 Dongxu Zhao
Dongxu Zhao Zhixia Teng
Zhixia Teng Yanjuan Li
Yanjuan Li Dong Chen2
Dong Chen2- 1College of Information and Computer Engineering, Northeast Forestry University, Harbin, China
- 2College of Electrical and Information Engineering, Quzhou University, Quzhou, China
Recently, several anti-inflammatory peptides (AIPs) have been found in the process of the inflammatory response, and these peptides have been used to treat some inflammatory and autoimmune diseases. Therefore, identifying AIPs accurately from a given amino acid sequences is critical for the discovery of novel and efficient anti-inflammatory peptide-based therapeutics and the acceleration of their application in therapy. In this paper, a random forest-based model called iAIPs for identifying AIPs is proposed. First, the original samples were encoded with three feature extraction methods, including g-gap dipeptide composition (GDC), dipeptide deviation from the expected mean (DDE), and amino acid composition (AAC). Second, the optimal feature subset is generated by a two-step feature selection method, in which the feature is ranked by the analysis of variance (ANOVA) method, and the optimal feature subset is generated by the incremental feature selection strategy. Finally, the optimal feature subset is inputted into the random forest classifier, and the identification model is constructed. Experiment results showed that iAIPs achieved an AUC value of 0.822 on an independent test dataset, which indicated that our proposed model has better performance than the existing methods. Furthermore, the extraction of features for peptide sequences provides the basis for evolutionary analysis. The study of peptide identification is helpful to understand the diversity of species and analyze the evolutionary history of species.
1 Introduction
As a part of the nonspecific immune response, inflammation response usually occurs in response to any type of bodily injury (Ferrero-Miliani et al., 2007). When the inflammatory response occurs in the condition of no obvious infection, or when the response continues despite the resolution of the initial insult, the process may be pathological and leads to chronic inflammation (Patterson et al., 2014). At present, the therapy for inflammatory and autoimmune diseases usually uses nonspecific anti-inflammatory drugs or other immunosuppressants, which may produce some side effects (Tabas and Glass, 2013; Yu et al., 2021). Several endogenous peptides found in the process of inflammatory response have become anti-inflammatory agents and can be used as new therapies for autoimmune diseases and inflammatory disorders (Gonzalez-Rey et al., 2007; Yu et al., 2020a). Compared with small-molecule drugs, the therapy based on peptides has minimal toxicity and high specificity under normal conditions, which is a better choice for inflammatory and autoimmune disorders and has been widely used in treatment (de la Fuente-Núñez et al., 2017; Shang et al., 2021).
Due to the biological importance of AIPs, many biochemical experimental methods have been developed for identifying AIPs. However, these biochemical methods usually need a long experimental cycle and have a high experimental cost. In recent years, machine learning has increasingly become the most popular tool in the field of bioinformatics (Zhao et al., 2017; Liu et al., 2020; Luo et al., 2020; Sun et al., 2020; Zhao et al., 2020; Jin et al., 2021; Wang et al., 2021a). Many researchers have tried to adopt machine learning algorithms to identify AIPs only based on peptide amino acid sequence information. In 2017, Gupta et al. proposed a predictor of AIPs based on the machine learning method. They constructed the combined features and inputted them in the SVM classifier to construct the prediction model (Gupta et al., 2017).
In 2018, Manavalan et al. proposed a novel prediction model called AIPpred. They encoded the original peptide sequence by the dipeptide composition (DPC) feature representation method, and then, they developed a random forest-based model to identify AIPs (Manavalan et al., 2018). AIEpred is a novel prediction model and is proposed by Zhang et al. AIEpred encodes peptide sequences based on three feature representations. Based on various feature representations, it constructed many base classifiers, which are the basis of ensemble classifier (Zhang et al., 2020a).
In this paper, we proposed a novel identification model of AIPs for further improving the identification ability. First, we encoded the samples with multiple features consisting of AAC, DDE, and GDC. It has been proven that multiple features can effectively discriminate positive instances from negative ones in various biological problems. Second, we selected the optimal features based on a feature selection strategy, which has achieved better performance in many biological problems. Finally, we used the random forest classifier to construct an identification model based on the optimal features. The experimental result shows that our proposed method in this paper has better performance than the existing methods.
2 Materials and Methods
Figure 1 gives the general framework of iAIPs proposed in this paper. The framework consists of four steps as follows: 1) Dataset preparation—It collects the data required for the experiment. 2) Feature extraction—It converts the collected sequence data from step 1 into numerical features. 3) Feature selection—removes redundant features from a feature set. 4) Prediction model construction. Each step of the framework will be described as follows.
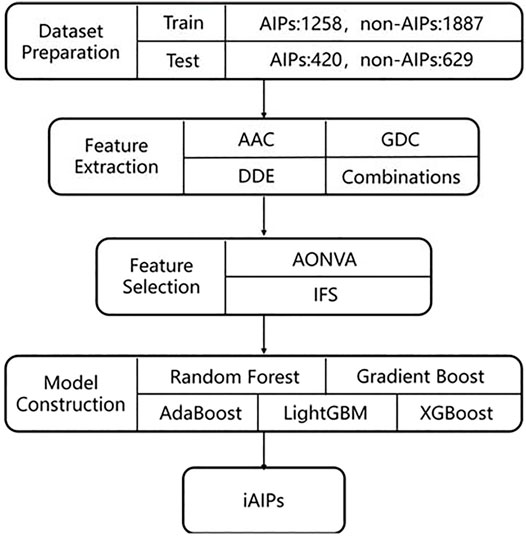
FIGURE 1. The framework of iAIPs.
2.1 Dataset Preparation
A high-quality dataset is critical to construct an effective and reliable prediction model. To measure the performance of our model by comparing it with other existing machine learning-based prediction models, we used the dataset with no change proposed in AIPpred (Manavalan et al., 2018). The dataset was first retrieved from the IEDB database (Kim et al., 2012; Vita et al., 2019), and then the samples with sequence identity >80% (Zou et al., 2020) are excluded by using CD-HIT (Huang et al., 2010). The dataset contains 1,678 AIPs and 2,516 non-AIPs. For this dataset, it is randomly selected as the training dataset, which is inputted into the classifier and used to construct the identification model. The training dataset is also used to measure the cross-validation performance of our model. The remaining dataset is used as an independent dataset, which will be used to evaluate the generalization capability of our identification model. In detail, the training dataset consists of 1,258 AIPs and 1,887 non-AIPs, and the independent dataset consists of 420 AIPs and 629 non-AIPs.
2.2 Feature Extraction Methods
In the process of peptide identification, finding an effective feature extraction method is the most important step (Liu, 2019; Fu et al., 2020; Cai et al., 2021). In this study, we tried a variety of feature extraction methods and used the random forest classifier to evaluate the performance of those methods. Finally, we chose three efficient feature extraction methods to encode peptide amino acid sequences, including amino acid composition, dipeptide deviation from expected mean, and g-gap dipeptide composition. The details of each feature extraction method are described as follows.
2.2.1 Amino Acid Composition
Different peptide sequences consist of different amino acid sequences. AAC tried to count the composition information of peptides. In detail, AAC calculates the frequency of occurrence of each amino acid type (Wei et al., 2018a; Liu et al., 2019; Ning et al., 2020; Yang et al., 2020; Zhang and Zou, 2020; Wu and Yu, 2021). The computation formula of AAC is as follows:
where L denotes the length of the peptide, which is the number of characters in the peptide, AAC (j) denotes the percentage of amino acid j, N (j) denotes the total number of amino acid j. The dimension of AAC is 20.
2.2.2 Dipeptide Deviation From the Expected Mean
According to the dipeptide composition information, DDE computes deviation frequencies from expected mean values (Saravanan and Gautham, 2015). The feature vector extracted by DDE is generated by three parameters: theoretical variance (TV), dipeptide composition (DC), and theoretical mean (TM). The formulas of the three parameters are as follows:
where
Cj1 denotes the number of codons that encode for the first amino acid, and Cj2 denotes the number of codons that encode for the second amino acid in the dipeptide j. CN denotes the total number of possible codons.
The formula of DDE(i) is as follows.
2.2.3 G-Gap Dipeptide Composition
GDC is used to measure the correlation of two non-adjacent residues; its dimension is 400 (Wei et al., 2018b). GDC can be represented as follows:
where
where
2.3 Feature Selection
In the Feature extraction methods section, we introduced the feature extraction method used in this paper. However, like other feature representation methods, our feature representation may also produce many noises (Wei et al., 2014; Wang et al., 2020a; Li et al., 2020; Tang et al., 2020; Wang et al., 2021b). Recently, many feature selection methods for eliminating noise has been used to solve many bioinformatics problems (He et al., 2020), such as TATA-binding protein prediction (Zou et al., 2016), DNA 4mc site prediction (Manavalan et al., 2019), antihypertensive peptide prediction (Manayalan et al., 2019), drug-induced hepatotoxicity prediction (Su et al., 2019), and enhance-promoter interaction prediction (Hong et al., 2020; Min et al., 2021).
Likewise, we will use a two-step feature selection method to solve the noise of features. In detail, the feature is first ranked based on the ANOVA score. Then, based on the orderly features, we use the incremental feature selection (IFS) strategy to generate different feature subsets, the feature subset with optimal performance is selected as the optimal feature subset. In the Result and discussion section, we will give the experiments about feature extraction, in which we will verify the effectiveness of our feature representation.
2.3.1 Analysis of Variance
In this work, the feature is first ranked based on the ANOVA score. For every feature, ANOVA calculated the ratio of the variance between groups and the variance within groups, which can test the mean difference between groups effectively (Ding et al., 2014). The score is calculated as follows:
where S (t) is the score of the feature t,
where K denotes the number of groups, and N denotes the total number of instances;
2.3.2 Incremental Feature Selection
Based on the orderly features, we use the incremental feature selection strategy to generate different feature subsets; the feature subset with optimal performance is selected as the optimal feature subset. In the incremental feature selection method, the feature set is constructed as empty at first, and then the feature vector is added one by one from the ranked feature set. Meanwhile, the new feature set is inputted into a classifier, and then a prediction model is constructed. We evaluate the performance of the model according to some indicators. Finally, the feature subset with the optimal performance is considered as the optimal feature set.
2.4 Machine Learning Methods
In this paper, we utilized various ensemble learning classification algorithms to develop identification models, which contain random forest (Ru et al., 2019; Wang et al., 2020b; Ao et al., 2021), AdaBoost, Gradient Boost Decision Tree (Yu et al., 2020b), LightGBM, and XGBoost. In addition, we also tried some traditional machine learning classification algorithms, such as logistic regression and Naïve Bayes. The description of these methods is as follows.
2.4.1 Random Forest
As one of the most powerful ensemble learning methods, random forest was proposed by Breiman (2001). Due to its effectiveness, random forest has been widely used in bioinformatics areas. Random forest can solve regression and classification tasks. To solve the problem, random forest uses the random feature selection method to construct hundreds or thousands of decision trees (Akbar et al., 2020). By voting on these decision trees, the final identification result is obtained. The random forest algorithm used in this paper is from WEKA (Hall et al., 2008), and all parameters are default.
2.4.2 AdaBoost
The AdaBoost algorithm is an iterative algorithm, which was proposed by Freund (1990). For a benchmark dataset, AdaBoost will train various weak classifiers and combine these weak classifiers by sample weight to construct a stronger final classifier. Among samples, low weights are assigned to easy samples that are classified correctly by the weak learner, while high weights are for the hard or misclassified samples. By constantly adjusting the weight of samples, AdaBoost will focus more on the samples that are classified incorrectly.
2.4.3 Gradient Boost Decision Tree
Similar to AdaBoost, Gradient Boost Decision Tree (GBDT) also combines weak learners to construct a prediction model (Friedman, 2001). Different from AdaBoost, GBDT will constantly adapt to the new model when the weak learners are learned. In detail, based on the negative gradient information of the loss function of the current model, the new weak classifier is trained. The training result is accumulated into the existing model to improve its performance (Basith et al., 2018).
2.4.4 LightGBM and XGBoost
Both LightGBM and XGBoost are improved algorithms based on GBDT. LightGBM is mainly optimized in three aspects. The histogram algorithm is used to convert continuous features into discrete features, the gradient-based one-side sampling (GOSS) method is used to adjust the sample distribution and reduce the numbers of samples, and the exclusive feature bundling (EFB) is used to merge multiple independent features. XGBoost adds the second-order Taylor expansion and regularization term to the loss function.
2.4.5 Naïve Bayes
Naïve Bayes is a probabilistic classification algorithm based on Bayes’ theorem, which assumes that the features are independent of each other. According to this theorem, the probability of a given sample classified into class k can be calculated as
where the sample has the expression formula of {X, C}.
2.4.6 Other Machine Learning Methods
Other traditional machine learning methods used for performance comparison include J48, logistic, SMO, and SGD. J48 is a decision tree algorithm provided in Weka, which is implemented based on the C4.5 idea. Logistic is a probability-based classification algorithm. Based on linear regression, Logistic introduces sigmoid function to limit the output value to [0,1] interval. SMO and SGD are optimization algorithms provided in Weka. SMO (sequential minimal optimization) is based on support vector machine (SVM), and SGD is based on linear regression.
2.5 Performance Evaluation
To measure the performance of our proposed model, we chose four commonly used measurements: SN, SP, ACC, and MCC (Jiang et al., 2013; Wei et al., 2017a; Ding et al., 2019; Shen et al., 2019; Huang et al., 2020). These measurements are calculated as follows.
where FP, FN, TN, and TP show the number of false-positive, false-negative, true-negative, and true-positive, respectively. These are widely used in bioinformatics studies, such as protein fold recognition (Shao et al., 2021), DNA-binding protein prediction (Wei et al., 2017b), protein–protein interaction prediction (Wei et al., 2017c), and drug–target interaction identification (Ding et al., 2020; Ding and JijunGuo, 2020).
Furthermore, we also used the receiver operating characteristic (ROC) curve (Hanley and McNeil, 1982; Fushing and Turnbull, 1996) to evaluate the performance of our proposed model. ROC computes the true-positive rate and low false-positive rate by setting various possible thresholds (Gribskov and Robinson, 1996). The area under the ROC curve (AUC) also shows the performance of the proposed model, which is more accurate in the aspect of evaluating the performance of the prediction model constructed by an imbalanced dataset.
3 Results and Discussion
To verify the effectiveness of our proposed model, we will measure the performance of our model from different perspectives. The detailed process of these experiments is presented as follows.
3.1 Performance of Different Features
In this study, we use a variety of feature extraction methods and their combinations to encode peptide sequences. At first, we measure the effectiveness of single features. The comparison results of the fivefold cross-validation on the training dataset are shown in Table 1.
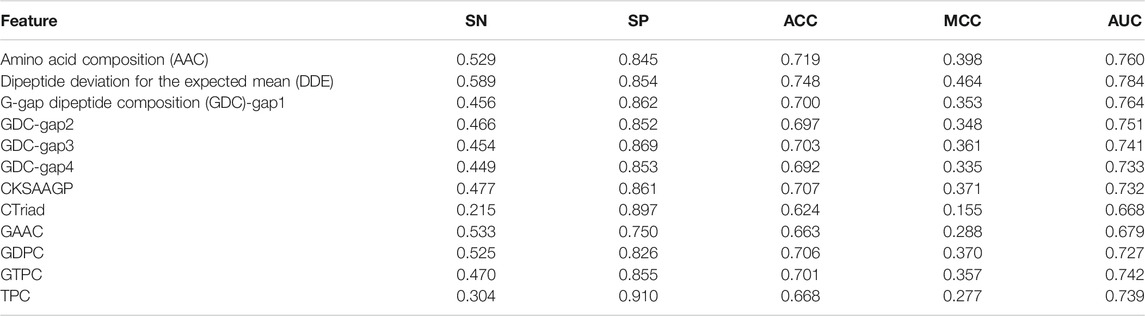
TABLE 1. Performance comparison of various single features.
Table 1 shows that DDE is much better than other features according to the indicators of AUC, MCC, ACC, SP, and SN. In detail, the AUC value reaches 0.784, which is 2%–11.6% higher than other features. Based on the indicator of AUC, the features of DDE, GDC-gap1, and AAC have the best performance.
To achieve better performance, we further test the performance of multiple features on the basis of DDE, GDC, and AAC. In detail, the GDC feature adopts four different parameters, that is, gap1, gap2, gap3, and gap4. The corresponding feature is GDC-gap1, GDC-gap2, GDC-gap3, and GDC-gap4. The performance comparison of the fivefold cross-validation on the training dataset is shown in Table 2.
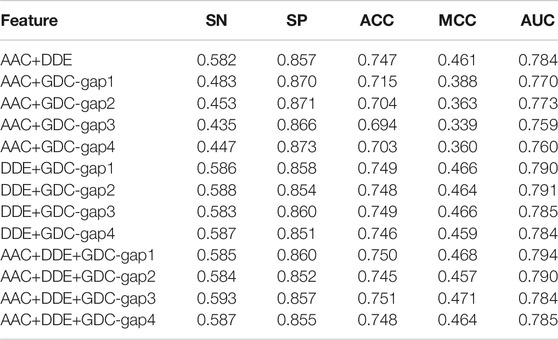
TABLE 2. Performance comparison of various combined features of fivefold cross-validation on the training dataset.
According to Table 2, the multiple features of AAC + DDE + GDC-gap1 has the best performance. Its value of SN, SP, ACC, MCC, and AUC are 0.585, 0.860, 0.750, 0.468, and 0.794, respectively.
To verify the performance of these combined features, we tested them on the independent test set. Table 3 shows the experimental results on the independent dataset. The results show that the combined features of AAC + DDE + GDC-gap1 have the best performance on the independent dataset.
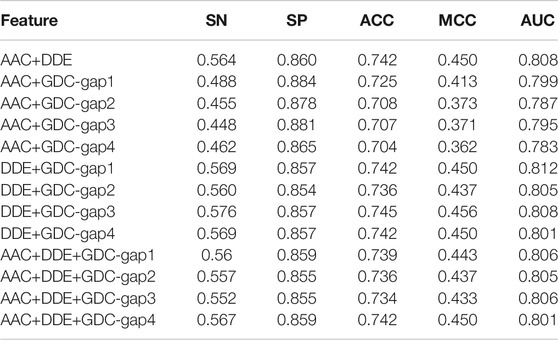
TABLE 3. Performance comparison of various combined features on the independent dataset.
3.2 Performance of Different Classifiers
In this study, we chose the random forest algorithm to construct the classifier. To verify the effectiveness of the random forest classifier, we compared its performance with other classifiers. We chose several ensemble classifiers that are similar to the random forest classifier, including AdaBoost, GBDT, LightGBM, and XGBoost. In addition, we also chose some machine learning classifiers, including J48, Logistic, SMO, SGD, and Naïve Bayes.
Based on the best feature combination, which is obtained from previous experiments, we constructed different identification models using different classifiers. The performance of these classifiers on the training dataset is shown in Table 4.
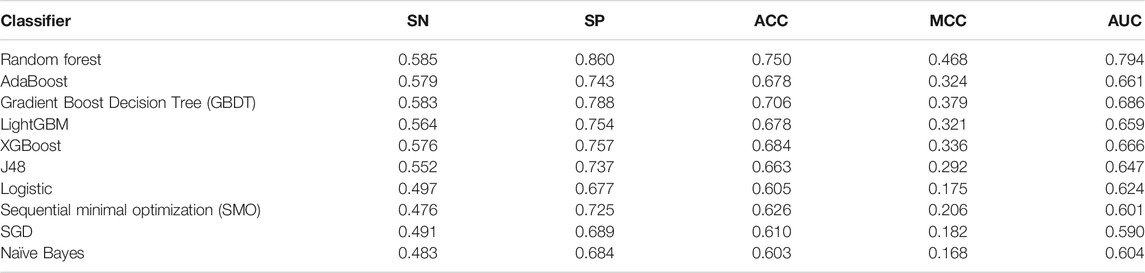
TABLE 4. Performance of various classifiers utilizing AAC-DDE-GDC-gap1 feature and fivefold cross-validation on the training dataset.
The results in Table 4 show that the performance of the random forest classifier is the best, and its AUC value is 10.8%–20.4% higher than other classifiers. To further compare the generalization ability of these classifiers, we test those models on the independent dataset. Table 5 shows the experimental results. The results showed that the random forest classifier is also better than other classifiers on the independent dataset.
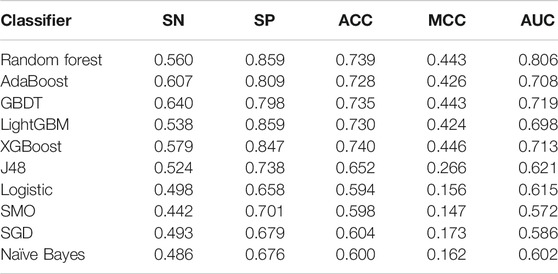
TABLE 5. Performance of various classifiers based on AAC-DDE-GDC-gap1 feature on the independent dataset.
3.3 The Analysis of Feature Selection
In the extracted features, some feature vectors may be noisy or redundant. To further improve the identification performance, we try to find optimal features by feature selection methods in this section. In this paper, the two-step feature selection strategy is used as the feature selection strategy to eliminate noise. In detail, we first used the ANOVA method to rank feature vectors, and then we used the IFS strategy to filter the optimal feature set.
The comparison of performance before and after dimensionality reduction is shown in Figure 2. All indicators of the selected features have higher values than the original ones. The results suggest that the optimal feature set can improve the overall performance of our identification model and our fewer selected features can still accurately describe AIPs.
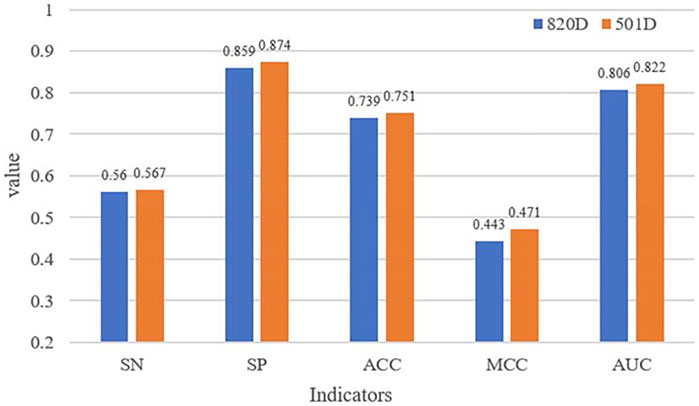
FIGURE 2. Comparison of identification performance before and after dimensionality reduction.
3.4 Comparison With Existing Methods
Independent dataset test plays an important role in testing the generalization ability of the identification model. Therefore, the independent dataset was used to measure our identification model; the performance of our identification model was compared with existing methods, which contains AntiInflam (Ferrero-Miliani et al., 2007), AIPpred, and AIEpred. Table 6 shows the detailed results of the different methods for identifying AIPs, where the results are ranked according to AUC.
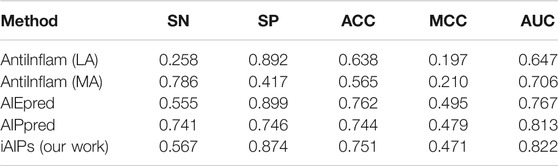
TABLE 6. Performance of different identification models on the independent dataset.
As shown in Table 6, the value of our proposed identification model iAIPs in SN, SP, ACC, AUC, and MCC are 0.567, 0.874, 0.751, 0.822, and 0.471, respectively. Furthermore, the same independent dataset-based experimental results showed that the ACC of iAIPs was 0.007–0.186 higher than that of AntiInflam and AIPpred, which is similar to AIEpred. Moreover, according to AUC, our performance is better than the other methods, which is 0.009–0.175 higher than the others. The results indicate that our method has better performance than other existing prediction models.
4 Conclusion
In this paper, an identifying AIP model based on peptide sequence is proposed. We tried various features and their combinations, utilized various commonly used ensemble learning classification algorithms and the two-step feature selection strategy. After trying a large number of experiments, we finally constructed an effective AIP prediction model. By conducting a large number of experiments on the training dataset and independent dataset, we verified that our proposed prediction model iAIPs could efficiently identify AIPs from the newly synthesized and discovered peptide sequences, which is better than the existing AIP prediction models.
In the future, the optimization of the feature representation method is a research direction. Especially, the research on a new feature representation method that can adaptively encode peptide sequences is of great significance. Furthermore, other optimization methods and computational intelligence models will be considered for identifying anti-inflammatory peptides. Deep learning (Lv et al., 2019; Zeng et al., 2020a; Zeng et al., 2020b; Zhang et al., 2020b; Du et al., 2020; Pang and Liu, 2020), unsupervised learning (Zeng et al., 2020c), and ensemble learning (Sultana et al., 2020; Zhong et al., 2020; Li et al., 2021; Niu et al., 2021; Shao and Liu, 2021) will be employed when the dataset is large enough.
Data Availability Statement
Publicly available datasets were analyzed in this study. These data can be found here: http://www.thegleelab.org/AIPpred/.
Author Contributions
DZ and ZT conceptualized the study. DZ and YL formulated the methodology. DZ validated the study and wrote the original draft. DC and YL reviewed and edited the manuscript. ZT supervised the study and acquired the funding. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by the Fundamental Research Funds for the Central Universities (2572018BH05, 2572017CB33), the National Natural Science Foundation of China (61901103, 28961671189), and the Natural Science Foundation of Heilongjiang Province (LH2019F002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akbar, S., Ateeq Ur, R., Maqsood, H., and Mohammad, S. (2020). cACP: Classifying Anticancer Peptides Using Discriminative Intelligent Model via Chou’s 5-step Rules and General Pseudo Components. Chemometrics Intell. Lab. Syst. 196, 103912. doi:10.1016/j.chemolab.2019.103912
Ao, C., Zou, Q., and Yu, L. (2021). RFhy-m2G: Identification of RNA N2-Methylguanosine Modification Sites Based on Random forest and Hybrid Features. Methods (San Diego, Calif.): Elsevier.
Basith, S., Manavalan, B., Shin, T. H., and Lee, G. (2018). iGHBP: Computational Identification of Growth Hormone Binding Proteins from Sequences Using Extremely Randomised Tree. Comput. Struct. Biotechnol. J. 16, 412–420. doi:10.1016/j.csbj.2018.10.007
Cai, L., Wang, L., Fu, X., Xia, C., Zeng, X., and Zou, Q. (2021). ITP-pred: an Interpretable Method for Predicting, Therapeutic Peptides with Fused Features Low-Dimension Representation. Brief Bioinform 22 (4), bbaa367. doi:10.1093/bib/bbaa367
de la Fuente-Núñez, C., Silva, O. N., Lu, T. K., and Franco, O. L. (2017). Antimicrobial Peptides: Role in Human Disease and Potential as Immunotherapies. Pharmacol. Ther. 178, 132–140. doi:10.1016/j.pharmthera.2017.04.002
Ding, H., Feng, P.-M., Chen, W., and Lin, H. (2014). Identification of Bacteriophage Virion Proteins by the ANOVA Feature Selection and Analysis. Mol. Biosyst. 10 (8), 2229–2235. doi:10.1039/c4mb00316k
Ding, Y. T., and JijunGuo, F. (2020). Identification of Drug-Target Interactions via Dual Laplacian Regularized Least Squares with Multiple Kernel Fusion. Knowledge-Based Syst., 204. doi:10.1016/j.knosys.2020.106254
Ding, Y., Tang, J., and Guo, F. (2019). Identification of Drug-Side Effect Association via Multiple Information Integration with Centered Kernel Alignment. Neurocomputing 325, 211–224. doi:10.1016/j.neucom.2018.10.028
Ding, Y., Tang, J., and Guo, F. (2020). Identification of Drug-Target Interactions via Fuzzy Bipartite Local Model. Neural Comput. Applic 32, 10303–10319. doi:10.1007/s00521-019-04569-z
Du, Z., Xiao, X., and Uversky, V. N. (2020). Classification of Chromosomal DNA Sequences Using Hybrid Deep Learning Architectures. Curr. Bioinformatics 15 (10), 1130–1136. doi:10.2174/1574893615666200224095531
Ferrero-Miliani, L., Nielsen, O. H., Andersen, P. S., and Girardin, S. E. (2007). Chronic Inflammation: Importance of NOD2 and NALP3 in Interleukin-1beta Generation. Clin. Exp. Immunol. 147 (2), 227–235. doi:10.1111/j.1365-2249.2006.03261.x
Freund, Y. (1990). Boosting a Weak Learning Algorithm by Majority. Inf. Comput. 121 (2), 256–285. doi:10.1006/inco.1995.1136
Friedman, J. H. (2001). Greedy Function Approximation: A Gradient Boosting Machine. Ann. Stat. 29 (5), 1189–1232. doi:10.1214/aos/1013203451
Fu, X., Cai, L., Zeng, X., and Zou, Q. (2020). StackCPPred: a Stacking and Pairwise Energy Content-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency. Bioinformatics 36 (10), 3028–3034. doi:10.1093/bioinformatics/btaa131
Fushing, H., and Turnbull, B. W. (1996). Nonparametric and Semiparametric Estimation of the Receiver Operating Characteristic Curve. Ann. Stat. 24 (1), 25–40. doi:10.1214/aos/1033066197
Gonzalez-Rey, E., Anderson, P., and Delgado, M. (2007). Emerging Roles of Vasoactive Intestinal Peptide: a New Approach for Autoimmune Therapy. Ann. Rheum. Dis. 66 (3), iii70–6. doi:10.1136/ard.2007.078519
Gribskov, M., and Robinson, N. L. (1996). Use of Receiver Operating Characteristic (ROC) Analysis to Evaluate Sequence Matching. Comput. Chem. 20 (1), 25–33. doi:10.1016/s0097-8485(96)80004-0
Gupta, S., Sharma, A. K., Shastri, V., Madhu, M. K., and Sharma, V. K. (2017). Prediction of Anti-inflammatory Proteins/peptides: an Insilico Approach. J. Transl Med. 15 (1), 7. doi:10.1186/s12967-016-1103-6
Hall, M., Eibe, F., Geoffrey, H., Bernhard, P., Peter, R., and Witten, I. H. (2008). The WEKA Data Mining Software: An Update. ACM SIGKDD Explorations Newsl. 11 (1), 10–18. doi:10.1145/1656274.1656278
Hanley, J. A., and McNeil, B. J. (1982). The Meaning and Use of the Area under a Receiver Operating Characteristic (ROC) Curve. Radiology 143 (1), 29–36. doi:10.1148/radiology.143.1.7063747
He, S., Fei, G., Quan, Z., and Hui, D. (2020). MRMD2.0: A Python Tool for Machine Learning with Feature Ranking and Reduction. Curr. Bioinformatics 15 (10), 1213–1221. doi:10.2174/1574893615999200503030350
Hong, Z., Zeng, X., Wei, L., and Liu, X. (2020). Identifying Enhancer-Promoter Interactions with Neural Network Based on Pre-trained DNA Vectors and Attention Mechanism. Bioinformatics 36 (4), 1037–1043. doi:10.1093/bioinformatics/btz694
Huang, Y., Niu, B., Gao, Y., Fu, L., and Li, W. (2010). CD-HIT Suite: a Web Server for Clustering and Comparing Biological Sequences. Bioinformatics 26 (5), 680–682. doi:10.1093/bioinformatics/btq003
Huang, Y., Zhou, D., Wang, Y., Zhang, X., Su, M., Wang, C., et al. (2020). Prediction of Transcription Factors Binding Events Based on Epigenetic Modifications in Different Human Cells. Epigenomics 12 (16), 1443–1456. doi:10.2217/epi-2019-0321
Jiang, Q., Wang, G., Jin, S., Li, Y., and Wang, Y. (2013). Predicting Human microRNA-Disease Associations Based on Support Vector Machine. Ijdmb 8 (3), 282–293. doi:10.1504/ijdmb.2013.056078
Jin, S., Zeng, X., Xia, F., Huang, W., and Liu, X. (2021). Application of Deep Learning Methods in Biological Networks. Brief. Bioinform. 22 (2), 1902–1917. doi:10.1093/bib/bbaa043
Kim, Y., Ponomarenko, J., Zhu, Z., Tamang, D., Wang, P., Greenbaum, J., et al. (2012). Immune Epitope Database Analysis Resource. Nucleic Acids Res. 40, W525–W530. doi:10.1093/nar/gks438
Li, J., Pu, Y., Tang, J., Zou, Q., and Guo, F. (2020). DeepATT: a Hybrid Category Attention Neural Network for Identifying Functional Effects of DNA Sequences. Brief Bioinform 21, 8. doi:10.1093/bib/bbaa159
Li, J., Wei, L., Guo, F., and Zou, Q. (2021). EP3: An Ensemble Predictor that Accurately Identifies Type III Secreted Effectors. Brief. Bioinform. 22 (2), 1918–1928. doi:10.1093/bib/bbaa008
Liu, B. (2019). BioSeq-Analysis: a Platform for DNA, RNA and Protein Sequence Analysis Based on Machine Learning Approaches. Brief. Bioinform. 20 (4), 1280–1294. doi:10.1093/bib/bbx165
Liu, B., Gao, X., and Zhang, H. (2019). BioSeq-Analysis2.0: an Updated Platform for Analyzing DNA, RNA and Protein Sequences at Sequence Level and Residue Level Based on Machine Learning Approaches. Nucleic Acids Res. 47(20): p. e127. doi:10.1093/nar/gkz740
Liu, Y., Yalin, H., Guohua, W., and Yadong, W. (2020). A Deep Learning Approach for Filtering Structural Variants in Short Read Sequencing Data. Brief Bioinform 22 (4). doi:10.1093/bib/bbaa370
Luo, X., Wang, F., Wang, G., and Zhao, Y. (2020). Identification of Methylation States of DNA Regions for Illumina Methylation BeadChip. BMC Genomics 21(Suppl. 1): p. 672. doi:10.1186/s12864-019-6019-0
Lv, Z., Ao, C., and Zou, Q. (2019). Protein Function Prediction: From Traditional Classifier to Deep Learning. Proteomics 19 (14), e1900119. doi:10.1002/pmic.201900119
Manavalan, B., Basith, S., Shin, T. H., Wei, L., and Lee, G. (2019). Meta-4mCpred: A Sequence-Based Meta-Predictor for Accurate DNA 4mC Site Prediction Using Effective Feature Representation. Mol. Ther. - Nucleic Acids 16, 733–744. doi:10.1016/j.omtn.2019.04.019
Manavalan, B., Shin, T. H., Kim, M. O., and Lee, G. (2018). AIPpred: Sequence-Based Prediction of Anti-inflammatory Peptides Using Random Forest. Front. Pharmacol. 9, 276. doi:10.3389/fphar.2018.00276
Manayalan, B., Shaherin, B., Tae Hwan, S., Leyi, W., and Gwang, L. (2019). mAHTPred: a Sequence-Based Meta-Predictor for Improving the Prediction of Anti-hypertensive Peptides Using Effective Feature Representation. Bioinformatics 35 (16), 2757–2765. doi:10.1093/bioinformatics/bty1047
Min, X., Ye, C., Liu, X., and Zeng, X. (2021). Predicting Enhancer-Promoter Interactions by Deep Learning and Matching Heuristic. Brief. Bioinform. 22. doi:10.1093/bib/bbaa254
Ning, L., Huang, J., He, B., and Kang, J. (2020). An In Silico Immunogenicity Analysis for PbHRH: An Antiangiogenic Peptibody by Fusing HRH Peptide and Human IgG1 Fc Fragment. Cbio 15 (6), 547–553. doi:10.2174/1574893614666190730104348
Niu, M., Lin, Y., and Zou, Q. (2021). sgRNACNN: Identifying sgRNA On-Target Activity in Four Crops Using Ensembles of Convolutional Neural Networks. Plant Mol. Biol. 105 (4-5), 483–495. doi:10.1007/s11103-020-01102-y
Pang, Y., and Liu, B., (2020). SelfAT-Fold: Protein Fold Recognition Based on Residue-Based and Motif-Based Self-Attention Networks. Ieee/acm Trans. Comput. Biol. Bioinf. 1, 1. doi:10.1109/TCBB.2020.3031888
Patterson, H., Nibbs, R., McInnes, I., and Siebert, S. (2014). Protein Kinase Inhibitors in the Treatment of Inflammatory and Autoimmune Diseases. Clin. Exp. Immunol. 176 (1), 1–10. doi:10.1111/cei.12248
Ru, X., Li, L., and Zou, Q. (2019). Incorporating Distance-Based Top-N-Gram and Random Forest to Identify Electron Transport Proteins. J. Proteome Res. 18 (7), 2931–2939. doi:10.1021/acs.jproteome.9b00250
Saravanan, V., and Gautham, N. (2015). Harnessing Computational Biology for Exact Linear B-Cell Epitope Prediction: A Novel Amino Acid Composition-Based Feature Descriptor. OMICS: A J. Integr. Biol. 19 (10), 648–658. doi:10.1089/omi.2015.0095
Shang, Y., Gao, L., Zou, Q., and Yu, L. (2021). Prediction of Drug-Target Interactions Based on Multi-Layer Network Representation Learning. Neurocomputing 434, 80–89. doi:10.1016/j.neucom.2020.12.068
Shao, J., and Liu, B. (2021). ProtFold-DFG: Protein Fold Recognition by Combining Directed Fusion Graph and PageRank Algorithm. Brief. Bioinform. 22, 32–40. doi:10.1093/bib/bbaa192
Shao, J., Yan, K., and Liu, B. (2021). FoldRec-C2C: Protein Fold Recognition by Combining Cluster-To-Cluster Model and Protein Similarity Network. Brief. Bioinform. 22, 32–40. doi:10.1093/bib/bbaa144
Shen, Y., Tang, J., and Guo, F. (2019). Identification of Protein Subcellular Localization via Integrating Evolutionary and Physicochemical Information into Chou's General PseAAC. J. Theor. Biol. 462, 230–239. doi:10.1016/j.jtbi.2018.11.012
Su, R., Wu, H., Xu, B., Liu, X., and Wei, L. (2019). Developing a Multi-Dose Computational Model for Drug-Induced Hepatotoxicity Prediction Based on Toxicogenomics Data. Ieee/acm Trans. Comput. Biol. Bioinf. 16 (4), 1231–1239. doi:10.1109/tcbb.2018.2858756
Sultana, N., Sharma, N., Sharma, K. P., and Verma, S. (2020). A Sequential Ensemble Model for Communicable Disease Forecasting. Cbio 15 (4), 309–317. doi:10.2174/1574893614666191202153824
Sun, S., Lei, X., Quan, Z., and Guohua, W. (2020). BP4RNAseq: a Babysitter Package for Retrospective and Newly Generated RNA-Seq Data Analyses Using Both Alignment-Based and Alignment-free Quantification Method. Bioinformatics 37 (9), 1319–1321. doi:10.1093/bioinformatics/btaa832
Tabas, I., and Glass, C. K. (2013). Anti-inflammatory Therapy in Chronic Disease: Challenges and Opportunities. Science 339 (6116), 166–172. doi:10.1126/science.1230720
Tang, Y.-J., Pang, Y.-H., Liu, B., and Idp-Seq2Seq, (2020). IDP-Seq2Seq: Identification of Intrinsically Disordered Regions Based on Sequence to Sequence Learning. Bioinformaitcs 36 (21), 5177–5186. doi:10.1093/bioinformatics/btaa667
Vita, R., Mahajan, S., Overton, J. A., Dhanda, S. K., Martini, S., Cantrell, J. R., et al. (2019). The Immune Epitope Database (IEDB): 2018 Update. Nucleic Acids Res. 47 (D1), D339–D343. doi:10.1093/nar/gky1006
Wang, C., Zhang, Y., and Han, S. (2020). Its2vec: Fungal Species Identification Using Sequence Embedding and Random Forest Classification. Biomed. Res. Int. 2020, 1–11. doi:10.1155/2020/2468789
Wang, H. D., Tang, J., Zou, Q., and Guo, F. (2021). Identify RNA-Associated Subcellular Localizations Based on Multi-Label Learning Using Chou's 5-steps Rule. BMC Genomics 22(56): p. 1-1.doi:10.1186/s12864-020-07347-7
Wang, H., Ding, Y., Tang, J., and Guo, F. (2020). Identification of Membrane Protein Types via Multivariate Information Fusion with Hilbert-Schmidt Independence Criterion. Neurocomputing 383, 257–269. doi:10.1016/j.neucom.2019.11.103
Wang, X., Yang, Y., Jian, L., and Guohua, W. (2021). The Stacking Strategy-Based Hybrid Framework for Identifying Non-coding RNAs. Brief Bioinform 22 (5), 32–40. doi:10.1093/bib/bbab023
Wei, L., Zhou, C., Chen, H., Song, J., and Su, R. (2018). ACPred-FL: a Sequence-Based Predictor Using Effective Feature Representation to Improve the Prediction of Anti-cancer Peptides. Bioinformatics 34 (23), 4007–4016. doi:10.1093/bioinformatics/bty451
Wei, L., Jie, H., Fuyi, Li., Jiangning, S., Ran, S., and Quan, Z. (2018). Comparative Analysis and Prediction of Quorum-Sensing Peptides Using Feature Representation Learning and Machine Learning Algorithms, 21. Brief Bioinform, 106–119. doi:10.1093/bib/bby107
Wei, L., Liao, M., Gao, Y., Ji, R., He, Z., and Zou, Q. (2014). Improved and Promising Identification of Human MicroRNAs by Incorporating a High-Quality Negative Set. Ieee/acm Trans. Comput. Biol. Bioinf. 11 (1), 192–201. doi:10.1109/tcbb.2013.146
Wei, L., Tang, J., and Zou, Q. (2017). Local-DPP: An Improved DNA-Binding Protein Prediction Method by Exploring Local Evolutionary Information. Inf. Sci. 384, 135–144. doi:10.1016/j.ins.2016.06.026
Wei, L., Wan, S., Guo, J., and Wong, K. K. (2017). A Novel Hierarchical Selective Ensemble Classifier with Bioinformatics Application. Artif. Intelligence Med. 83, 82–90. doi:10.1016/j.artmed.2017.02.005
Wei, L., Xing, P., Zeng, J., Chen, J., Su, R., and Guo, F. (2017). Improved Prediction of Protein-Protein Interactions Using Novel Negative Samples, Features, and an Ensemble Classifier. Artif. Intelligence Med. 83, 67–74. doi:10.1016/j.artmed.2017.03.001
Wu, X., and Yu, L. (2021). EPSOL: Sequence-Based Protein Solubility Prediction Using Multidimensional Embedding. Oxford, England): Bioinformatics. doi:10.1093/bioinformatics/btab463
Zhong, Y., Lin, W., and Zou, Q. (2020). Predicting Disease-Associated Circular RNAs Using Deep Forests Combined with Positive-Unlabeled Learning Methods. Brief. Bioinformatics 21 (4), 1425–1436. doi:10.1093/bib/bbz080
Yang, L., Gao, H., Wu, K., Zhang, H., Li, C., and Tang, L. (2020). Identification of Cancerlectins by Using Cascade Linear Discriminant Analysis and Optimal G-gap Tripeptide Composition. Cbio 15 (6), 528–537. doi:10.2174/1574893614666190730103156
Yu, L., Wang, M., Yang, Y., Xu, F., Zhang, X., Xie, F., et al. (2021). Predicting Therapeutic Drugs for Hepatocellular Carcinoma Based on Tissue-specific Pathways. Plos Comput. Biol. 17 (2), e1008696. doi:10.1371/journal.pcbi.1008696
Yu, L., Shi, Q., Wang, S., Zheng, L., and Gao, L. (2020). Exploring Drug Treatment Patterns Based on the Action of Drug and Multilayer Network Model. Ijms 21 (14), 5014. doi:10.3390/ijms21145014
Yu, X., Jianguo, Z., Mingming, Z., Chao, Y., Qing, D., Wei, Z., et al. (2020). Exploiting XGBoost for Predicting Enhancer-Promoter Interactions. Curr. Bioinformatics 15 (9), 1036–1045. doi:10.2174/1574893615666200120103948
Zeng, X., Wang, W., Chen, C., and Yen, G. G. (2020). A Consensus Community-Based Particle Swarm Optimization for Dynamic Community Detection. IEEE Trans. Cybern. 50 (6), 2502–2513. doi:10.1109/tcyb.2019.2938895
Zeng, X., Yinglai, L., Yuying, H., Linyuan, L., Xiaoping, M., and Rodriguez-Paton, A. (2020). Deep Collaborative Filtering for Prediction of Disease Genes. IEEE/ACM Trans. Comput. Biol. Bioinform. 17 (5), 1639–1647. doi:10.1109/tcbb.2019.2907536
Zeng, X., Zhu, S., Lu, W., Liu, Z., Huang, J., Zhou, Y., et al. (2020). Target Identification Among Known Drugs by Deep Learning from Heterogeneous Networks. Chem. Sci. 11 (7), 1775–1797. doi:10.1039/c9sc04336e
Zhang, J., Zehua, Z., Lianrong, P., Jijun, T., and Fei, G. (2020). AIEpred: An Ensemble Predictive Model of Classifier Chain to Identify Anti-inflammatory Peptides, 18. IEEE/ACM Trans Comput Biol Bioinform, 1831–1840. doi:10.1109/tcbb.2020.2968419
Zhang, Y., Jianrong, Y., Siyu, C., Meiqin, G., Dongrui, G., Min, Z., et al. (2020). Review of the Applications of Deep Learning in Bioinformatics. Curr. Bioinformatics 15 (8), 898–911. doi:10.2174/1574893615999200711165743
Zhang, Y. P., and Zou, Q. (2020). PPTPP: A Novel Therapeutic Peptide Prediction Method Using Physicochemical Property Encoding and Adaptive Feature Representation Learning. Bioinformatics 36 (13), 3982–3987. doi:10.1093/bioinformatics/btaa275
Zhao, X., Jiao, Q., Li, H., Wu, Y., Wang, H., Huang, S., et al. (2020). ECFS-DEA: an Ensemble Classifier-Based Feature Selection for Differential Expression Analysis on Expression Profiles. BMC Bioinformatics 21 (1), 43. doi:10.1186/s12859-020-3388-y
Zhao, Y., Wang, F., Chen, S., Wan, J., and Wang, G. (2017). Methods of MicroRNA Promoter Prediction and Transcription Factor Mediated Regulatory Network. Biomed. Res. Int. 2017, 7049406. doi:10.1155/2017/7049406
Zou, Q., Wan, S., Ju, Y., Tang, J., and Zeng, X. (2016). Pretata: Predicting TATA Binding Proteins with Novel Features and Dimensionality Reduction Strategy. BMC Syst. Biol. 10, 114. doi:10.1186/s12918-016-0353-5
Keywords: anti-inflammatory peptides, random forest, feature extraction, evolutionary information, evolutionary analysis
Citation: Zhao D, Teng Z, Li Y and Chen D (2021) iAIPs: Identifying Anti-Inflammatory Peptides Using Random Forest. Front. Genet. 12:773202. doi: 10.3389/fgene.2021.773202
Received: 09 September 2021; Accepted: 08 October 2021;
Published: 30 November 2021.
Edited by:
Juan Wang, Inner Mongolia University, ChinaReviewed by:
Haiying Zhang, Xiamen University, ChinaJiajie Peng, Northwestern Polytechnical University, China
Copyright © 2021 Zhao, Teng, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhixia Teng, dGVuZ3poaXhpYUBuZWZ1LmVkdS5jbg==