 Somayah Albaradei
Somayah Albaradei Mahmut Uludag
Mahmut Uludag Maha A. Thafar
Maha A. Thafar Takashi Gojobori
Takashi Gojobori Magbubah Essack
Magbubah Essack Xin Gao
Xin Gao- 1Computer, Electrical and Mathematical Sciences and Engineering Division (CEMSE), Computational Bioscience Research Center (CBRC), King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
- 2Faculty of Computing and Information Technology, King Abdulaziz University, Jeddah, Saudi Arabia
- 3College of Computers and Information Technology, Taif University, Taif, Saudi Arabia
Bone is the most common site of distant metastasis from malignant tumors, with the highest prevalence observed in breast and prostate cancers. Such bone metastases (BM) cause many painful skeletal-related events, such as severe bone pain, pathological fractures, spinal cord compression, and hypercalcemia, with adverse effects on life quality. Many bone-targeting agents developed based on the current understanding of BM onset’s molecular mechanisms dull these adverse effects. However, only a few studies investigated potential predictors of high risk for developing BM, despite such knowledge being critical for early interventions to prevent or delay BM. This work proposes a computational network-based pipeline that incorporates a ML/DL component to predict BM development. Based on the proposed pipeline we constructed several machine learning models. The deep neural network (DNN) model exhibited the highest prediction accuracy (AUC of 92.11%) using the top 34 featured genes ranked by betweenness centrality scores. We further used an entirely separate, “external” TCGA dataset to evaluate the robustness of this DNN model and achieved sensitivity of 85%, specificity of 80%, positive predictive value of 78.10%, negative predictive value of 80%, and AUC of 85.78%. The result shows the models’ way of learning allowed it to zoom in on the featured genes that provide the added benefit of the model displaying generic capabilities, that is, to predict BM for samples from different primary sites. Furthermore, existing experimental evidence provides confidence that about 50% of the 34 hub genes have BM-related functionality, which suggests that these common genetic markers provide vital insight about BM drivers. These findings may prompt the transformation of such a method into an artificial intelligence (AI) diagnostic tool and direct us towards mechanisms that underlie metastasis to bone events.
Introduction
Cancer-related morbidity and mortality are primarily associated with metastasis, and the most frequent site for tumor metastasis is the bone, particularly for breast and prostate cancers (Coleman, 1997; Landemaine et al., 2008). Also, cancer cells present in the bone marrow called disseminated tumor cells (DTCs) were shown to correlate with increased risk of disease recurrence and poor prognosis in early breast cancer (BCa) patients (Braun et al., 2005; Bidard et al., 2008). We now know that cancer metastasizing to the bone (BM), called osteotropism, requires stepwise processes that include tumor cells acquiring specific molecular characteristics to one/detach from the primary tumor, two/enter the bone, and three/home within the bone niche. However, the molecular pathways of metastases are still unknown despite the substantial advancements made in cancer-related therapies. Moreover, adjuvant treatment with bisphosphonates or denosumab only benefits specific patient subgroups (Paterson et al., 2012; Gnant et al., 2015; Jacobs et al., 2015). Thus, a number of groups have been attempting to unravel BM mechanisms using molecular biology methods (Kingsley et al., 2007).
Recent works (Josefsson et al., 2018; Rizzo et al., 2019; Pantano et al., 2020) used circulating tumor cells’ protein or gene expression profiles to suggest biomarkers for predicting BM. However, primary tumors’ protein or gene expression profiles are more commonly studied and recommended biomarkers for predicting BM. For example, high or elevated levels of CAPG, GIPC1 (Westbrook et al., 2016), ITGBL1 (Li et al., 2015), IL-1B (Li et al., 2015), DOCK-4 (Westbrook et al., 2019), nPAK4 (Li Y. et al., 2019), PRDX4 (Tiedemann et al., 2019), LPC1 (Tiedemann et al., 2019), and PRL (Sutherland et al., 2016) are all suggested BM biomarkers based on different studies. Also, several works (Kang et al., 2003; Smid et al., 2006; Sanz-Pamplona et al., 2012; Dean-Colomb et al., 2013; Zhou and Liu, 2014) have attempted to identify panels of BM-related genes from gene expression data. Few studies, such as (Smid et al., 2006; Zhou and Liu, 2014), used the identified genes as signatures to construct a model for predicting BM risk in breast cancer. Developing more such models that can predict BM from a disease specific and generic perspective with high performance accuracy could be used to support the physician’s work. Additionally, exploring the mechanism of BM from different primary sites and determining if this mechanism has common features despite originating from various primary sites is necessary, as it may provide a better understanding of the biological underpinnings of BM (Albaradei et al., 2021b).
In this study we performed a meta-analysis of three breast cancer and two prostate cancer gene expression profiles, to identify metastasis-related genes common to both cancer types. We started this process by identifying the differentially expressed genes (DEGs) between primary and metastasized tumors, then used these genes to construct a protein-protein interaction (PPI) network. We then calculated betweenness centrality (BC) to determine the hub genes which we used as input to develop machine learning models that can predict BM with high prediction accuracy. We developed support vector machine (SVM), random forest (RF), and deep learning network (DNN) models. The DNN model produced the highest prediction accuracy using only 34 top-ranked hub genes. Next, the robustness of the DNN model was validated using independent datasets from the cancer genome atlas (TCGA) and the metastasis-related functionality of the 34 top-ranked hub genes were validated by experimental evidence in existing literature.
Method and Materials
Gene Expression Datasets
We searched for gene expression datasets in Gene Expression Omnibus (GEO) (Edgar et al., 2002) using the following query: "metastas* AND bone AND Homo sapiens” filtered by “Expression profiling by array” on July 19th, 2021. We retrieved 241 entries that we sifted through but only found breast or prostate cancer samples with microarray gene expression data for primary tumors (without metastasis) and tumors with BM (metastasis to bone). The data used in this study include breast cancer data (GSE103357, GSE137842, GSE 2034) and prostate cancer data (GSE32269, GSE43332) (see Table 1). We fed this data to the ImaGEO tool (Toro-Domínguez et al., 2019) to perform the initial differential expression analysis, including background correction, normalization, and batch effect correction.
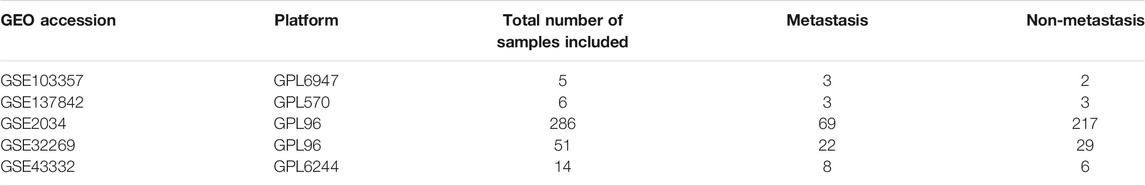
TABLE 1. Information of the gene expression datasets from GEO.
Meta-Analysis of Gene Expression Data
We used ImaGEO software, with default settings and the effect size method for the gene expression data meta-analysis. The tool transforms expression values to the logarithmic scale where needed, annotates the probe identifiers with unique Entrez Gene identifiers, merges the data, and provides data quality control checking. The tool further computes median values for duplicate gene expression profiles in each dataset, filters out genes with missing values in more than 10% of samples, and imputes missing values for the remaining genes using the average expression values in the respective primary or metastasis group.
We identified the DEGs using MetaDE.ES in the MetaDE package. This method tested the heterogeneity of gene expression value using three statistical parameters: τ2, Q-value, and Qpval. Then, we tested for differential expression of genes between the primary and metastasized groups using p-value. To ensure the homogeneity of featured genes, τ2 = 0, Qpval >0.05, and p < 0.05 were set as the cut-offs. The criteria for DEGs were false discovery rate (FDR) p-value < 0.05 and log2fold change >2. Thus, the MetaDE package performs heterogeneity tests first to determine if genuine differences underlie the results of the studies (heterogeneity) as opposed to variation based on chance alone, then selects DEGs successively (Wei et al., 2018), unlike commonly used limma, which selects DEGs based on p-value and fold-change thresholds.
Constructing the PPI Network and Identifying Hub Genes
Many recent studies use GeneMANIA (Gene Ontology molecular function-based weighting) to analyze the gene lists and prioritize genes for functional assays (Taye et al., 2017). The reason being, it offers several advantages over other PPI networks in terms of flexibility, data representation, and predictive accuracy as it is a collection of many datasets and different interactions from GEO, BioGRID (Stark et al., 2006), IRefIndex (Razick et al., 2008), and I2D (Brown and Jurisica, 2007). Thus, we used the GeneMANIA Cytoscape 3.6.0 plugin (Montojo et al., 2014) to generate a physical protein-protein interaction network using the 534 DEGs. Briefly, we uploaded our 534 DEGs to Cytoscape, then selected the physical interactions option and removed the nodes with no connections. Next, we used the Cytoscape CytoHubba plugin to identify hub genes in the constructed PPI network via the BC scoring technique. Genes/proteins were ranked based on the BC score. DEGs among the top 100 hub genes were shortlisted and subsequently used to develop ML/DL models that distinguish between primary and metastasized samples.
Using the Hub Genes as Features to Develop ML/DL Models
We created a parameter search space to evaluate different configurations for the SVM, RF, and DNN models (see Table 2). We implemented the SVM SVC class from the Scikit-learn Python library (Pedregosa et al., 2011). We employed the standard parameters, radial basis function kernel with degree = 3 and gamma = auto. We also implemented an RF model from the Scikit-learn Python library with 100 trees in the forest and max depth = 2. Also, we implemented DNN, a neural network with two hidden layers with 12 and eight nodes using the Python Keras library (https://github.com/fchollet/keras). We employed the SGD algorithm with the default parameters as the optimizer and used cross-entropy to compute the loss between actual and predicted labels. We set the number of epochs to 500 and the batch size to 8. We used the early stopping technique and the dropout technique with a drop rate of 0.3 to avoid overfitting. Because the number of samples is imbalanced, we also used the synthetic minority oversampling technique (SMOTE) to oversample the minority class using the imbalanced-learn python library (Chawla et al., 2002).
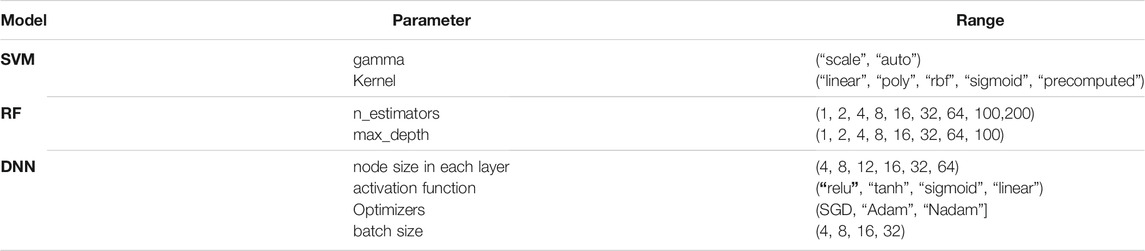
TABLE 2. Parameter search space for optimizing SVM, RF, and DNN models (Bold fond indicates the selected value).
We previously developed ML/DL models that successfully distinguish between primary and metastasis samples (Albaradei et al., 2019; Albaradei et al., 2021a). Thus, we here too iteratively added ten top-ranked genes based upon their BC value to train SVM, RF, and DNN models to mine the top essential genes that distinguish the primary and BM tumors.
We used the GEO integrated datasets (samples) for model training and computed the area under the curve (AUC) to evaluate the prediction performance of all the models. Using stratified random sampling technique (Pedregosa et al., 2011), we split the data into 80% training (296 samples) and 20% validation (74 samples). In addition, we used external testing data from the TCGA datasets to test the robustness of the best-performing model. The external set was extracted from the human cancer metastasis database (HCMDB) (Zheng et al., 2018), where we found 117 samples in which 38 were metastasized to bone (see the complete list of TCGA IDs in Supplementary Table S1). We computed sensitivity (Se), specificity (Sp), positive predictive value (PPV), negative predictive value (NPV), and AUC to evaluate the model on the test set.
Validating the Metastasis-Related Functionality
To validate the metastasis-related functionality of the 34 featured hub genes, we conducted a literature review and used the R package to explore the diseases associated with the 34 featured genes based on the disease gene network (DisGeNet). The enrichment significance was calculated using gene set enrichment analysis (GSEA), a computational method determining if a predefined set of genes exhibit a statistically significant or concordant difference between two biological states (Subramanian et al., 2005).
Results
Study Design
The study design comprises six steps, depicted in a flowchart in Figure 1. First, we used ImaGEO to integrate and analyze the five GEO datasets and obtain DEGs (Step 1). Then, the DEGs were used to construct a gene-gene functional interaction network in GeneMANIA (Step 2). Next, we calculated network nodes’ betweenness centrality and degree centrality to determine the hub genes (Step 3). We then used the hub genes to develop ML/DL models that distinguish primary from metastasized samples (Step 4). Next, we validated the best-performing model using an independent test set from TCGA (Step 5). Finally, we conducted a literature review to validate the metastasis-related functionality of the 34 hub genes (Step 6).
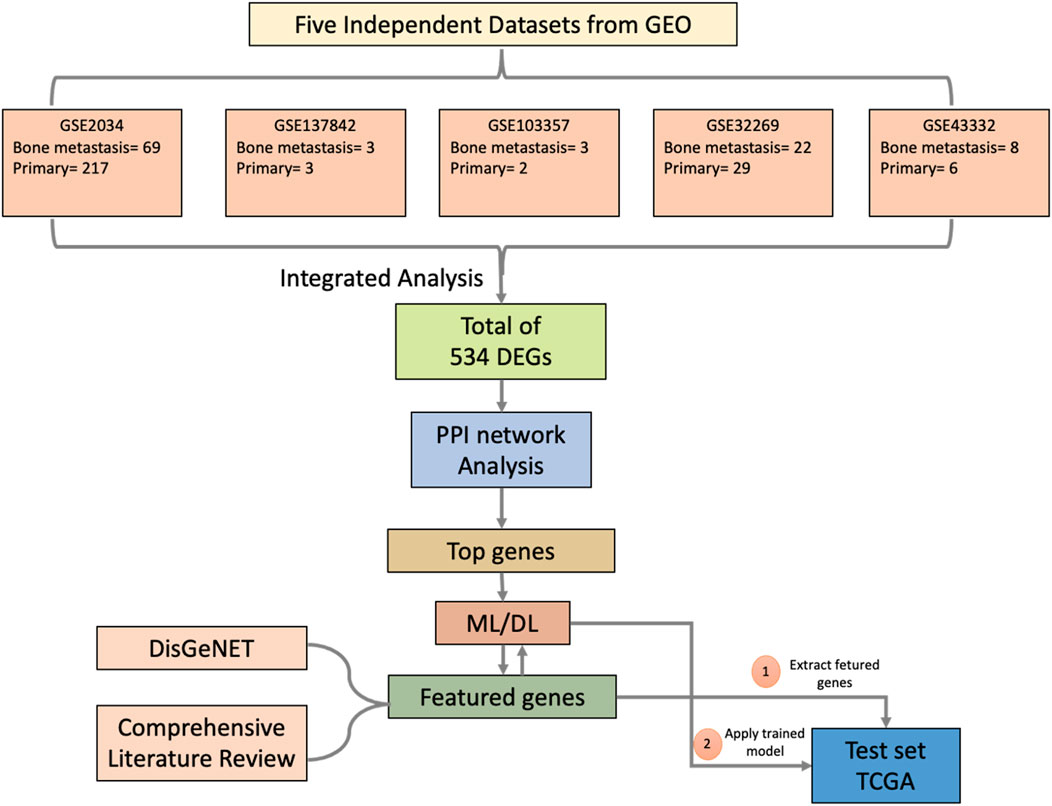
FIGURE 1. A Flowchart description of the study design.
Differentially Expressed Genes (DEGs) Between Primary and Bone Metastasized Tumours
Table 3 provides the ImaGEO tool’s quality control test results for the five gene expression datasets. The test shows the data used in this study is of good quality. The ImaGEO tool further annotated the probes with gene identifiers, merged and normalized data to provide the DEGs. The tool identified 534 DEGs, which include 365 up-regulated DEGs and 170 down-regulated DEGs. We provide the complete list of DEGs in Supplementary Table S2. A visual representation of the top 100 DEGs in the form of a heatmap shows the expression of more of the genes in the primary group is consistent in all the samples compared to the metastasized group (see Figure 2). Also, about 25% of these clearly down-regulated genes in the primary group are consistently up-regulated in the metastasized samples.
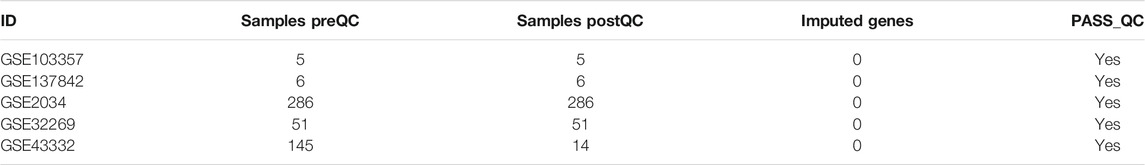
TABLE 3. The dataset quality control results generated by the ImaGEO tool.
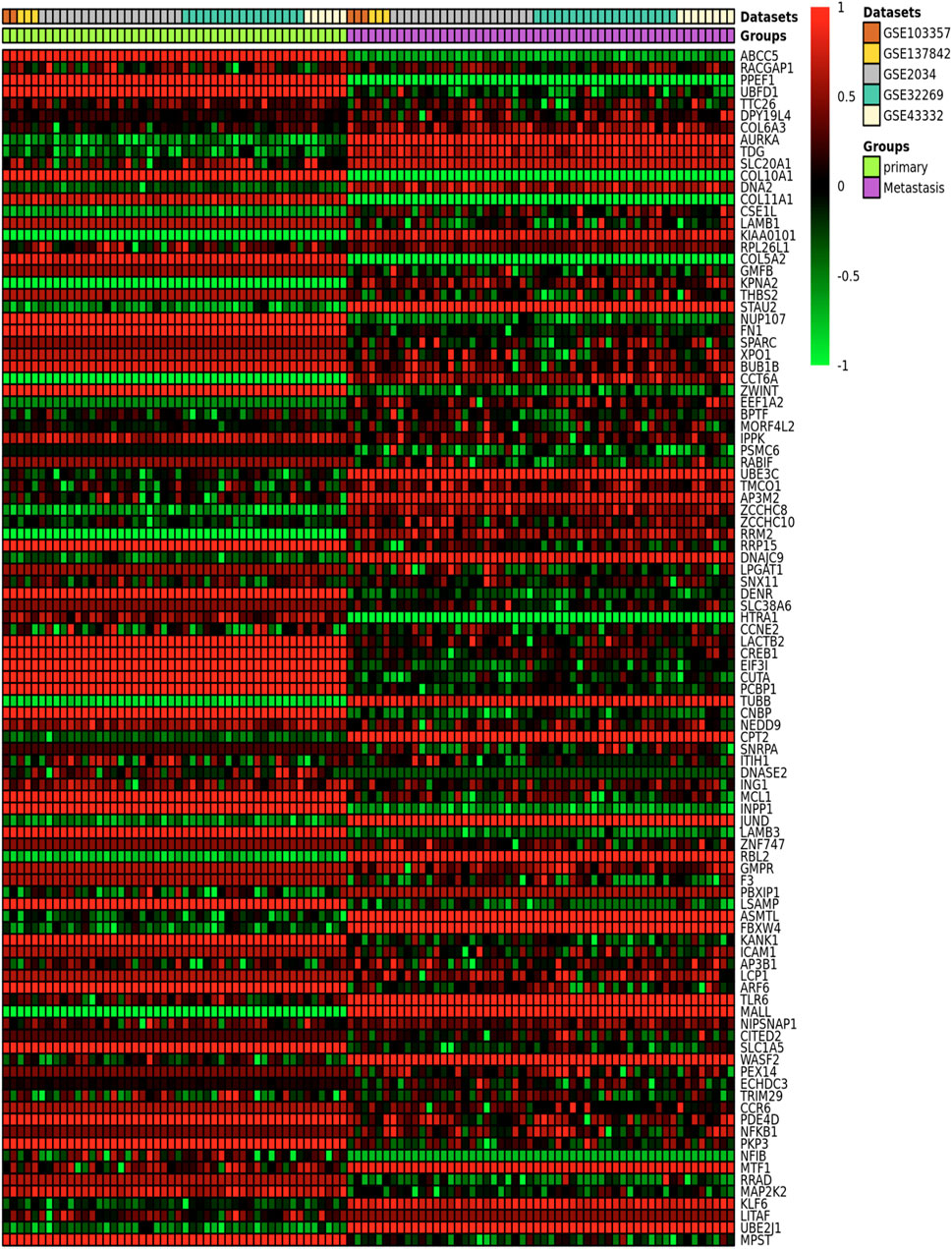
FIGURE 2. An ImaGEO generated heatmap of the top 100 DEGs based on the five microarray profiles.
Determining which DEGs are Hub Genes
The previous step provides us with the 534 DEGs but does not provide a means to identify the genes with the most functional impact, i.e., the so-called “hub” genes. Hub genes, according to research, are nodes that are highly connected to other nodes and are responsible for the majority of diseases such as cancer (Wachi et al., 2005). To identify the hub genes, we generated a gene-gene functional interaction network using GeneMANIA. First, the GeneMANIA software generates an interactive functional association network, comprising 634 nodes (which include the 534 genes and genes added based on the guilt-by-association approach) and 3,024 edges representing only direct physical protein-protein interaction (see Table 4). Then, we removed all the genes with no connected edges, leaving a network with 549 nodes and 3,005 connections. Next, we used the Cytoscape cytoHubba plugin to estimate the topological parameters, specifically, the betweenness centrality. Based on the BC score, we found 80 genes/proteins from the 534 DEGs among the top 100 hub genes. These 80 genes/proteins were subsequently used to develop ML/DL models that distinguish between primary and metastasized samples.
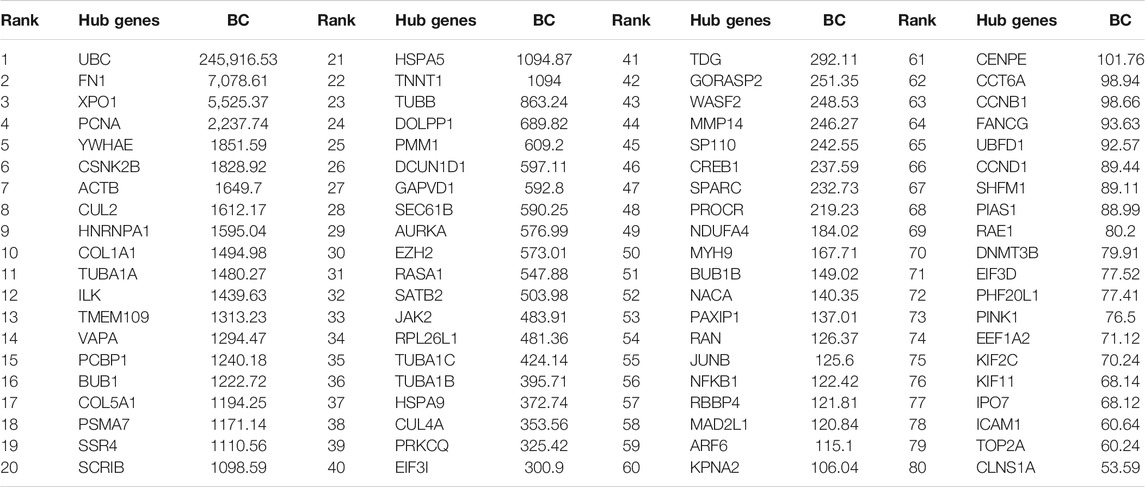
TABLE 4. The 80 DEGs ranked among the top 100 hub genes.
Evaluating if the Hub Genes Can Be Used to Develop Robust ML/DL Models that Distinguish Primary and Metastasized Tumours
We fed the 80 hub genes to each model (SVM, RF, and DNN) for training. That is, we iteratively added ten of the top-ranked genes based upon their BC value to train the models. The DNN model achieved the best AUC when including the 30 top-ranked genes (see Figure 3). We then evaluated the effect of adding some genes surrounding the 30 top-ranked genes to get the optimized performance. The 34 top-ranked featured genes (see Table 5) achieved the best performance with AUC of 92.11% and were selected to construct the final DNN model. To evaluate the robustness of this DNN model, we further used the model to distinguish primary and BM samples in a completely separate, “external” TCGA dataset. The DNN model achieved Se of 85%, Sp of 80%, PPV of 78.10%, NPV of 80%, and AUC of 85.78%. This result shows that the DNN model provides a more than satisfactory performance. Also, the models’ way of learning allowed it to zoom in on the featured genes that provide the added benefit of the model displaying generic capabilities in terms of the phenotype under investigation (primary versus BM).
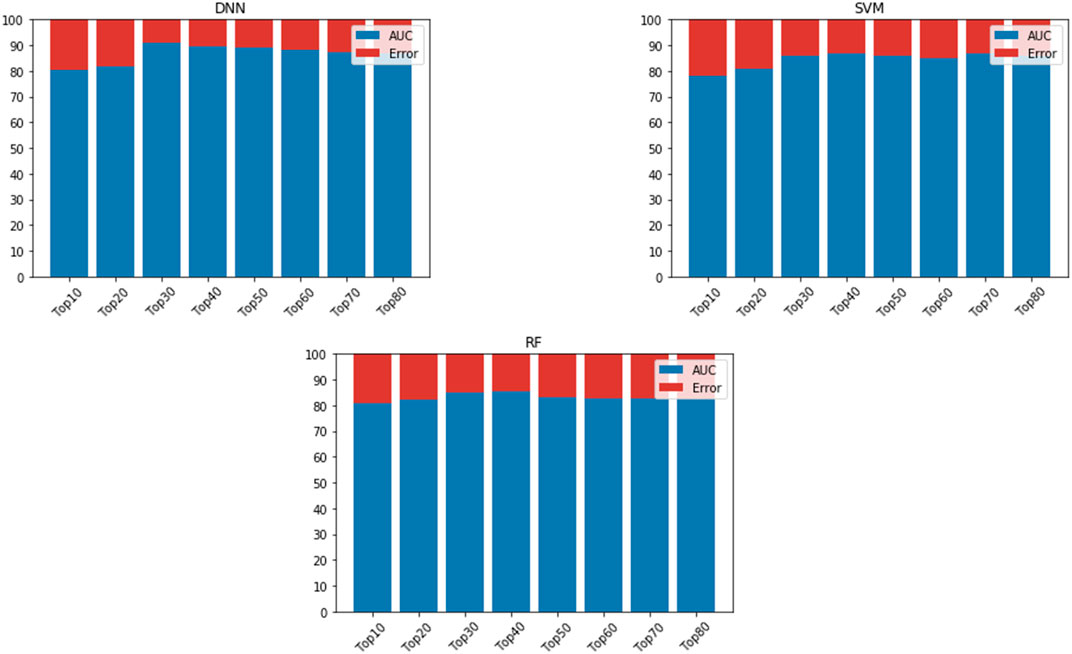
FIGURE 3. AUC is based on different numbers of featured genes using DNN, SVM, and RF. AUC is indicated in blue, while error rate is indicated in red.

TABLE 5. Metrics and literature linking the 34 feature genes to metastasis or specifically BM.
Validating the Metastasis-Related Functionality of the 34 Top-Ranked Hub Genes
Thus far, the gene-gene functional interaction network allowed us to predict several of the critical metastasis-related genes based on diverse metrics, including FN1 with the lowest FDR value (0.001) and highest BC value (7,078.61), and XPO1 with a similarly low FDR value (0.001) and high BC value (5,525.37). Therefore, FN1 and XPO1 were the most important hub genes among DEGs across five microarray studies, followed by UBC (FDR 0.038, BC 245916.54), PCNA (FDR 0.0127, BC 2237.75), and YWHAE (FDR 0.0233, BC 1851.59).
However, we still do not know the gene-disease associations of the 34 hub genes or if available experimental evidence links the genes to metastasis-related functionality. Thus, we evaluated the gene-disease associations of the 34 hub genes using DisGeNET (see Figure 4). DisGeNET indicates that the genes are associated with numerous types of cancer, autoimmunity, and bone disorders. For example, featured genes such as COL1A1, COL5A1, FN1, and ACTB are involved in invasive breast carcinoma and osteogenesis imperfecta, a heritable bone fragility disorder associated with short stature and abnormalities. This links these genes to breast cancer and bone softening, which is a feature of BM. In addition, genes such as COL1A1, HSPA5, FN1, ACTB, HNRNPA1, COL5A1, JAK2, and RASA1 are involved in Carcinomatosis and Mastitis, which shows these genes are involved in cancer spread throughout the body and inflammation in breast tissue. Also, FN1, PCNA, ACTB, COL1A1, EZH2, JAK2, and HSPA5 are involved in ureteric obstruction, an outcome of long-term invasive prostate cancer (Deng, Liu et al., 2015). This is interesting as a 2006 case report indicates that ureteric obstruction is a rare manifestation of metastatic breast cancer and that the obstruction may be due to retroperitoneal fibrosis, retroperitoneal or ureteric metastases. Furthermore, gastric cancer and renal cell carcinoma can also cause similar manifestations (Jani, 2006). We also conducted a literature review to provide a type of verification that the genes pinpointed in this study are indeed involved in metastasis-related functionality (see Table 5). As a result, we found literature supporting 17 of the 34 hub genes having known metastasis-related functionality. These results provide confidence that about 50% of the 34 hub genes have BM-related functionality and provide a birds-eye-view of the knowledge or lack of knowledge related to underlying BM mechanisms.

FIGURE 4. Represent the significantly over-expressed and under expressed genes present in the DisGeNET disease and genes involved in the significantly enriched DisGeNET disease. The depth of the color represents the fold change and the names of DisGeNET disease displayed vertically.
Discussion
Certain types of cancer, such as breast and prostate, migrate to and grow in the bone microenvironment due to specific conditions. However, the number of large-scale gene expression research undertaken to identify the shared genetic markers responsible for BM is low. Therefore, this study aims to perform a meta-analysis of the primary site and BM-related gene expression profiles from breast and prostate cancers to identify BM-related genes common to both cancer types. First, we identified the DEGs and the subset of hub genes that we can use as features in the ML/DL models to distinguish between the primary tumors and the BM. Then, we tested how generic the best-performing model is with respect to predicting BM for samples from different primary sites, but could not compare our model related models because all previous works are based on predicting BM from one primary site. However, we could not compare our model to related models because all previous works predict BM from one primary site. Additionally, we are exploring BM from different primary sites to determine common features despite originating from various primary sites. Thus, this work is different from previous works. Nonetheless, the developed model predicts BM from a disease-specific and generic perspective with high-performance accuracy, which could support the physician’s work if transformed into an AI tool.
To recap, we set out to perform a BM-related meta-analysis across different cancer types but only found five GEO gene expression datasets associated with prostate and breast cancers that fulfil this criterion. Briefly, the methodology we implemented allowed us to identify 534 DEGs (p-value <0.05) shortlisted to a subset of 80 hub genes based on betweenness centrality. Next, we fed the 80 top-ranked hub genes as features to each machine learning model, including SVM, RF, DNN models. In this manner, we filtered the genes to prioritize the most significant hub genes based on AUC using ML/DL models. Then, to test the robustness of the best-performing model, we used an external set (Zheng et al., 2018) comprising 117 samples, of which 38 were metastasized to bone. The DNN model achieved Se of 85%, Sp of 80%, PPV of 78.10%, NPV of 80%, and AUC of 85.78%. These results provide a good indication of the potential power of the selected 34 featured genes combined with a DNN to predict BM for samples from different primary sites, promoting the development of artificial intelligence (AI) diagnostic tools to enhance BM treatment.
Beyond that, these findings point out key genes involved in the metastasis process. Specifically, we further validated that more than 50% of the 34 hub genes have metastasis-related functionality. Here we mention the metastasis-related functionality exhibited by the products of a few of the top-ranked hub genes. Soikkeli and others demonstrated that the transforming growth factor-β signaling pathway is activated during metastatic outgrowth, and transforming growth factor-β inducible genes, including POSTN, FN1, and COL-I and VCAN, are up-regulated (Soikkeli et al., 2010). Moreover, they showed that POSTN, FN1, VCAN, and pro-collagen-I (PCOL-I, newly synthesized COL-I) colocalize in extracellular strand and ring structures, visible inside the metastases and at the tumor-stroma interface. Later findings supported this work, as Li and others demonstrated that small interfering RNA (siRNA)-mediated downregulation of FN1 suppress the migration, invasion, adhesion, proliferation capabilities, and induced apoptosis of melanoma cells (Li B. et al., 2019). Additionally, Armstrong and others also demonstrated that depletion of fibronectin (FN1) by siRNA knockdown markedly reduce the invasive capacity of prostate cancer (PCa) cells (Armstrong et al., 2018). Then, we have Exportin 1 (XPO1), one of the few exportins involved in transporting several tumor suppressor proteins (including p53, BRCA1, Survivin, NPM, APC, and FOXO) out of the nucleus. Gravina and others used a selective inhibition of XPO1, Selinexor (KPT-330), to demonstrate that XPO1 inhibition affects the metastatic potential of PCa cells using one model of intraprostatic tumor growth and two models of bone metastasis (Gravina et al., 2014). Concerning PCNA, Cui and others demonstrated that small hairpin RNA(shRNA)-mediated knockdown of a nuclear effector of the Hippo pathway, Yes-associated protein 1 (YAP1), down-regulate the expression of AxI, PCNA, and MMP-9, and inhibit the proliferation and invasion of human lung adenocarcinomas and gastric adenocarcinoma cells (Cui et al., 2012). Also, Zuo and others wanted to examine the role of circ-SMAD7 in glioma progression (Zuo et al., 2020). They demonstrated that downregulated Circ-SMAD7 inhibits cell proliferation, migration, and invasion in glioma cells. In addition, repressed PCNA mRNA and protein expression was observed after circ-SMAD7 was knocked down in the glioma cells, suggesting circ-SMAD7 promotes proliferation and metastasis of glioma via upregulating PCNA. In another study, Meng and others aimed to investigate how the key epithelial-mesenchymal transition (EMT) protein, Twist 1, increases vimentin expression (Meng et al., 2018). They reported that Twist1 binds to the Cullin2 (Cul2) promoter to activate the selective transcription of Cul2 circular RNA (circ-10720), but not mRNA. The circ-10720 absorb miRNAs that target the vimentin, and it is in this indirect manner that Twist1 promoted vimentin expression. They further demonstrated that circ-10720 knockdown represses the tumor-promoting activity of Twist1 in vitro and patient-derived xenograft.
Overall, the experimental evidence shows that downregulation of several of the upregulated top-ranked hub genes suppresses the metastasis-related process, including migration, invasion, adhesion, and proliferation capabilities. Additionally, their functionality extends from being structurally-related to affecting the transportation of tumour suppressor genes and even eliminating miRNA that suppresses genes with metastasis functionality. Moreover, experimental evidence shows that silencing of the downregulated top-ranked hub genes such as YWHAE (Leal et al., 2016), ILK (Zhu et al., 2012) and PCBP1 (Wang et al., 2010) induces cell proliferation, migration, and/or invasion.
The present work yields the common genetic markers between breast and prostate cancer and provides vital insight about BM drivers. Additionally, more research focused on the subset of genes with no experimental evidence may yield new biomarkers or treatment targets.
Concluding Remarks
To our knowledge, this is among the few studies to consolidate data on various cancer types, allowing us to understand the shared or consistent biological features of BM. In addition, this research unveiled several new and previously unknown genes related to BM. The last thing to mention is that, in this study, we developed a high-accuracy DNN model with 34 featured or hub genes. As far as we know, the primary site associated with BM does not hamper the models’ prediction performance. Therefore, we will focus our future work on identifying the unknown but “standard” molecular mechanisms that underlie BM from any primary site and transforming the model into an AI diagnostic tool.
Availability
We also developed a web server to serve the scientific community. The web-based tool, bone metastasis predictor https://www.cbrc.kaust.edu.sa/bonemetastasis/, implements the DNN model developed in the current study to allow the users to predict the BM state of their sample using gene expression quantification values. The user needs to provide the gene expression of the genes for every sample. The number of samples corresponds to the number of rows in a file. The output includes a list of samples and indicates if the prediction is primary or BM.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: Gene Expression Omnibus (GSE103357; GSE137842; GSE 2034; GSE32269; GSE43332).
Author Contributions
SA, ME and XG. conceived and designed the study; S.A. performed the experiments; S.A, M.T, and M.E. analyzed the results; M.U. designed the web tool; S.A, M.U, M.T, T.G, M.E, and X.G. contributed to writing and reviewing the manuscript. All authors read and approved the final manuscript.
Funding
The research reported in this publication was supported by King Abdullah University of Science and Technology (KAUST) through the Awards Nos. BAS/1/1059-01-01, BAS/1/1624-01-01, FCC/1/1976-20-01, and FCC/1/1976-26-01.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.771092/full#supplementary-material
References
Albaradei, S., Napolitano, F., Thafar, M. A., Gojobori, T., Essack, M., and Gao, X. (2021a). MetaCancer: A Deep Learning-Based Pan-Cancer Metastasis Prediction Model Developed Using Multi-Omics Data. Comput. Struct. Biotechnol. J. 19, 4404–4411. doi:10.1016/j.csbj.2021.08.006
Albaradei, S., Thafar, M. A., Alsaedi, A., Van Neste, C., Gojobori, T., Essack, M., and Gao, X. (2021b). Machine Learning and Deep Learning Methods That Use Omics Data for Metastasis Prediction. Comput. Struct. Biotechnol. J. 19, 5008–5018. doi:10.1016/j.csbj.2021.09.001
Albaradei, S., Thafar, M., Van Neste, C., Essack, M., and Bajic, V. B. (2019). “Metastatic State of Colorectal Cancer Can Be Accurately Predicted with Methylome,” in Proceedings of the 2019 6th International Conference on Bioinformatics Research and Applications, University of Seoul, December 19, 2019. doi:10.1145/3383783.3383792
Armstrong, H. K., Gillis, J. L., Johnson, I. R. D., Nassar, Z. D., Moldovan, M., Levrier, C., et al. (2018). Dysregulated Fibronectin Trafficking by Hsp90 Inhibition Restricts Prostate Cancer Cell Invasion. Sci. Rep. 8 (1), 2090. doi:10.1038/s41598-018-19871-4
Bidard, F.-C., Vincent-Salomon, A., Gomme, S., Nos, C., de Rycke, Y., Thiery, J. P., et al. (2008). Disseminated Tumor Cells of Breast Cancer Patients: A Strong Prognostic Factor for Distant and Local Relapse. Clin. Cancer Res. 14 (11), 3306–3311. doi:10.1158/1078-0432.ccr-07-4749
Braun, S., Vogl, F. D., Naume, B., Janni, W., Osborne, M. P., Coombes, R. C., et al. (2005). A Pooled Analysis of Bone Marrow Micrometastasis in Breast Cancer. N. Engl. J. Med. 353 (8), 793–802. doi:10.1056/nejmoa050434
Brown, K. R., and Jurisica, I. (2007). Unequal Evolutionary Conservation of Human Protein Interactions in Interologous Networks. Genome Biol. 8 (5), R95–R11. doi:10.1186/gb-2007-8-5-r95
Chawla, N. V., Bowyer, K. W., Hall, L. O., and Kegelmeyer, W. P. (2002). SMOTE: Synthetic Minority Over-Sampling Technique. J. Artif. Intelligence Res. 16, 321–357. doi:10.1613/jair.953
Chen, C., Song, G., Xiang, J., Zhang, H., Zhao, S., and Zhan, Y. (2017). AURKA Promotes Cancer Metastasis by Regulating Epithelial-Mesenchymal Transition and Cancer Stem Cell Properties in Hepatocellular Carcinoma. Biochem. Biophysical Res. Commun. 486 (2), 514–520. doi:10.1016/j.bbrc.2017.03.075
Chen, Y., Liu, J., Wang, W., Xiang, L., Wang, J., Liu, S., et al. (2018). High Expression of hnRNPA1 Promotes Cell Invasion by Inducing EMT in Gastric Cancer. Oncol. Rep. 39 (4), 1693–1701. doi:10.3892/or.2018.6273
Coleman, R. E. (1997). Skeletal Complications of Malignancy. Cancer 80 (8 Suppl. l), 1588–1594. doi:10.1002/(sici)1097-0142(19971015)80:8+<1588:aid-cncr9>3.3.co;2-z
Cotul, E. K., Zuo, Q., Santaliz-Casiano, A., Imir, O. B., Mogol, A. N., Tunc, E., et al. (2020). Combined Targeting of Estrogen Receptor Alpha and Exportin 1 in Metastatic Breast Cancers. Cancers (Basel) 12 (9), 2397. doi:10.3390/cancers12092397
Cui, Z.-L., Han, F.-F., Peng, X.-H., Chen, X., Luan, C.-Y., Han, R.-C., et al. (2012). YES-Associated Protein 1 Promotes Adenocarcinoma Growth and Metastasis through Activation of the Receptor Tyrosine Kinase Axl. Int. J. Immunopathol. Pharmacol. 25 (4), 989–1001. doi:10.1177/039463201202500416
Dean-Colomb, W., Hess, K. R., Young, E., Gornet, T. G., Handy, B. C., Moulder, S. L., et al. (2013). Elevated Serum P1NP Predicts Development of Bone Metastasis and Survival in Early-Stage Breast Cancer. Breast Cancer Res. Treat. 137 (2), 631–636. doi:10.1007/s10549-012-2374-0
Edgar, R., Domrachev, M., and Lash, A. E. (2002). Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 30 (1), 207–210. doi:10.1093/nar/30.1.207
Feng, G., Ma, H.-M., Huang, H.-B., Li, Y.-W., Zhang, P., Huang, J.-J., et al. (2019). Overexpression of COL5A1 Promotes Tumor Progression and Metastasis and Correlates with Poor Survival of Patients with clear Cell Renal Cell Carcinoma. Cancer Manag. Res. 11, 1263–1274. doi:10.2147/cmar.s188216
Gnant, M., Mlineritsch, B., Stoeger, H., Luschin-Ebengreuth, G., Knauer, M., Moik, M., et al. (2015). Zoledronic Acid Combined with Adjuvant Endocrine Therapy of Tamoxifen versus Anastrozol Plus Ovarian Function Suppression in Premenopausal Early Breast Cancer: Final Analysis of the Austrian Breast and Colorectal Cancer Study Group Trial 12. Ann. Oncol. 26 (2), 313–320. doi:10.1093/annonc/mdu544
Gravina, G. L., Tortoreto, M., Mancini, A., Addis, A., Di Cesare, E., Lenzi, A., et al. (2014). XPO1/CRM1-Selective Inhibitors of Nuclear Export (SINE) Reduce Tumor Spreading and Improve Overall Survival in Preclinical Models of Prostate Cancer (PCa). J. Hematol. Oncol. 7, 46. doi:10.1186/1756-8722-7-46
Hirukawa, A., Smith, H. W., Zuo, D., Dufour, C. R., Savage, P., Bertos, N., et al. (2018). Targeting EZH2 Reactivates a Breast Cancer Subtype-Specific Anti-Metastatic Transcriptional Program. Nat. Commun. 9 (1), 2547. doi:10.1038/s41467-018-04864-8
Hussein, U. K., Ahmed, A. G., Choi, W. K., Kim, K. M., Park, S. H., Park, H. S., et al. (2021). SCRIB Is Involved in the Progression of Ovarian Carcinomas in Association with the Factors Linked to Epithelial-To-Mesenchymal Transition and Predicts Shorter Survival of Diagnosed Patients. Biomolecules 11 (3), 405. doi:10.3390/biom11030405
Jacobs, C., Amir, E., Paterson, A., Zhu, X., and Clemons, M. (2015). Are Adjuvant Bisphosphonates Now Standard of Care of Women with Early Stage Breast Cancer? A Debate from the Canadian Bone and the Oncologist New Updates Meeting. J. Bone Oncol. 4 (2), 54–58. doi:10.1016/j.jbo.2015.06.001
Jani, K. (2006). Ureteric Obstruction Secondary to Metastatic Breast Carcinoma. Pakistan J. Med. Sci. 22 (2), 197. https://pjms.com.pk/issues/aprjun06/pdf/ureteric_obstruction.pdf.
Josefsson, A., Larsson, K., Månsson, M., Björkman, J., Rohlova, E., Åhs, D., et al. (2018). Circulating Tumor Cells Mirror Bone Metastatic Phenotype in Prostate Cancer. Oncotarget 9 (50), 29403–29413. doi:10.18632/oncotarget.25634
Kang, Y., Siegel, P. M., Shu, W., Drobnjak, M., Kakonen, S. M., Cordón-Cardo, C., et al. (2003). A Multigenic Program Mediating Breast Cancer Metastasis to Bone. Cancer Cell 3 (6), 537–549. doi:10.1016/s1535-6108(03)00132-6
Kingsley, L. A., Fournier, P. G. J., Chirgwin, J. M., and Guise, T. A. (2007). Molecular Biology of Bone Metastasis. Mol. Cancer Ther. 6 (10), 2609–2617. doi:10.1158/1535-7163.mct-07-0234
Landemaine, T., Jackson, A., Bellahcène, A., Rucci, N., Sin, S., Abad, B. M., et al. (2008). A Six-Gene Signature Predicting Breast Cancer Lung Metastasis. Cancer Res. 68 (15), 6092–6099. doi:10.1158/0008-5472.can-08-0436
Leal, M. F., Ribeiro, H. F., Rey, J. A., Pinto, G. R., Smith, M. C., Moreira-Nunes, C. A., et al. (2016). YWHAE Silencing Induces Cell Proliferation, Invasion and Migration through the Up-Regulation of CDC25B and MYC in Gastric Cancer Cells: New Insights about YWHAE Role in the Tumor Development and Metastasis Process. Oncotarget 7 (51), 85393–85410. doi:10.18632/oncotarget.13381
Li, B., Shen, W., Peng, H., Li, Y., Chen, F., Zheng, L., et al. (2019b). Fibronectin 1 Promotes Melanoma Proliferation and Metastasis by Inhibiting Apoptosis and Regulating EMT. Onco. Targets Ther. 12, 3207–3221. doi:10.2147/ott.s195703
Li, J., Yu, T., Yan, M., Zhang, X., Liao, L., Zhu, M., et al. (2019c). DCUN1D1 Facilitates Tumor Metastasis by Activating FAK Signaling and Up-Regulates PD-L1 in Non-small-cell Lung Cancer. Exp. Cel. Res. 374 (2), 304–314. doi:10.1016/j.yexcr.2018.12.001
Li, X.-Q., Du, X., Li, D.-M., Kong, P.-Z., Sun, Y., Liu, P.-F., et al. (2015). ITGBL1 Is a Runx2 Transcriptional Target and Promotes Breast Cancer Bone Metastasis by Activating the TGFβ Signaling Pathway. Cancer Res. 75 (16), 3302–3313. doi:10.1158/0008-5472.can-15-0240
Li, Y., Zhang, H., Zhao, Y., Wang, C., Cheng, Z., Tang, L., et al. (2019a). A Mandatory Role of Nuclear PAK4-LIFR axis in Breast-To-Bone Metastasis of ERα-Positive Breast Cancer Cells. Oncogene 38 (6), 808–821. doi:10.1038/s41388-018-0456-0
Liu, J., Shen, J. X., Wu, H. T., Li, X. L., Wen, X. F., Du, C. W., et al. (2018a). Collagen 1A1 (COL1A1) Promotes Metastasis of Breast Cancer and Is a Potential Therapeutic Target. Discov. Med. 25 (139), 211–223. https://www.discoverymedicine.com/Jing-Liu/2018/05/collagen-col1a1-promotes-metastasis-of-breast-cancer-potential-therapeutic-target/.
Liu, W., Wei, H., Gao, Z., Chen, G., Liu, Y., Gao, X., et al. (2018b). COL5A1 May Contribute the Metastasis of Lung Adenocarcinoma. Gene 665, 57–66. doi:10.1016/j.gene.2018.04.066
Loh, T. J., Moon, H., Cho, S., Jang, H., Liu, Y. C., Tai, H., et al. (2015). CD44 Alternative Splicing and hnRNP A1 Expression Are Associated with the Metastasis of Breast Cancer. Oncol. Rep. 34 (3), 1231–1238. doi:10.3892/or.2015.4110
Luo, L.-J., Yang, F., Ding, J.-J., Yan, D.-L., Wang, D.-D., Yang, S.-J., et al. (2016). MiR-31 Inhibits Migration and Invasion by Targeting SATB2 in Triple Negative Breast Cancer. Gene 594 (1), 47–58. doi:10.1016/j.gene.2016.08.057
Meng, J., Chen, S., Han, J.-X., Qian, B., Wang, X.-R., Zhong, W.-L., et al. (2018). Twist1 Regulates Vimentin through Cul2 Circular RNA to Promote EMT in Hepatocellular Carcinoma. Cancer Res. 78 (15), 4150–4162. doi:10.1158/0008-5472.can-17-3009
Montojo, J., Zuberi, K., Rodriguez, H., Bader, G. D., and Morris, Q. (2014). GeneMANIA: Fast Gene Network Construction and Function Prediction for Cytoscape. F1000Res. 3, 153. doi:10.12688/f1000research.4572.1
Pantano, F., Rossi, E., Iuliani, M., Facchinetti, A., Simonetti, S., Ribelli, G., et al. (2020). Dynamic Changes of Receptor Activator of Nuclear Factor-Κb Expression in Circulating Tumor Cells during Denosumab Predict Treatment Effectiveness in Metastatic Breast Cancer. Sci. Rep. 10 (1), 1288. doi:10.1038/s41598-020-58339-2
Paterson, A. H., Anderson, S. J., Lembersky, B. C., Fehrenbacher, L., Falkson, C. I., King, K. M., et al. (2012). Oral Clodronate for Adjuvant Treatment of Operable Breast Cancer (National Surgical Adjuvant Breast and Bowel Project Protocol B-34): A Multicentre, Placebo-Controlled, Randomised Trial. Lancet Oncol. 13 (7), 734–742. doi:10.1016/s1470-2045(12)70226-7
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., et al. (2011). Scikit-Learn: Machine Learning in Python. J. Machine Learn. Res. 12, 2825–2830. doi:10.5555/1953048.2078195
Razick, S., Magklaras, G., and Donaldson, I. M. (2008). iRefIndex: A Consolidated Protein Interaction Database with Provenance. BMC Bioinformatics 9 (1), 405–419. doi:10.1186/1471-2105-9-405
Rizzo, F. M., Vesely, C., Childs, A., Marafioti, T., Khan, M. S., Mandair, D., et al. (2019). Circulating Tumour Cells and Their Association with Bone Metastases in Patients with Neuroendocrine Tumours. Br. J. Cancer 120 (3), 294–300. doi:10.1038/s41416-018-0367-4
Sanz-Pamplona, R., García-García, J., Franco, S., Messeguer, X., Driouch, K., Oliva, B., et al. (2012). A Taxonomy of Organ-Specific Breast Cancer Metastases Based on a Protein-Protein Interaction Network. Mol. Biosyst. 8 (8), 2085–2096. doi:10.1039/c2mb25104c
Seong, B. K. A., Lau, J., Adderley, T., Kee, L., Chaukos, D., Pienkowska, M., et al. (2015). SATB2 Enhances Migration and Invasion in Osteosarcoma by Regulating Genes Involved in Cytoskeletal Organization. Oncogene 34 (27), 3582–3592. doi:10.1038/onc.2014.289
Shin, Y. J., and Kim, J.-H. (2012). The Role of EZH2 in the Regulation of the Activity of Matrix Metalloproteinases in Prostate Cancer Cells. PLoS One 7 (1), e30393. doi:10.1371/journal.pone.0030393
Shuang, Y., Li, C., Zhou, X., Huang, Y., and Zhang, L. (2017). MicroRNA-195 Inhibits Growth and Invasion of Laryngeal Carcinoma Cells by Directly Targeting DCUN1D1. Oncol. Rep. 38 (4), 2155–2165. doi:10.3892/or.2017.5875
Smid, M., Wang, Y., Klijn, J. G. M., Sieuwerts, A. M., Zhang, Y., Atkins, D., et al. (2006). Genes Associated with Breast Cancer Metastatic to Bone. J. Clin. Oncol. 24 (15), 2261–2267. doi:10.1200/jco.2005.03.8802
Soikkeli, J., Podlasz, P., Yin, M., Nummela, P., Jahkola, T., Virolainen, S., et al. (2010). Metastatic Outgrowth Encompasses COL-I, FN1, and POSTN Up-Regulation and Assembly to Fibrillar Networks Regulating Cell Adhesion, Migration, and Growth. Am. J. Pathol. 177 (1), 387–403. doi:10.2353/ajpath.2010.090748
Stark, C., Breitkreutz, B. J., Reguly, T., Boucher, L., Breitkreutz, A., and Tyers, M. (2006). BioGRID: A General Repository for Interaction Datasets. Nucleic Acids Res. 34 (Suppl. l_1), D535–D539. doi:10.1093/nar/gkj109
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. 102 (43), 15545–15550. doi:10.1073/pnas.0506580102
Sutherland, A., Forsyth, A., Cong, Y., Grant, L., Juan, T. H., Lee, J. K., et al. (2016). The Role of Prolactin in Bone Metastasis and Breast Cancer Cell-Mediated Osteoclast Differentiation. J. Natl. Cancer Inst. 108 (3), djv338. doi:10.1093/jnci/djv338
Talati, P. G., Gu, L., Ellsworth, E. M., Girondo, M. A., Trerotola, M., Hoang, D. T., et al. (2015). Jak2-Stat5a/b Signaling Induces Epithelial-To-Mesenchymal Transition and Stem-Like Cell Properties in Prostate Cancer. Am. J. Pathol. 185 (9), 2505–2522. doi:10.1016/j.ajpath.2015.04.026
Taye, B., Vaz, C., Tanavde, V., Kuznetsov, V. A., Eisenhaber, F., Sugrue, R. J., et al. (2017). Benchmarking Selected Computational Gene Network Growing Tools in Context of Virus-Host Interactions. Sci. Rep. 7 (1), 5805–5811. doi:10.1038/s41598-017-06020-6
Tiedemann, K., Sadvakassova, G., Mikolajewicz, N., Juhas, M., Sabirova, Z., Tabariès, S., et al. (2019). Exosomal Release of L-Plastin by Breast Cancer Cells Facilitates Metastatic Bone Osteolysis. Translational Oncol. 12 (3), 462–474. doi:10.1016/j.tranon.2018.11.014
Toro-Domínguez, D., Martorell-Marugán, J., López-Domínguez, R., García-Moreno, A., González-Rumayor, V., Alarcón-Riquelme, M. E., et al. (2019). ImaGEO: Integrative Gene Expression Meta-Analysis from GEO Database. Bioinformatics 35 (5), 880–882. doi:10.1093/bioinformatics/bty721
Wachi, S., Yoneda, K., and Wu, R. (2005). Interactome-Transcriptome Analysis Reveals the High Centrality of Genes Differentially Expressed in Lung Cancer Tissues. Bioinformatics 21 (23), 4205–4208. doi:10.1093/bioinformatics/bti688
Wang, H., Vardy, L. A., Tan, C. P., Loo, J. M., Guo, K., Li, J., et al. (2010). PCBP1 Suppresses the Translation of Metastasis-Associated PRL-3 Phosphatase. Cancer Cell 18 (1), 52–62. doi:10.1016/j.ccr.2010.04.028
Wang, S., Liang, K., Hu, Q., Li, P., Song, J., Yang, Y., et al. (2017). JAK2-Binding Long Noncoding RNA Promotes Breast Cancer Brain Metastasis. J. Clin. Invest. 127 (12), 4498–4515. doi:10.1172/jci91553
Wei, L., Wang, Q., Zhang, Y., Yang, C., Guan, H., Jiang, J., et al. (2018). Integrated Analysis of Microarray Data to Identify the Genes Critical for the Rupture of Intracranial Aneurysm. Oncol. Lett. 15 (4), 4951–4957. doi:10.3892/ol.2018.7935
Westbrook, J. A., Cairns, D. A., Peng, J., Speirs, V., Hanby, A. M., Holen, I., et al. (2016). CAPG and GIPC1: Breast Cancer Biomarkers for Bone Metastasis Development and Treatment. J. Natl. Cancer Inst. 108 (4), djv360. doi:10.1093/jnci/djv360
Westbrook, J. A., Wood, S. L., Cairns, D. A., McMahon, K., Gahlaut, R., Thygesen, H., et al. (2019). Identification and Validation of DOCK4 as a Potential Biomarker for Risk of Bone Metastasis Development in Patients with Early Breast Cancer. J. Pathol. 247 (3), 381–391. doi:10.1002/path.5197
Yang, L., Zhou, Q., Chen, X., Su, L., Liu, B., and Zhang, H. (2016). Activation of the FAK/PI3K Pathway Is Crucial for AURKA-Induced Epithelial-Mesenchymal Transition in Laryngeal Cancer. Oncol. Rep. 36 (2), 819–826. doi:10.3892/or.2016.4872
Zhang, X., Xu, Y., Yamaguchi, K., Hu, J., Zhang, L., Wang, J., et al. (2020). Circular RNA circVAPA Knockdown Suppresses Colorectal Cancer Cell Growth Process by Regulating miR-125a/CREB5 axis. Cancer Cel. Int. 20, 103. doi:10.1186/s12935-020-01178-y
Zheng, G., Ma, Y., Zou, Y., Yin, A., Li, W., and Dong, D. (2018). HCMDB: The Human Cancer Metastasis Database. Nucleic Acids Res. 46 (D1), D950–D955. doi:10.1093/nar/gkx1008
Zhou, S.-Y., Chen, W., Yang, S.-J., Li, J., Zhang, J.-Y., Zhang, H.-D., et al. (2020). Circular RNA circVAPA Regulates Breast Cancer Cell Migration and Invasion via Sponging miR-130a-5p. Epigenomics 12 (4), 303–317. doi:10.2217/epi-2019-0124
Zhou, X., and Liu, J. (2014). A Computational Model to Predict Bone Metastasis in Breast Cancer by Integrating the Dysregulated Pathways. BMC Cancer 14, 618. doi:10.1186/1471-2407-14-618
Zhu, X.-Y., Liu, N., Liu, W., Song, S.-W., and Guo, K.-J. (2012). Silencing of the Integrin-Linked Kinase Gene Suppresses the Proliferation, Migration and Invasion of Pancreatic Cancer Cells (Panc-1). Genet. Mol. Biol. 35 (2), 538–544. doi:10.1590/s1415-47572012005000028
Keywords: metastasis, bone, gene experession, machine learining, hub genes, genetic diagnostic tool, deep learning
Citation: Albaradei S, Uludag M, Thafar MA, Gojobori T, Essack M and Gao X (2021) Predicting Bone Metastasis Using Gene Expression-Based Machine Learning Models. Front. Genet. 12:771092. doi: 10.3389/fgene.2021.771092
Received: 05 September 2021; Accepted: 20 October 2021;
Published: 10 November 2021.
Edited by:
Othman Soufan, St. Francis Xavier University, CanadaCopyright © 2021 Albaradei, Uludag, Thafar, Gojobori, Essack and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Gao, eGluLmdhb0BrYXVzdC5lZHUuc2E=; Magbubah Essack, bWFnYnViYWguZXNzYWNrQGthdXN0LmVkdS5zYQ==