shagimardanova, E. (2021). The Novel HLA‐A*02:941 Allele Was Identified during High‐resolution HLA Typing. Hla 97 (2), 136–138. doi:10.1111/tan.14088
Antigens, T. (1987). Nomenclature for Factors of the HLA System. Tissue Antigens 32 (4), 177–187. Available at: http://www.ncbi.nlm.nih.gov/pubmed/3217934.
Apps, R., Qi, Y., Carlson, J. M., Chen, H., Gao, X., Thomas, R., et al. (2013). Influence of HLA-C Expression Level on HIV Control. Science (80- ) 340 (6128), 87–91. doi:10.1126/science.1232685
Baier, D. M., Hofmann, J. A., Fischer, H., Rall, G., Stolze, J., Ruhner, K., et al. (2019). Very Low Error Rates of NGS-Based HLA Typing at Stem Cell Donor Recruitment Question the Need for a Standard Confirmatory Typing Step before Donor Work-Up. Bone Marrow Transpl. 54 (6), 928–930. doi:10.1038/s41409-018-0411-2
Bashirova, A. A., Viard, M., Naranbhai, V., Grifoni, A., Garcia-Beltran, W., Akdag, M., et al. (2020). HLA Tapasin independence: Broader Peptide Repertoire and HIV Control. Proc. Natl. Acad. Sci. USA 117 (45), 28232–28238. doi:10.1073/pnas.2013554117
Bauer, D. C., Zadoorian, A., Wilson, L. O. W., and Thorne, N. P. (2016). Evaluation of Computational Programs to Predict HLA Genotypes from Genomic Sequencing Data. Brief Bioinform 19 (2), bbw097. doi:10.1093/bib/bbw097
Beck, S., Geraghty, D., Inoko, H., Rowen, L., Aguado, B., Bahram, S., et al. (1999). Complete Sequence and Gene Map of a Human Major Histocompatibility Complex. The MHC Sequencing Consortium. Nature 401 (6756), 921. doi:10.1038/44853
Boegel, S., Löwer, M., Schäfer, M., Bukur, T., de Graaf, J., Boisguérin, V., et al. (2012). HLA Typing from RNA-Seq Sequence Reads. Genome Med. 4 (12), 102. doi:10.1186/gm403
Brandt, D. Y. C., Aguiar, V. R. C., Bitarello, B. D., Nunes, K., Goudet, J., and Meyer, D. (2015). Mapping Bias Overestimates Reference Allele Frequencies at the HLA Genes in the 1000 Genomes Project Phase I Data. G3 (Bethesda) 5 (5), 931–941. doi:10.1534/g3.114.015784
Brandt, D. Y. C., César, J., Goudet, J., and Meyer, D. (2018). The Effect of Balancing Selection on Population Differentiation: A Study with HLA Genes. G3 (Bethesda) 8 (8), 2805–2815. doi:10.1534/g3.118.200367
Browning, B. L., and Browning, S. R. (2009). A Unified Approach to Genotype Imputation and Haplotype-phase Inference for Large Data Sets of Trios and Unrelated Individuals. Am. J. Hum. Genet. 84 (2), 210–223. doi:10.1016/j.ajhg.2009.01.005
Browning, B. L., Zhou, Y., and Browning, S. R. (2018). A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 103 (3), 338–348. doi:10.1016/j.ajhg.2018.07.015
Carrington, M., Martin, M. P., and van Bergen, J. (2008). KIR-HLA intercourse in HIV Disease. Trends Microbiol. 16 (12), 620–627. doi:10.1016/j.tim.2008.09.002
Castelli, E. C., de Castro, M. V., Naslavsky, M. S., Scliar, M. O., Silva, N. S. B., Andrade, H. S., et al. (2021). MHC Variants Associated with Symptomatic versus Asymptomatic SARS-CoV-2 Infection in Highly Exposed Individuals. Front. Immunol. 12 (September), 1–11. doi:10.3389/fimmu.2021.742881
Castelli, E. C., Paz, M. A., Souza, A. S., Ramalho, J., and Mendes-Junior, C. T. (2018). Hla-mapper: An Application to Optimize the Mapping of HLA Sequences Produced by Massively Parallel Sequencing Procedures. Hum. Immunol. 79 (9), 678–684. doi:10.1016/j.humimm.2018.06.010
Chen, J., Madireddi, S., Nagarkar, D., Migdal, M., Vander Heiden, J., Chang, D., et al. (2021). In Silico tools for Accurate HLA and KIR Inference from Clinical Sequencing Data Empower Immunogenetics on Individual-Patient and Population Scales. Brief Bioinform 22 (3), 1–11. doi:10.1093/bib/bbaa223
Cheranev, V., Loginova, M., Jankevic, T., Rebrikov, D., and Korostin, D. (2021). HLA‐A *11: 382N , a Novel HLA‐A Null Allele Identified by Next‐generation Sequencing. Hla 97 (5), 448–449. doi:10.1111/tan.14185
Choi, W., Luo, Y., Raychaudhuri, S., and Han, B. (2021). HATK: HLA Analysis Toolkit. Bioinformatics 37 (3), 416–418. doi:10.1093/bioinformatics/btaa684
Claussnitzer, M., Cho, J. H., Collins, R., Cox, N. J., Dermitzakis, E. T., Hurles, M. E., et al. (2020). A Brief History of Human Disease Genetics. Nature 577 (7789), 179–189. doi:10.1038/s41586-019-1879-7
Colombo, S., Rauch, A., Rotger, M., Fellay, J., Martinez, R., Fux, C., et al. (2008). TheHCP5Single‐Nucleotide Polymorphism: A Simple Screening Tool for Prediction of Hypersensitivity Reaction to Abacavir. J. Infect. Dis. 198 (6), 864–867. doi:10.1086/591184
Cook, S., Choi, W., Lim, H., Luo, Y., Kim, K., Jia, X., et al. (2021). Accurate Imputation of Human Leukocyte Antigens with CookHLA. Nat. Commun. 12, 1–11. doi:10.1038/s41467-021-21541-5
Cook, S., and Han, B. (2017). MergeReference: A Tool for Merging Reference Panels for HLA Imputation. Genomics Inform. 15 (3), 108–111. doi:10.5808/gi.2017.15.3.108
COVID-19 Host Genetics Initiative (2021). Mapping the Human Genetic Architecture of COVID-19. Nature. Available at: http://www.nature.com/articles/s41586-021-03767-x.
Dausset, J. (1981). The Major Histocompatibility Complex in Man: Past, Present and Futur Concepts. Science (80- ) 213 (September), 55–97. Available at: http://linkinghub.elsevier.com/retrieve/pii/B9780124169746000065.
de Bakker, P. I. W., McVean, G., Sabeti, P. C., Miretti, M. M., Green, T., Marchini, J., et al. (2006). A High-Resolution HLA and SNP Haplotype Map for Disease Association Studies in the Extended Human MHC. Nat. Genet. 38 (10), 1166–1172. doi:10.1038/ng1885
De Santis, D., Dinauer, D., Duke, J., Erlich, H. A., Holcomb, C. L., Lind, C., et al. (2013). 16 Th IHIW : Review of HLA Typing by NGS. Int. J. Immunogenet. 40 (1), 72–76. doi:10.1111/iji.12024
De Santis, D., Truong, L., Martinez, P., and D'Orsogna, L. (2020). Rapid High‐resolution HLA Genotyping by MinION Oxford Nanopore Sequencing for Deceased Donor Organ Allocation. Hla 96 (2), 141–162. doi:10.1111/tan.13901
Degenhardt, F., Wendorff, M., Wittig, M., Ellinghaus, E., Datta, L. W., Schembri, J., et al. (2019). Construction and Benchmarking of a Multi-Ethnic Reference Panel for the Imputation of HLA Class I and II Alleles. Hum. Mol. Genet. 28 (12), 2078–2092. doi:10.1093/hmg/ddy443
del Guercio, M. F., Sidney, J., Hermanson, G., Perez, C., Grey, H. M., Kubo, R. T., et al. (1995). Binding of a Peptide Antigen to Multiple HLA Alleles Allows Definition of an A2-like Supertype. J. Immunol. 154 (2), 685–693. Available at: http://www.ncbi.nlm.nih.gov/pubmed/7529283.
Dendrou, C. A., Petersen, J., Rossjohn, J., and Fugger, L. (2018). HLA Variation and Disease. Nat. Rev. Immunol. 18 (5), 325–339. doi:10.1038/nri.2017.143
Di, D., Nunes, J. M., Jiang, W., and Sanchez-Mazas, A. (2021). Like Wings of a Bird: Functional Divergence and Complementarity between HLA-A and HLA-B Molecules. Mol. Biol. Evol. 38 (4), 1580–1594. doi:10.1093/molbev/msaa325
Dilthey, A. T., Moutsianas, L., Leslie, S., and McVean, G. (2011). HLA*IMP-an Integrated Framework for Imputing Classical HLA Alleles from SNP Genotypes. Bioinformatics 27 (7), 968–972. doi:10.1093/bioinformatics/btr061
Donadi, E. A., Castelli, E. C., Arnaiz-Villena, A., Roger, M., Rey, D., and Moreau, P. (2011). Implications of the Polymorphism of HLA-G on its Function, Regulation, Evolution and Disease Association. Cell. Mol. Life Sci. 68 (3), 369–395. doi:10.1007/s00018-010-0580-7
Douillard, V., Castelli, E., Mack, S. J., Hollenbach, J., Gourraud, P-A., Vince, N., et al. (2021). Approaching Genetics through the MHC Lens: Current HLA Investigations on SARS-CoV-2 and Perspectives. Front. Genet. In review.
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin Suite Ver 3.5: a New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 10 (3), 564–567. doi:10.1111/j.1755-0998.2010.02847.x
Fan, Y., and Song, Y.-Q. (2017). PyHLA: Tests for the Association between HLA Alleles and Diseases. BMC Bioinformatics 18 (1), 90. doi:10.1186/s12859-017-1496-0
Faner, R., James, E., Huston, L., Pujol-Borrel, R., Kwok, W. W., and Juan, M. (2009). Reassessing the Role of HLA-DRB3 T-Cell Responses: Evidence for Significant Expression and Complementary Antigen Presentation. Eur. J. Immunol. 40 (1), 91–102. doi:10.1002/eji.200939225
Geffard, E., Limou, S., Walencik, A., Daya, M., Watson, H., Torgerson, D., et al. (2020). Easy-HLA: a Validated Web Application Suite to Reveal the Full Details of HLA Typing. Bioinformatics 36 (7), 2157–2164. doi:10.1093/bioinformatics/btz875
Ghadially, H., Brown, L., Lloyd, C., Lewis, L., Lewis, A., Dillon, J., et al. (2017). MHC Class I Chain-Related Protein A and B (MICA and MICB) Are Predominantly Expressed Intracellularly in Tumour and normal Tissue. Br. J. Cancer 116 (9), 1208–1217. doi:10.1038/bjc.2017.79
Hayashi, S., Moriyama, T., Yamaguchi, R., Mizuno, S., Komura, M., Miyano, S., et al. (2019). ALPHLARD-NT: Bayesian Method for Human Leukocyte Antigen Genotyping and Mutation Calling through Simultaneous Analysis of Normal and Tumor Whole-Genome Sequence Data. J. Comput. Biol. 26 (9), 923–937. doi:10.1089/cmb.2018.0224
Horton, R., Wilming, L., Rand, V., Lovering, R. C., Bruford, E. A., Khodiyar, V. K., et al. (2004). Gene Map of the Extended Human MHC. Nat. Rev. Genet. 5 (12), 889–899. doi:10.1038/nrg1489
Hosomichi, K., Shiina, T., Tajima, A., and Inoue, I. (2015). The Impact of Next-Generation Sequencing Technologies on HLA Research. J. Hum. Genet. 60 (11), 665–673. doi:10.1038/jhg.2015.102
Hughes, A. L., and Nei, M. (1988). Pattern of Nucleotide Substitution at Major Histocompatibility Complex Class I Loci Reveals Overdominant Selection. Nature 335 (6186), 167–170. doi:10.1038/335167a0
Hurley, C. K. (2021). Naming HLA Diversity: A Review of HLA Nomenclature. Hum. Immunol. 82 (7), 457–465. doi:10.1016/j.humimm.2020.03.005
Jeanmougin, M., Noirel, J., Coulonges, C., and Zagury, J.-F. (2017). HLA-check: Evaluating HLA Data from SNP Information. BMC Bioinformatics 18 (1), 334. doi:10.1186/s12859-017-1746-1
Jekarl, D. W., Lee, G. D., Yoo, J. Bin., Kim, J. R., Yu, H., Yoo, J., et al. (2021). HLA-A, -B, -C, -DRB1 Allele and Haplotype Frequencies of the Korean Population and Performance Characteristics of HLA Typing by Next‐generation Sequencing. HLA 97 (3), 188–197. doi:10.1111/tan.14167
Jia, X., Han, B., Onengut-Gumuscu, S., Chen, W.-M., Concannon, P. J., Rich, S. S., et al. (2013). Imputing Amino Acid Polymorphisms in Human Leukocyte Antigens. PLoS One 8 (6), e64683. doi:10.1371/journal.pone.0064683
Juhos, S., Vágó, T., Ferriola, D., Duke, J., Vörös, S., Brown, B. O., et al. (2015). Deriving HLA Genotyping from Whole Genome Sequencing Data Using Omixon HLA Twin(tm) in G3's Global Clinical Study. Hum. Immunol. 76, 131. doi:10.1016/j.humimm.2015.07.183
Jurtz, V., Paul, S., Andreatta, M., Marcatili, P., Peters, B., and Nielsen, M. (2017). NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 199 (9), 3360–3368. doi:10.4049/jimmunol.1700893
Karnes, J. H., Shaffer, C. M., Bastarache, L., Gaudieri, S., Glazer, A. M., Steiner, H. E., et al. (2017). Comparison of HLA Allelic Imputation Programs. PLoS ONE 12 (2), e0172444. doi:10.1371/journal.pone.0172444
Kaufman, J. (2018). Generalists and Specialists: A New View of How MHC Class I Molecules Fight Infectious Pathogens. Trends Immunol. 39 (5), 367–379. doi:10.1016/j.it.2018.01.001
Kennedy, A. E., Ozbek, U., and Dorak, M. T. (2017). What Has GWAS Done for HLA and Disease Associations? Int. J. Immunogenet. 44 (5), 195–211. doi:10.1111/iji.12332
Khor, S.-S., Yang, W., Kawashima, M., Kamitsuji, S., Zheng, X., Nishida, N., et al. (2015). High-Accuracy Imputation for HLA Class I and II Genes Based on High-Resolution SNP Data of Population-specific References. Pharmacogenomics J. 15 (6), 530–537. doi:10.1038/tpj.2015.4
Kim, H. J., and Pourmand, N. (2013). HLA Haplotyping from RNA-Seq Data Using Hierarchical Read Weighting. PLoS ONE 8 (6), e67885. doi:10.1371/journal.pone.0067885
Klasberg, S., Surendranath, V., Lange, V., and Schöfl, G. (2019). Bioinformatics Strategies, Challenges, and Opportunities for Next Generation Sequencing-Based HLA Genotyping. Transfus. Med. Hemother 46 (5), 312–325. doi:10.1159/000502487
Kramer, C. S. M., Koster, J., Haasnoot, G. W., Roelen, D. L., Claas, F. H. J., and Heidt, S. (2020). HLA‐EMMA : A User‐friendly Tool to Analyse HLA Class I and Class II Compatibility on the Amino Acid Level. Hla 96 (1), 43–51. doi:10.1111/tan.13883
Kulkarni, S., Martin, M. P., and Carrington, M. (2008). The Yin and Yang of HLA and KIR in Human Disease. Semin. Immunol. 20 (6), 343–352. doi:10.1016/j.smim.2008.06.003
Kulkarni, S., Savan, R., Qi, Y., Gao, X., Yuki, Y., Bass, S. E., et al. (2011). Differential microRNA Regulation of HLA-C Expression and its Association with HIV Control. Nature 472 (7344), 495–498. doi:10.1038/nature09914
Kuniholm, M. H., Xie, X., Anastos, K., Xue, X., Reimers, L., French, A. L., et al. (2016). Human Leucocyte Antigen Class I and II Imputation in a Multiracial Population. Int. J. Immunogenet. 43 (6), 369–375. doi:10.1111/iji.12292
Lancaster, A. K., Single, R. M., Solberg, O. D., Nelson, M. P., and Thomson, G. (2007). PyPop Update - a Software Pipeline for Large-Scale Multilocus Population Genomics. Tissue Antigens 69 (3), 192–197. doi:10.1111/j.1399-0039.2006.00769.x
Lima, T. H. A., Souza, A. S., Porto, I. O. P., Paz, M. A., Veiga-Castelli, L. C., Oliveira, M. L. G., et al. (2019). HLA-A Promoter, Coding, and 3'UTR Sequences in a Brazilian Cohort, and Their Evolutionary Aspects. HLA 93 (2–3), 65–79. doi:10.1111/tan.13474
Loginova, M., Smirnova, D., Kutyavina, S., Paramonov, I., and Zarubin, M. (2021). The Novel HLA‐A Allele, HLA‐A*01:354 , Identified in a Buryat Individual. Hla 97 (5), 435–436. doi:10.1111/tan.14170
Loginova, M., Smirnova, D., Paramonov, I., and Kozhemyako, O. (2020). The Novel HLA‐DRB1*14:221 Allele Was Identified during High‐resolution HLA Typing. Hla 96 (2), 231–232. doi:10.1111/tan.13868
Luo, Y., Kanai, M., Choi, W., Li, X., Yamamoto, K., Ogawa, K., et al. (2020). A High-Resolution HLA Reference Panel Capturing Global Population Diversity Enables Multi-Ethnic fine-mapping in HIV Host Response. medRxiv, 1–46. doi:10.1101/2020.07.16.20155606
Mack, S. J., and Hollenbach, J. A. (2010). Allele Name Translation Tool and Update NomenCLature: Software Tools for the Automated Translation of HLA Allele Names between Successive Nomenclatures. Tissue Antigens 75 (5), 457–461. doi:10.1111/j.1399-0039.2010.01477.x
Mack, S. J., Milius, R. P., Gifford, B. D., Sauter, J., Hofmann, J., Osoegawa, K., et al. (2015). Minimum Information for Reporting Next Generation Sequence Genotyping (MIRING): Guidelines for Reporting HLA and KIR Genotyping via Next Generation Sequencing. Hum. Immunol. 76 (12), 954–962. doi:10.1016/j.humimm.2015.09.011
Marsh, S. G. E., Albert, E. D., Bodmer, W. F., Bontrop, R. E., Dupont, B., Erlich, H. A., et al. (2010). Nomenclature for Factors of the HLA System, 2010. Tissue Antigens 75 (4), 291–455. doi:10.1111/j.1399-0039.2010.01466.x
Martin, M. P., and Carrington, M. (2013). Immunogenetics of HIV Disease. Immunol. Rev. 254 (1), 245–264. doi:10.1111/imr.12071
Mayor, N. P., Robinson, J., McWhinnie, A. J. M., Ranade, S., Eng, K., Midwinter, W., et al. (2015). HLA Typing for the Next Generation. PLoS One 10 (5), e0127153. doi:10.1371/journal.pone.0127153
McCarthy, S., Das, S., Kretzschmar, W., Delaneau, O., Wood, A. R., Teumer, A., et al. (2016). A Reference Panel of 64,976 Haplotypes for Genotype Imputation. Nat. Genet. 48 (10), 1279–1283. doi:10.1038/ng.3643
McLaren, P. J., Ripke, S., Pelak, K., Weintrob, A. C., Patsopoulos, N. A., Jia, X., et al. (2012). Fine-mapping Classical HLA Variation Associated with Durable Host Control of HIV-1 Infection in African Americans. Hum. Mol. Genet. 21 (19), 4334–4347. doi:10.1093/hmg/dds226
Mei, S., Li, F., Leier, A., Marquez-Lago, T. T., Giam, K., Croft, N. P., et al. (2019). A Comprehensive Review and Performance Evaluation of Bioinformatics Tools for HLA Class I Peptide-Binding Prediction. Brief. Bioinform. 21, 1119–1135. Available at: https://academic.oup.com/bib/article/21/4/1119/5511798.
Meral, B. (2007). “Bone Marrow and Stem Cell Transplantation,” in Methods in Molecular Biology. Editor M. Beksaç (New Jersey: Humana Press), Vol. 134, 313. doi:10.1007/978-1-4614-9437-9
Meyer, D., C. Aguiar, V. R., Bitarello, B. D., C. Brandt, D. Y., and Nunes, K. (2018). A Genomic Perspective on HLA Evolution. Immunogenetics 70 (1), 5–27. doi:10.1007/s00251-017-1017-3
Meyer, D., and Nunes, K. (2017). HLA Imputation, what Is it Good for? Hum. Immunol. 78 (3), 239–241. doi:10.1016/j.humimm.2017.02.007
Meyer, D., and Thomson, G. (2001). How Selection Shapes Variation of the Human Major Histocompatibility Complex: a Review. Ann. Hum. Genet 65 (1), 1–26. doi:10.1046/j.1469-1809.2001.6510001.x
Middleton, D., Menchaca, L., Rood, H., and Komerofsky, R. (2003). New Allele Frequency Database: http://www.allelefrequencies.Net. Tissue Antigens 61 (5), 403–407. doi:10.1034/j.1399-0039.2003.00062.x
Milius, R. P., Heuer, M., Valiga, D., Doroschak, K. J., Kennedy, C. J., Bolon, Y.-T., et al. (2015). Histoimmunogenetics Markup Language 1.0: Reporting Next Generation Sequencing-Based HLA and KIR Genotyping. Hum. Immunol. 76 (12), 963–974. doi:10.1016/j.humimm.2015.08.001
Mimori, T., Yasuda, J., Kuroki, Y., Shibata, T. F., Katsuoka, F., Saito, S., et al. (2019). Construction of Full-Length Japanese Reference Panel of Class I HLA Genes with Single-Molecule, Real-Time Sequencing. Pharmacogenomics J. 19 (2), 136–146. doi:10.1038/s41397-017-0010-4
Montgomery, R. A., Tatapudi, V. S., Leffell, M. S., and Zachary, A. A. (2018). HLA in Transplantation. Nat. Rev. Nephrol. 14, 558–570. doi:10.1038/s41581-018-0039-x
Mosbruger, T. L., Dinou, A., Duke, J. L., Ferriola, D., Mehler, H., Pagkrati, I., et al. (2020). Utilizing Nanopore Sequencing Technology for the Rapid and Comprehensive Characterization of Eleven HLA Loci; Addressing the Need for Deceased Donor Expedited HLA Typing. Hum. Immunol. 81 (8), 413–422. doi:10.1016/j.humimm.2020.06.004
Motyer, A., Vukcevic, D., Dilthey, A., Donnelly, P., McVean, G., and Leslie, S. (2016). Practical Use of Methods for Imputation of HLA Alleles from SNP Genotype Data. bioRxiv, 091009.
Murdoch, D. J., Tsai, Y.-L., and Adcock, J. (2008). P-values Are Random Variables. The Am. Statistician 62 (3), 242–245. doi:10.1198/000313008x332421
Naito, T., Suzuki, K., Hirata, J., Kamatani, Y., Matsuda, K., Toda, T., et al. (2021). A Deep Learning Method for HLA Imputation and Trans-ethnic MHC fine-mapping of Type 1 Diabetes. Nat. Commun. 12 (1), 1–14. doi:10.1038/s41467-021-21975-x
Nielsen, M., and Andreatta, M. (2016). NetMHCpan-3.0; Improved Prediction of Binding to MHC Class I Molecules Integrating Information from Multiple Receptor and Peptide Length Datasets. Genome Med. 8 (1), 1–9. doi:10.1186/s13073-016-0288-x
Nilsson, L. L., Funck, T., Kjersgaard, N. D., and Hviid, T. V. F. (2018). Next-generation Sequencing ofHLA-Gbased on Long-Range Polymerase Chain Reaction. Hla 92 (3), 144–153. doi:10.1111/tan.13342
Nunes, J. M., Buhler, S., Roessli, D., and Sanchez-Mazas, A. (2014). TheHLA-net GENE[RATE]pipeline for Effective HLA Data Analysis and its Application to 145 Population Samples from Europe and Neighbouring Areas. Tissue Antigens 83 (5), 307–323. doi:10.1111/tan.12356
Osoegawa, K., Mack, S. J., Udell, J., Noonan, D. A., Ozanne, S., Trachtenberg, E., et al. (2016). HLA Haplotype Validator for Quality Assessments of HLA Typing. Hum. Immunol. 77 (3), 273–282. doi:10.1016/j.humimm.2015.10.018
Pappas, D. J., Lizee, A., Paunic, V., Beutner, K. R., Motyer, A., Vukcevic, D., et al. (2018). Significant Variation between SNP-Based HLA Imputations in Diverse Populations: the Last Mile Is the Hardest. Pharmacogenomics J. 18 (3), 367–376. doi:10.1038/tpj.2017.7
Pappas, D. J., Marin, W., Hollenbach, J. A., and Mack, S. J. (2016). Bridging ImmunoGenomic Data Analysis Workflow Gaps (BIGDAWG): An Integrated Case-Control Analysis Pipeline. Hum. Immunol. 77 (3), 283–287. doi:10.1016/j.humimm.2015.12.006
Parham, P., Norman, P. J., Abi-Rached, L., and Guethlein, L. A. (2012). Human-specific Evolution of Killer Cell Immunoglobulin-like Receptor Recognition of Major Histocompatibility Complex Class I Molecules. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367 (1590), 800. doi:10.1098/rstb.2011.0266
Park, I., and Terasaki, P. (2000). Origins of the First HLA Specificities. Hum. Immunol. 61 (3), 185–189. doi:10.1016/s0198-8859(99)00154-8
Praest, P., Luteijn, R. D., Brak-Boer, I. G. J., Lanfermeijer, J., Hoelen, H., Ijgosse, L., et al. (2018). The Influence of TAP1 and TAP2 Gene Polymorphisms on TAP Function and its Inhibition by Viral Immune Evasion Proteins. Mol. Immunol. 101 (May), 55–64. doi:10.1016/j.molimm.2018.05.025
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi:10.1086/519795
Ralazamahaleo, M., Andreani, M., Giustiniani, P., Guidicelli, G., and Visentin, J. (2019). Characterization of the Novel HLA‐DQA1*01:01:05 Allele by Sequencing‐based Typing. Hla 94 (2), 172–173. doi:10.1111/tan.13569
Ritari, J., Hyvärinen, K., Clancy, J., Partanen, J., and Koskela, S. (2020). Increasing Accuracy of HLA Imputation by a Population-specific Reference Panel in a FinnGen Biobank Cohort. NAR Genomics Bioinforma 2 (2), 1–9. doi:10.1093/nargab/lqaa030
Robinson, J., Barker, D. J., Georgiou, X., Cooper, M. A., Flicek, P., and Marsh, S. G. E. (2019). IPD-IMGT/HLA Database. Nucleic Acids Res. 48 (D1), D948–D955. doi:10.1093/nar/gkz950
Robinson, J., Halliwell, J. A., Hayhurst, J. D., Flicek, P., Parham, P., and Marsh, S. G. (2015). The IPD and IMGT/HLA Database: Allele Variant Databases. Nucleic Acids Res. 43, D423–D431. doi:10.1093/nar/gku1161
Rock, K. L., Reits, E., and Neefjes, J. (2016). Present Yourself! by MHC Class I and MHC Class II Molecules. Trends Immunol. 37, 724–737. doi:10.1016/j.it.2016.08.010
Sacchi, N., Castagnetta, M., Miotti, V., Garbarino, L., and Gallina, A. (2019). High‐resolution Analysis of the HLA‐A, ‐B, ‐C and ‐DRB1 Alleles and National and Regional Haplotype Frequencies Based on 120 926 Volunteers from the Italian Bone Marrow Donor Registry. Hla 94 (3), 285–295. doi:10.1111/tan.13613
Schmidt, A. H., Sauter, J., Baier, D. M., Daiss, J., Keller, A., Klussmeier, A., et al. (2020). Immunogenetics in Stem Cell Donor Registry Work: The DKMS Example (Part 1). Int. J. Immunogenet. 47 (1), 13–23. doi:10.1111/iji.12471
Shiina, T., Hosomichi, K., Inoko, H., and Kulski, J. K. (2009). The HLA Genomic Loci Map: Expression, Interaction, Diversity and Disease. J. Hum. Genet. 54 (1), 15–39. doi:10.1038/jhg.2008.5
Sidney, J., del Guercio, M. F., Southwood, S., Engelhard, V. H., Appella, E., Rammensee, H. G., et al. (1995). Several HLA Alleles Share Overlapping Peptide Specificities. J. Immunol. 154 (1), 247–259. Available at: http://www.ncbi.nlm.nih.gov/pubmed/7527812.
Spurgin, L. G., and Richardson, D. S. (2010). How Pathogens Drive Genetic Diversity: MHC, Mechanisms and Misunderstandings. Proc. Biol. Sci. 277, 979–988. doi:10.1098/rspb.2009.2084
Squire, D. M., Motyer, A., Ahn, R., Nititham, J., Huang, Z.-M., Oksenberg, J. R., et al. (2020). MHC*IMP - Imputation of Alleles for Genes in the Major Histocompatibility Complex. bioRxiv. doi:10.1101/2020.01.24.919191
The COVID-19 HLA and Immunogenetics Consortium (2020a). HLA|COVID-19. Available at: http://www.hlacovid19.org/.
The COVID-19 HLA and Immunogenetics Consortium (2020b). HLA|COVID-19 Database. Available at: https://database-hlacovid19.org/.
Trowsdale, J., and Knight, J. C. (2013). Major Histocompatibility Complex Genomics and Human Disease. Annu. Rev. Genom. Hum. Genet. 14 (1), 301–323. doi:10.1146/annurev-genom-091212-153455
Trowsdale, J., and Moffett, A. (2008). NK Receptor Interactions with MHC Class I Molecules in Pregnancy. Semin. Immunol. 20 (6), 317–320. doi:10.1016/j.smim.2008.06.002
van Tol, S., Hage, A., Giraldo, M., Bharaj, P., and Rajsbaum, R. (2017). The TRIMendous Role of TRIMs in Virus-Host Interactions. Vaccines 5 (3), 23. doi:10.3390/vaccines5030023
Vayntrub, T. A., Mack, S. J., and Fernandez-Viña, M. A. (2020). Preface: 17th International HLA and Immunogenetics Workshop. Hum. Immunol. 81, 52–58. doi:10.1016/j.humimm.2020.01.008
Vince, N., Bashirova, A. A., Lied, A., Gao, X., Dorrell, L., McLaren, P. J., et al. (2014). HLA Class I and KIR Genes Do Not Protect against HIV Type 1 Infection in Highly Exposed Uninfected Individuals with Hemophilia A. J. Infect. Dis. 210 (7), 1047–1051. doi:10.1093/infdis/jiu214
Vince, N., Douillard, V., Geffard, E., Meyer, D., Castelli, E. C., Mack, S. J., et al. (2020). SNP‐HLA Reference Consortium (SHLARC): HLA and SNP Data Sharing for Promoting MHC‐centric Analyses in Genomics. Genet. Epidemiol. 44 (7), 733–740. doi:10.1002/gepi.22334
Vince, N., Li, H., Ramsuran, V., Naranbhai, V., Duh, F.-M., Fairfax, B. P., et al. (2016). HLA-C Level Is Regulated by a Polymorphic Oct1 Binding Site in the HLA-C Promoter Region. Am. J. Hum. Genet. 99 (6), 1353–1358. doi:10.1016/j.ajhg.2016.09.023
Vince, N., Limou, S., Daya, M., Morii, W., Rafaels, N., Geffard, E., et al. (2020). Association of HLA-Drb1∗09:01 with tIgE Levels Among African-Ancestry Individuals with Asthma. J. Allergy Clin. Immunol. 146 (1), 147–155. doi:10.1016/j.jaci.2020.01.011
Wainberg, M., Sinnott-Armstrong, N., Mancuso, N., Barbeira, A. N., Knowles, D. A., Golan, D., et al. (2019). Opportunities and Challenges for Transcriptome-wide Association Studies. Nat. Genet. 51 (4), 592–599. doi:10.1038/s41588-019-0385-z
Wang, M., and Claesson, M. H. (2014). “Classification of Human Leukocyte Antigen (HLA) Supertypes,” in Methods in Molecular Biology (Clifton, NJ), 309–317. doi:10.1007/978-1-4939-1115-8_17
Xie, C., Yeo, Z. X., Wong, M., Piper, J., Long, T., Kirkness, E. F., et al. (2017). Fast and Accurate HLA Typing from Short-Read Next-Generation Sequence Data with xHLA. Proc. Natl. Acad. Sci. USA 114 (30), 8059–8064. doi:10.1073/pnas.1707945114
Keywords: Major Histocompatibility Complex (MHC), HLA, association analysis, imputation, immunogenetics
Citation: Douillard V, Castelli EC, Mack SJ, Hollenbach JA, Gourraud P-A, Vince N and Limou S (2021) Approaching Genetics Through the MHC Lens: Tools and Methods for HLA Research. Front. Genet. 12:774916. doi: 10.3389/fgene.2021.774916
Received: 13 September 2021; Accepted: 08 November 2021;
Published: 02 December 2021.
Edited by:
Ramcés Falfán-Valencia, Instituto Nacional de Enfermedades Respiratorias-México (INER), MexicoReviewed by:
Gilberto Vargas Alarcón, Instituto Nacional de Cardiologia Ignacio Chavez, MexicoPengyu Hong, Brandeis University, United States
Martha Perez-rodriguez, Mexican Social Security Institute (IMSS), Mexico
Copyright © 2021 Douillard, Castelli, Mack, Hollenbach, Gourraud, Vince and Limou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicolas Vince, bmljb2xhcy52aW5jZUB1bml2LW5hbnRlcy5mcg==; Sophie Limou, c29waGllLmxpbW91QHVuaXYtbmFudGVzLmZy
†These authors have contributed equally to this work and share last authorship.