
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 26 November 2021
Sec. Epigenomics and Epigenetics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.768913
This article is part of the Research TopicB cell Non-Hodgkin’s Lymphoma & Tumor Microenvironment Crosstalk: An Epigenetic Matter?View all 5 articles
 Esther K. Elliott1,2†
Esther K. Elliott1,2† Lloyd N. Hopkins1†
Lloyd N. Hopkins1† Robert Hensen2
Robert Hensen2 Heidi G. Sutherland1
Heidi G. Sutherland1 Larisa M. Haupt1
Larisa M. Haupt1 Lyn R. Griffiths1*
Lyn R. Griffiths1*MicroRNAs (miRNAs) are well known for their ability to regulate the expression of specific target genes through degradation or inhibition of translation of the target mRNA. In various cancers, miRNAs regulate gene expression by altering the epigenetic status of candidate genes that are implicated in various difficult to treat haematological malignancies such as non-Hodgkin lymphoma by acting as either oncogenes or tumour suppressor genes. Cellular and circulating miRNA biomarkers could also be directly utilised as disease markers for diagnosis and monitoring of non-Hodgkin lymphoma (NHL); however, the role of DNA methylation in miRNA expression regulation in NHL requires further scientific inquiry. In this study, we investigated the methylation levels of CpGs in CpG islands spanning the promoter regions of the miR-17–92 cluster host gene and the TET2 gene and correlated them with the expression levels of TET2 mRNA and miR-92a-3p and miR-92a-5p mature miRNAs in NHL cell lines, tumour samples, and the whole blood gDNA of an NHL case control cohort. Increased expression of both miR-92a-3p and miR-92a-5p and aberrant expression of TET2 was observed in NHL cell lines and tumour tissues, as well as disparate levels of dysfunctional promoter CGI methylation. Both miR-92a and TET2 may play a concerted role in NHL malignancy and disease pathogenesis.
Non-Hodgkin Lymphoma (NHL) is a class of cancer that originates in the lymphatic system, caused by an over-proliferation of malignant B-cells. NHL is one of the most common cancers in the United States, and lymphoma is the fifth most common cancer in Australasia (Abba et al., 2016), therefore posing a significant health burden (Calin et al., 2002; Cancer Council, 2020). The five-year survival rate is an estimated 71% (Allemani et al., 2018); however, both outcomes and pathogenesis vary greatly between NHL subtypes. Of the more than 40 subtypes of NHL, the two most common subtypes consist of the indolent follicular lymphoma (FL), and the more aggressive diffuse large B-cell lymphoma (DLBCL). Aggressive subtypes account for approximately 60% of cases, and a significant number of NHL patients suffer relapse on various treatments, with refractory NHL having a much poorer prognosis despite access to extensive chemotherapy and immunotherapy regimes (Michot et al., 2018; Sarkozy and Sehn, 2018; Ayers et al., 2020). Overall, this makes NHL an intractable disease to manage and treat, highlighting the need for reliable and subtype-specific diagnostic and prognostic biomarkers.
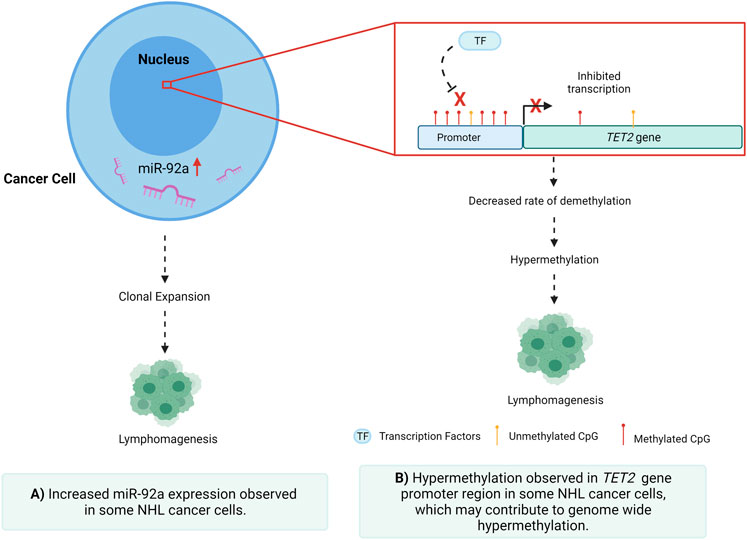
GRAPHICAL ABSTRACT
Understanding the cell of origin also plays a significant role in understanding NHL classification, progression, and prognosis (Alizadeh et al., 2000). B-cell lymphomas account for approximately 80% of NHL cases (Shankland et al., 2012), with DLBCL making up 25% of NHL diagnoses (Teras et al., 20162016; Siegel et al., 20192019). In 2000, gene expression profiling further defined DLBCL into two molecular subtypes; germinal centre B-cell like (GCB) and activated B-cell like (ABC) (Alizadeh et al., 2000). Also known as non-GCB, ABC demonstrated a poorer response to standard immunochemotherapy when compared to DLBCL cases with no specific subtype. This has been in part attributed to two oncogenic mechanisms seen in this subtype, encompassing the prevention of apoptosis and blockage of terminal differentiation (Blenk et al., 2007). These new subtypes of DLBCL, termed high-grade B-cell lymphoma, double-hit lymphoma, or triple hit lymphoma are associated with MYC and BCL2 and/or BCL6 rearrangements (Blenk et al., 2007) which are believed to contribute to their more aggressive oncogenesis.
Dysregulation of miRNAs is a hallmark of both cancer initiation and metastasis, through the regulation of gene expression via post-transcriptional repression and mRNA degradation (Di Lisio et al., 2012; Martin-Guerrero et al., 2015; Bradshaw et al., 2016; Getaneh et al., 2019). It is well established that miRNAs can act as both oncogenes and tumour suppressors in several cancers (Esquela-Kerscher and Slack, 2006; Di Lisio et al., 2012; Caramuta et al., 2013; Martin-Guerrero et al., 2015; Bradshaw et al., 2016; Getaneh et al., 2019), including NHL, via targeted repression of regulatory factors involved in processes such as cellular proliferation and migration (Xiao et al., 2008; Lawrie et al., 2009; Culpin, 2010). Previous studies have identified numerous miRNAs as having oncogenic potential in NHL (Amodio et al., 2015; Gado et al., 2019), as well as several being identified to be differentially expressed in tissues and fluids of individuals with these diseases. It has been previously established that miR-92a-3p has oncogenic potential, implicated in several cancers, including NHL, proposing that dysregulation of this miRNA may play a role in disease development (Culpin, 2010; Amodio et al., 2015; Gado et al., 2019). In both leukemia and DLBCL, miR-92 was seen to be downregulated, raising the question of whether this same downregulation is seen in indolent NHL subtypes such as FL (Amodio et al., 2015; Gado et al., 2019). The oncogenic miR-17–92 cluster, which includes miR-92a-3p, is a known driver of Burkitt lymphoma (BL), wherein miR-17 is associated with a poor prognosis and decreased overall survival rate (Robaina et al., 2016). miR-17–92 regulates multiple cellular pathways within NHL subtypes that favour malignant transformation, cellular proliferation, and cell survival (Dal Bo et al., 2015). In GCB DLBCL, the mir-17–92 microRNA cluster has been shown to be over-expressed when compared to ABC DLBCL (Lenz et al., 2008). Comparatively, the miR-92a-5p mature miRNA is not well described in NHL. With these relationships in mind, oncogenic or tumour suppressive miRNAs may be feasible targets for therapeutic agents, with the possibility of slowing or even halting malignancy (Wang and Wu, 2009), (Arif et al., 2020; Otoukesh et al., 2020). As recently reviewed in an earlier publication by our group, miRNAs can be epigenetically regulated, and even self-mediate these epigenetic modulations in various cancers (Arif et al., 2020). Aberrant DNA methylation in cytosines and adenines of mature miRNAs can lead to downregulation and suppression of the specific miRNA and, in NHL, a greater likelihood of lymphomagenesis (Calin and Croce, 2006; Esquela-Kerscher and Slack, 2006; Abba et al., 2017; Arif et al., 2020). These relationships support the possibility of miRNAs as being both functionally relevant in NHL and possible therapeutic targets.
The TET2 gene primarily functions as a tumour suppressor gene, immune regulator, and driver of DNA repair, as well as playing a key role in DNA demethylation (Fang et al., 2012). The TET2 protein catalyses the conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5-hmC), playing an integral role in transcriptional regulation. It is plausible that a hypermethylated genome may be due to a combination of factors that include high expression of DNA methyltransferases (DNMTs) or low expression of TET proteins (Fang et al., 2012; Asmar et al., 2013). TET2 is also involved in the recruitment of O-GlcNAc transferase OGT to CpG-rich sites to promote histone H2B GlcNAcylation (Asmar et al., 2013; Wang et al., 2018). TET2 is highly expressed in stem and progenitor cells, and in T-helper cells, whereas loss of TET2 gene function in bone marrow cells leads to increased immature B-cells resulting in lymphomagenesis (Fang et al., 2012; Asmar et al., 2013; Chiba, 2017). In NHL, TET2 expression is often reduced and regulated by methylation (Chiba, 2017). Various pathways are hypothesised to be regulated by miR-92a-TET2 including; JAK-STAT signalling, human pluripotent stem cells (hPSCs) differentiation, balanced B-cell terminal differentiation, and oxidative demethylation (miRTarBase, 2021; mimirna, 2021). To date, the prognostic significance of TET2 dysregulation in NHL is still not well characterised and further understanding of epigenetic drivers of TET2 regulation in lymphoma requires additional investigation.
A case-control cohort was previously recruited (Bradshaw et al., 2015), and a sub-cohort consisting of the peripheral blood gDNA of 80 retrospective case samples of Caucasian origin with Australian/British/European grandparents and with no family history of a haematological malignancy was assembled for this study. Details on the clinical diagnosis of individuals in the NHL patient cohort are listed in Table 1. The healthy control cohort consisted of 80 healthy individuals of similar age who had not been diagnosed with and cancer, nor did they have a family history of haematological malignancy. Cases were matched according to age (within ±5 years), sex, and ethnicity with the healthy control samples. For the 80 cases and controls, 26 (32.5%) were male and 54 (67.5%) were female. The mean age of the NHL diagnosed individuals was 66.56 years at collection in 2013, with a standard deviation of 12.40 years; the mean age of the control cohort was 64.54 years at collection in 2013, with a standard deviation of 11.61 years.

TABLE 1. GLP NHL case cohort with subtypes and number of cases for each subtype listed.
Snap-frozen lymph node tumour biopsies were obtained from BioOptions (California United States), with tumour tissue from 11 NHL cases (4 female and 7 female) with a confirmed diagnosis of NHL (5 DLBCL and 6 FL) (Table 2). A cohort of healthy volunteers was used for controls to compare with cell lines and tumours, using PB-derived leukocytes from 6 individuals aged between 23 and 38 years, with a mean age of 30 years and a standard deviation of 5.65, who had not been diagnosed with cancer nor did they have a family history of haematological malignancy. Of the 6 healthy controls, 3 (50%) were female.
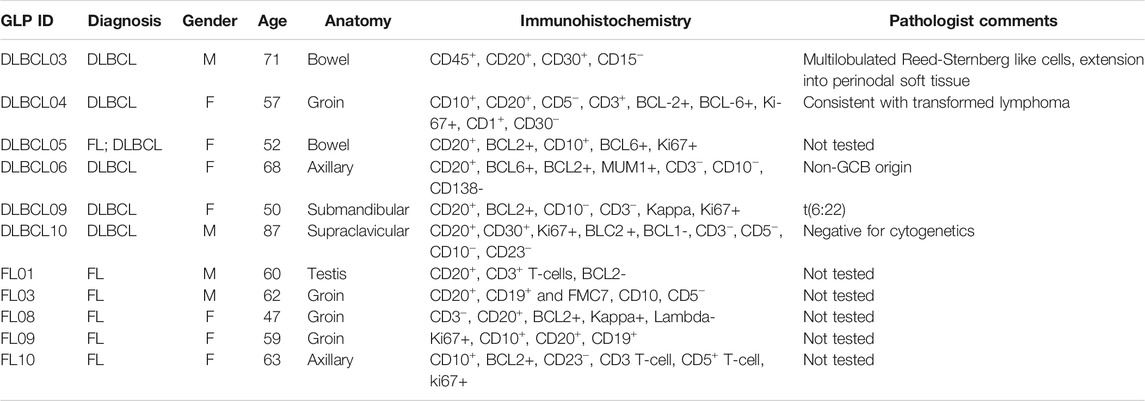
TABLE 2. Pathology of NHL tumour samples, including immunohistochemistry and comments from pathologist.
Studies were conducted using four commercial immortalised cell lines: Raji (BL), Toledo (DLBCL), SUDLH4 (DLBCL), and Mino (Mantle cell lymphoma (MCL)). Vials were stored in liquid nitrogen and rapidly thawed and seeded in a T-75 culture flask in 25 ml RPMI-1640, supplemented with 10% FBS and 1% Penicillin-Streptomycin antibiotics. The cells were incubated at 37°C in a humidified 5% CO2 incubator. Cell count and viability were assessed using Trypan Blue exclusion method in a 1:1 ratio of cell suspension to dye and measured on a TC10™ automated cell counter. Cells were split into a 1:2 ratio, with fresh media was added every second day until >80% confluence and 90% viability were achieved.
Computational analysis of miRNA and target gene prediction was performed using miRbase (miRBase, 2021), miRTarBase (miRTarBase, 2021), and Target Scan (Target Scan, 2021), identifying miR-92a-3p and 5p as candidates. DNA methylation analysis in the miR-17–92 cluster host-gene (MIR17HG) promoter region and the miR-92a to TET2 promoter was performed using MethHC 2.0 software.
gDNA was previously extracted from whole blood, collected into EDTA tubes, using an in-house salting-out method as evaluated by Chacon et al. (Chacon-Cortes et al., 2012). Both NHL cell line, control peripheral blood mononuclear cell (PBMC), and NHL tumour sample gDNA was extracted using the Wizard® SV Genomic DNA Purification System (Promega). DNA quality and quantity were quantified using a Nanodrop spectrophotometer, determined by A260/A280 ratio (ND1000 V3.8.1, ThermoFisher Scientific).
Bisulfite conversion of 500 ng of NHL B-cell line and control PBMC DNA and 750 ng of DNA from each patient/control DNA sample and NHL tumour sample was performed using the EZ DNA Methylation™ Kit (Zymo Research) according to the manufacturer’s protocol, with a modification to the elution volume, from 10 to 40 µL.
Total RNA was extracted using TRIzol® Reagent (ThermoFisher) and the Direct-zol RNA MiniPrep Kit (Zymo Research) from cell lines, control PBMCs, and NHL tumour samples. Total RNA, including miRNA, was reverse transcribed using the MiScript II RT Kit (Qiagen) for all samples.
Analyses were performed by real-time quantitative PCR (Q-PCR) to measure miR-92a mature miRNAs (MiScript SYBR® Green PCR, Qiagen) and mRNA transcripts (SYBR® Green PCR master mix, Promega). Customised forward and reverse primers (Integrated DNA Technologies, Inc) were designed for the miRNAs and TET2 mRNA transcripts of interest (Table 3). All assays were performed on the QuantStudio™ 7 Flex Real-Time PCR System (ThermoFisher) running the QuantStudio™ Software (v1.7.1) (ThermoFisher). miRNA assays were performed under the following conditions: 95°C for 2 min s (x1 cycle), 95°C for 10 s, 56°C for 60 s (x40 cycles), melt curve analysis 60–95°C. TET2 expression was measured under the following conditions: 50°C for 2 min (x1 cycle), 95°C for 3 min (x1 cycle), 95°C for 3 s, 60°C for 30 s (x50 cycles), melt curve analysis 60–95°C. Endogenous controls were miR103 for the miRNA assays and 18 S ribosomal RNA for the TET2 assays. Specific amplification of targets was confirmed via melt curve analysis and gene expression was calculated using the relative quantification method (ΔΔCt).

TABLE 3. Gene expression forward and reverse primer sequences with transcript accession numbers. miRNA expression assays utilised miScript universal reverse primer (Qiagen).
The UCSC genome browser was used to identify CGI regions in the MIR17HG and TET2 promoter regions (Figure 1). Pyromark Assay Design 2.0 software (Qiagen) was used to generate amplification and pyrosequencing primers to target regions in the CGI containing numerous CpGs (Table 4). Primers were designed to incorporate biotin on the reverse primer and an additional sequencing primer for the pyrosequencing, with one set targeting 7 CpGs in Exon 1 of TET2 and two regions of four CpGs were targeted within the promoter CGI of the MIR17HG gene, both regions that containing several transcription factor binding sites. Pyrosequencing was performed on a PyroMark Q48 Autoprep system (Qiagen) as per the manufacturer’s instructions using PyroMark Q96 Gold reagents (Qiagen). Pyrosequencing output analysis was performed using the Qseq software (BioMolecular Systems, V2.4.3).
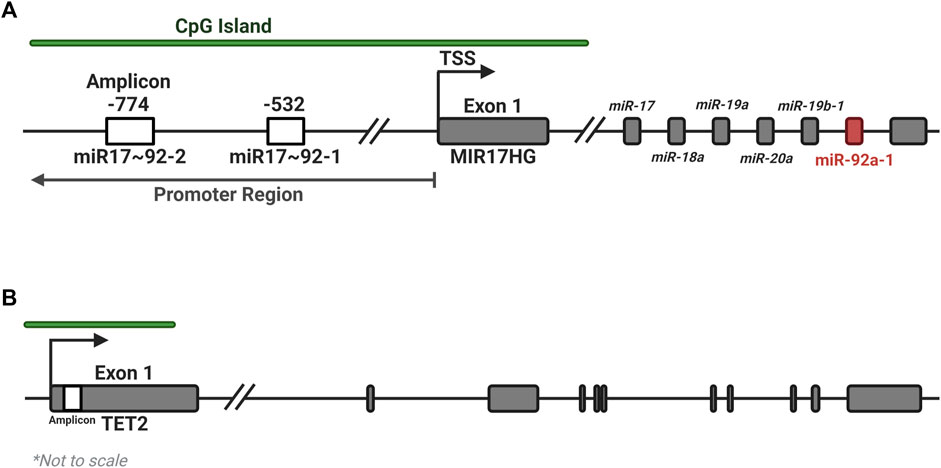
FIGURE 1. Location of amplicons for the MIR17HG and TET2 methylation assays. Two regions of four CpGs each were identified within the promoter CGI of the MIR17HG gene (A), and one region in the first exon of the TET2 gene (B), each containing several regulatory motifs and transcription factor binding sites. Created with BioRender.com.

TABLE 4. PCR and Pyrosequencing primers for TET2 and MIR17HG regulatory regions.
Statistical analysis of both expression and methylation data was performed in GraphPad Prism, with a Kruskal–Wallis (KW) test of significance and a post-hoc Dunn test for comparisons of individual groups.
The expression of miR-92a-3p and miR-92a-5p mature miRNAs and TET2 mRNA were assayed by RT-qPCR, and upstream promoter CGI methylation for each gene was assayed by bisulfite pyrosequencing, in 4 biological replicates of NHL cell lines SU-DHL-4, Mino, Raji, and Toledo, and in PBMCs of 6 healthy controls (Figure 2). miR-92a-3p expression was increased in Mino cells compared to controls (Figure 2A, p = 0.0145) and miR-92a-5p expression increased in Mino (p = 0.0013), Raji (p = 0.0016), and Toledo (p = 0.0200) cells compared to controls (Figure 2B). Differential methylation was observed in the MIR17HG upstream region, with Mino cells showing decreased methylation in region 1 when compared to controls (Figure 2D, p = 0.0460). Differential methylation was also observed between SU-DHL-4 and Raji cells (p = 0.0237). and between Mino cells and Raji (p = 0.0029) and Toledo (p = 0.0278). Toledo (p = 0.0329) cells showed increased methylation in region 2 when compared to controls (Figure 2E).
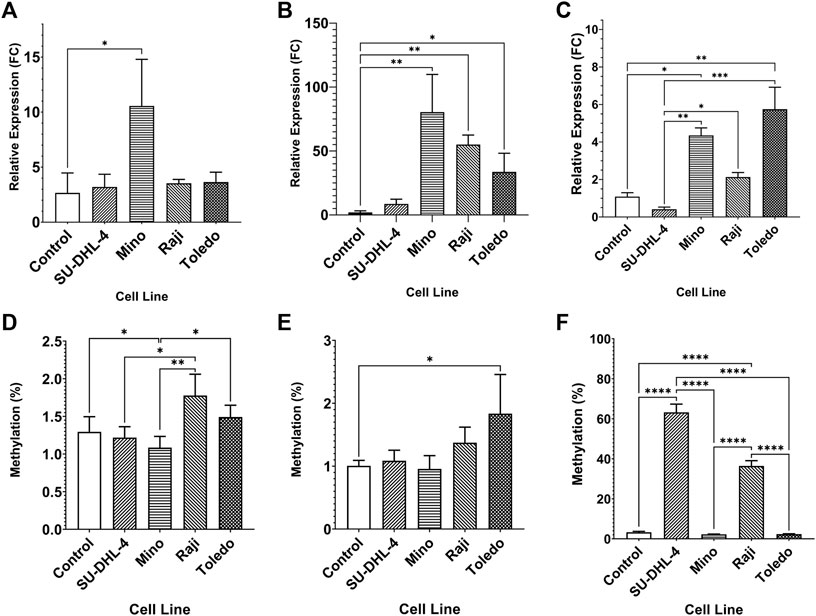
FIGURE 2. Expression miRNA 92a-3p, miRNA 92a-3p, and TET2 mRNA, and promoter DNA methylation of CpGs in two regions of the promoter CGI of the miR-17∼92 cluster and one region in the promoter CGI of the TET2 gene in NHL cell lines and in healthy control PBMCs. The expression of miR-92a-3p (A), miR-92a-5p (B) and TET2 mRNA (C) was assayed by RT-qPCR in commercially available NHL cell lines SU-DHL-4, Mino, Raji, and Toledo, and in healthy control PBMCs. Bisulfite pyrosequencing was performed on two regions in the promoter CGIs of the miR-17–92 cluster and in one region of the TET2 gene in the same cells and controls. Mean methylation levels of 4 CpGs in the two regions, 532 bp (D) and bp (E) upstream of the MIR17HG TSS were assayed, and mean methylation levels of 6 CpGs in the first exon of TET2 were also assayed (F). Bars denote mean and error bars denote SEM. Statistical significance calculated by KW test of significance and a post-hoc Dunn test for comparison of cell lines to controls, with significance denoted as: **** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.05.
TET2 was differentially expressed in Mino (p = 0.0153) and Toledo (p = 0.0045) cells compared to controls (Figure 2C). Differential expression was also observed between SU-DHL-4 cells and Mino (p = 0.0016), Raji (p = 0.0385), and Toledo (p = 0.0004) cells. Upstream promoter hypermethylation was observed in SU-DHL-4 and Raji cell lines when compared to controls. (Figure 2F, p < 0.0001). SU-DHL-4 cells were also significantly different to Mino and Toledo cells (p < 0.0001), and Raji cells were significantly different to Mino and Toledo cells (p < 0.0001).
The expression of miR-92a-3p and miR-92a-5p mature miRNAs and TET2 mRNA were assayed in NHL patient tumour biopsies and compared to healthy control PBMCs by RT-qPCR (Figure 3). Promoter methylation for each gene was assayed by bisulfite pyrosequencing in the same cohort. miR-92a-5p expression was increased in DLBCL tumours compared to controls (Figure 3B, p = 0.0017), and differential methylation was observed between controls and FL tumours in region 1 of the MIR17HG upstream promoter CGI (Figure 3D, p = 0.0203).
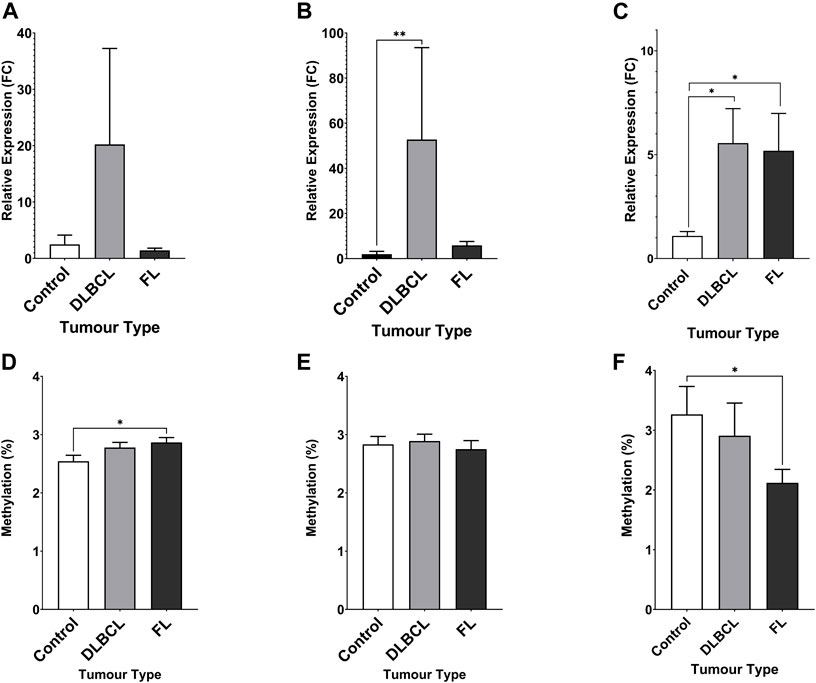
FIGURE 3. Expression miRNA 92a-3p, miRNA 92a-3p, and TET2 mRNA, and promoter DNA methylation of CpGs in two regions of the promoter CGI of the miR-17∼92 cluster and one region in the promoter CGI of the TET2 gene in NHL tumours compared to healthy control PBMCs. The expression of miR-92a-3p (A), miR-92a-5p (B) and TET2 mRNA (C) was assayed in by RT-qPCR in malignant lymphatic tissue of NHL patients and in healthy control PBMCs. Bisulfite pyrosequencing was performed on two regions in the promoter CGIs of the miR-17–92 cluster and in one region of the TET2 gene. Mean methylation levels of 4 CpGs in the two regions, 532 bp (D) and 774 bp (E) upstream of the MIR17HG TSS were assayed, and mean methylation levels of 6 CpGs in the first exon of TET2 were also assayed (F). Bars denote mean and error bars denote SEM. Statistical significance calculated by KW test of significance and a post-hoc Dunn test for comparison of cell lines to controls, with significance denoted as: **** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.05.
Increased TET2 mRNA expression was observed in both DLBCL (p = 0.0164) and FL (p = 0.0240) patient tumour biopsies compared to controls (Figure 3C), and decreased methylation was observed in FL tumours compared to controls (Figure 3F, p = 0.0300).
DNA methylation of CpGs in each region was assayed by bisulfite pyrosequencing of whole blood gDNA from an NHL case-control cohort, comprised of 80 cases with age and sex-matched controls (Figure 4). Increased DNA methylation of region 1 of the MIR17HG promoter CGI was observed in the NHL cohort compared to the controls (Figure 4A, p < 0.0001). Furthermore, differential DNA methylation was observed between healthy controls and NHL patient subtypes, with increased levels of DNA methylation observed in the FL (p = 0.0004), MCL (p = 0.0046), BL (p = 0.0062), and LBL (p = 0.0004) patient groups (Figure 4B). No differential methylation was observed between the cases and controls in region 2 of the MIR17HG upstream promoter CGI (Figure 4C); however, differential methylation was observed between controls and the DLBCL (p = 0.0041) and MCL (p = 0.0398) subtypes when the specific diagnosis was considered (Figure 4D). Decreased TET2 promoter CGI methylation was observed in NHL cases compared to controls (Figure 4E, p = 0.0159), and between healthy controls and FL subtype (Figure 4F, p = 0.0210).
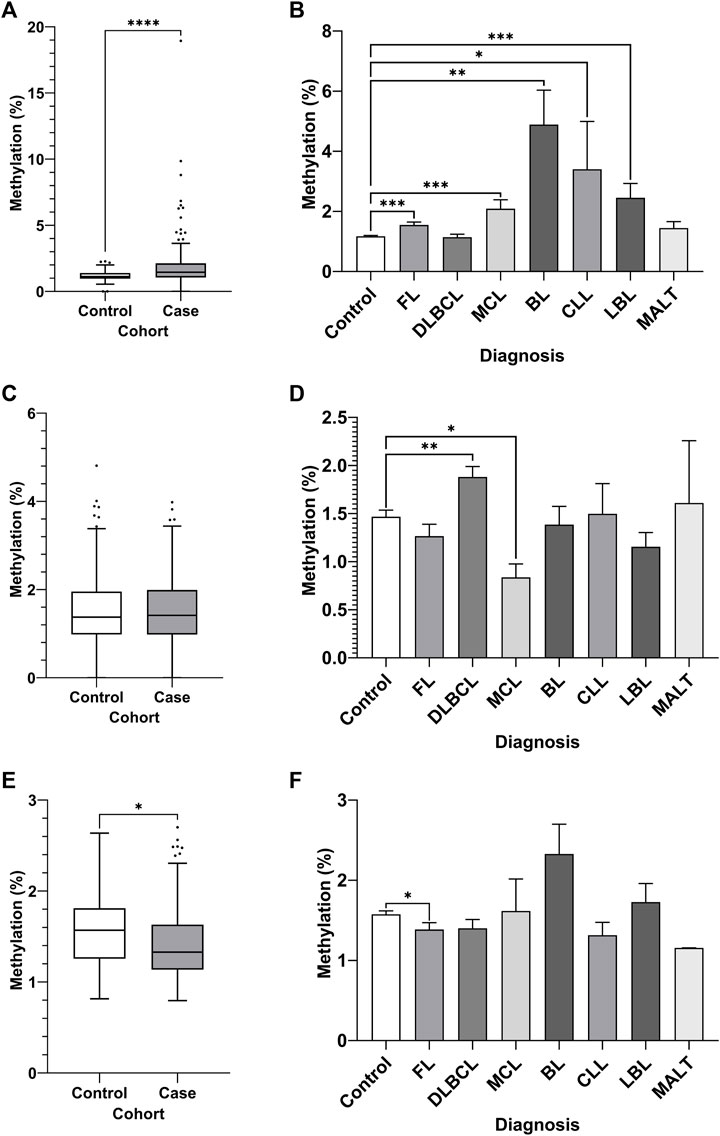
FIGURE 4. Promoter DNA methylation of CpGs in two regions of the promoter CGI of the miR-17∼92 cluster and one region in the promoter CGI of the TET2 gene in whole blood gDNA of a retrospective NHL case-control cohort. Bisulfite pyrosequencing was performed on two regions in the promoter CGIs of the miR-17–92 cluster and in one region of the TET2 gene. Mean methylation levels of 4 CpGs in the two regions, 532 bp (A–B) and 774 bp (C–D) upstream of the MIR17HG TSS were assayed, and mean methylation levels of 6 CpGs in the first exon of TET2 were also assayed (E–F). Patients were categorised by NHL subtype, with mean methylation levels compared between subtype and controls. Individuals without a specific subtype were not included in comparison between diagnosis. Bars denote mean and error bars denote SEM. Statistical significance calculated by KW test of significance and a post-hoc Dunn test for comparison of cell lines to controls, with significance denoted as: **** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.05.
In the last decade, both the role of miRNAs in malignancy and their viability as biomarkers for malignancy has been a rapidly expanding area of research. The mechanisms behind miRNA expression are not fully understood; however, aberrant methylation of promoter regions of miRNA genes has been previously implicated in aberrant miRNA expression in several cancers (Weber et al., 2007; Suzuki et al., 2010; Suzuki et al., 2012; Harada et al., 2015), including lymphoma (Stumpel et al., 2011; Yim et al., 2012). The expression profiles of miRNAs have been previously presented as novel biomarker panels for NHL (Khare et al., 2017; Solé et al., 2017), but the role of DNA methylation in miRNA expression regulation in NHL is not well explored. In this study, the methylation levels of CpGs in CGIs spanning the promoter regions of the miR-17–92 cluster host gene and the TET2 gene were quantified by bisulfite pyrosequencing and correlated with the expression levels of TET2 mRNA and mature miR-92a-3p and miR-92a-5p miRNAs. Gene expression was assayed in NHL B-cell lines, NHL tumour samples, and healthy PBMCs. Methylation of promoter CGIs was measured in the DNA of several NHL B-cell lines, in NHL tumour samples, in healthy PBMCs, and in whole blood gDNA of a retrospective NHL case-control cohort.
Increased expression of both the −3p and −5p mature miRNAs was observed in Mino cells when compared to controls, and additionally, upregulation of −5p was observed in Raji and Toledo cells lines. Previous studies have identified miR-92a overexpression in BL, MCL, and DLBCL cell lines, and our study replicates these findings (He et al., 2005; Ji et al., 2011). Higher expression of miR-92a has also been previously identified in MCL tumours (Husby et al., 2013; Roisman et al., 2016), reflecting the increased expression identified in Mino cells in this study. Increased expression of miR-92a-3p was also observed in our DLBCL tumour samples when compared to controls. Increased expression of miR-92a has previously been found to cause lymphatic malignancy in mice (Xiao et al., 2008), with downregulation of miR-92a implicated in the inhibition of tumour growth in an MCL mouse model (Rao et al., 2012). Overexpression of miR-92a and has been previously implicated in the reduction of overall survival and event-free survival in NHL patients (Yan et al., 2019), reinforcing the relevance of our findings.
Differential methylation was observed in the upstream promoter CGI of MIR17HG between cell lines, and between cell lines and healthy controls; however, this difference may not be biologically significant as the difference between mean methylation is within the detection limit margin of error (Gruber et al., 2002; Tost and Gut, 2007). Therefore, it seems unlikely that methylation of CpGs in these specific regions in the MIR17HG promoter region contribute to the differential miR-92a expression between NHL cell lines. A similar trend was identified in the NHL tumour samples; however, again the difference between mean methylation is within the detection limit margin of error. Aberrant methylation of the MIR17HG promoter has been previously correlated with dysfunctional expression of miRNAs in the miR-17–92 cluster in lung biopsies of individuals diagnosed with pulmonary fibrosis (Dakhlallah et al., 2013), and in both human samples and mouse models of bronchopulmonary dysplasia (Rogers et al., 2015; Robbins et al., 2016). The epigenetic mechanisms for the regulation of the miR-17–92 cluster expression in cancer, and in NHL specifically, are not well explored and further investigation is therefore required to identify the role of miR-92a in malignancy and specifically in lymphomagenesis. In NHL patient whole blood gDNA, statistically significant increased levels of mean methylation were observed in region 1 of the MIR17HG CGI when compared to healthy controls. Although the mean difference between the two cohorts was not substantial, several individuals in the NHL patient cohort presented high levels of genomic DNA methylation across these targeted regions. Aberrant methylation of levels were observed in individuals diagnosed with specific subtypes of NHL when compared to controls, BL being the most prominent. An aggressive subtype of NHL, the BL cell line Raji was identified to exhibit increased methylation in these same regions in the cell line populations, and previous studies have reported a similar increase in methylation levels to those identified in this study in other miRNA promoter regions, in both BL cell lines and BL tumours (Mazzoccoli et al., 2018; Mazzoccoli et al., 2019). Increased methylation was also observed between CLL individuals and controls in region 1 of the MIR17HG, and a previous study identified that aberrant methylation of miRNA promoter regions in circulating B-cells of CLL patients was associated with abnormal miRNA expression when compared to healthy B-cells. Increased expression of miR-92a has been implicated in CLL malignancy, specifically in lymph node proliferation centres (Szurián et al., 2017). As the case-control cohort in this study is comprised of whole blood gDNA rather than specifically PBMC or B-cell DNA these previous results cannot be replicated, but the similar rates of increased methylation of DNA can be considered, and further investigation into miRNA promoter methylation in CLL is required. It should be considered that, although differential methylation was observed between subtypes in this study, DNA hypermethylation has long been regarded as a hallmark of cancer, and the aberrant methylation observed in these regions may be a result of genome-wide increases in DNA methylation rather than a consequence of dysregulation of specific genes or promoter regions, endorsing further investigation into the role of DNA methylation in these specific NHL subtypes.
The findings of this study support previous conclusions regarding the significance of miR-92a in B-cell malignancy and reinforce the importance of investigating its specific role in NHL. Several previous studies have identified miR-92a as being implicated in dysregulation and suppression of tumour suppressors, including PTEN (Xiao et al., 2008) and PHLPP2 (Rao et al., 2012) in MCL models and VHL in CLL B-cells (Ghosh et al., 2009). This same may be true of TET2, wherein our studies identified a trend of dysregulation in some cells, along with additional novel findings which warrant further investigation. Differential expression of TET2 was seen across NHL cell lines and malignant tissues compared to controls, with specifically increased expression of TET2 in Mino, Raji, and Toledo cell lines alongside DLBCL and FL tumour tissue compared to controls. However, when taking into account the increased miR-92a-5p expression in the cell lines, NHL tumour tissue, and increased miR-92a-3p expression in the Mino cell line compared to controls, it is suggestive that miR-92a-5p and −3p may not be strong negative regulators of TET2 within these subtypes. Further assessment of the functional role of miR-92a on TET2 expression is required, feasibly with the utilisation of miRNA mimics and inhibitors, to determine the effect of miR-92a-5p and −3p expression and inhibition in NHL cell models.
SU-DHL-4 and Raji cell lines showed significant hypermethylation in the studied CpGs within the TET2 upstream promotor region when compared to controls and to Mino and Raji cell lines. This novel finding correlated with expected downregulation in TET2 expression in the SU-DHL-4 cells, along with a similar trend observed in the Raji cells compared to Mino and Toledo cell lines in previous studies (Chiba, 2016; Chiba, 2017). Although TET2 mRNA generally reported increased expression in cell lines and tumours, with the exception of SU-DHL-4, when compared to controls in our findings, TET2 methylation in our target region negatively correlated with expression in the NHL cell lines, indicating that the CpGs assayed in this region may be involved in the regulation of TET2 transcription and expression. TET2 overexpression in NHL is not well explored when compared to well-documented trends of downregulation in TET2 within various haematological malignancies (Chiba, 2017); however, previous studies have established this same trend of TET2 over-expression in CLL as observed in our study (Hernández-Sánchez et al., 2014). A possible explanation for this finding is TET2 overexpression in cells of some aggressive and indolent NHL subtypes is an innate immune response, involving tumour suppressor and DNA repair pathways, to the specific subtype of malignancy. Examination of TET2 protein and of downstream pathways of TET2 related expression regulation may assist in determining the translational consequence of TET2 overexpression in these NHL samples and cell lines.
Previous studies have identified TET2 variants in DLBCL as being associated with malignancy via the hypermethylation of pro-tumorigenic genes (Liu et al., 2017). Genotyping of the TET2 gene in the specific NHL cell models and tumours samples may therefore provide greater understanding into the potential presence of disease-causing TET2 mutations, which may contribute to disease phenotypes; as highlighted by differential expression DLBCL cell lines SU-DHL-4 and Toledo, both of which are DLBCL cell models. DNA methylation studies of CpGs in the TET2 upstream promotor region across both NHL tumour-derived and control PBMC samples showed consistently low levels of methylation, and a similar trend was observed in the methylation of genomic DNA in the NHL case-control cohort. Differential methylation was observed in the TET2 regulatory region between the case and control cohort; however, this difference may again not be biologically significant (Gruber et al., 2002; Tost and Gut, 2007). It is therefore unlikely that localised, as well as genome-wide, aberration of DNA methylation in CpGs in this region are directly involved in TET2 expression regulation in these NHL cohorts.
Clinical data from specific DLBCL tumour samples presented interesting characteristics and relationships when both TET2 and miR-92a expression were considered (Table 1). One tumour sample displayed Reed-Sternberg-like cells during the histological examination, while our studies showed significantly decreased TET2 expression compared to other DLBCL and FL tumour samples, as well as compared controls considered (Supplementary Figure S1E). Hypermethylation within our studied CpGs was not noted, therefore a likely cause of decreased TET2 expression in this outlier is the presence of a functional mutation in TET2. Reed-Sternberg-like cells have been previously associated with TET2 mutations (Venanzi et al., 2021) along with diminished 5 hmC levels (Siref et al., 2020). Genotyping the tumour samples would assist in determining whether a polymorphism or variant in TET2 is the cause of downregulated TET2 levels, thereby further supporting previous findings regarding TET2 as a potential marker and driver of malignancy in NHL.
The role of TET2 in NHL has been previously documented; however, the mechanisms driving TET2 expression regulation, such as DNA methylation, are not well explored. We identified in this study that aberrant methylation of several CpGs in the regulatory region of TET2 correlated with TET2 mRNA expression in NHL cell models. These findings are novel and may indicate that methylation of these regions may play a functional role in TET2 expression. This same relationship was not observed in NHL tumour samples or in NHL patient genomic DNA. Although a target of miR-92a, and aberrant miR-92a-3p and miR-92a-5p expression was observed in NHL cell models and NHL tumours, an association between miR-92a and TET2 was not observed in this study. Further assessment of the functional role of miR-92a-TET2 in human cell models is required; as is, ultimately, assessment in clinical cohorts. Although TET2 may not be comprehensively regulated by miR-92a, these novel findings regarding dysregulated methylation of TET2 regulatory regions in NHL is an intriguing relationship warranting further investigation and reinforces the prominence of TET2 as an element in NHL pathogenesis and malignancy.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation to any qualified researcher.
The case and control volunteers completed a questionnaire and provided written consent to participate in research. The QUT Research Ethics Committee approved ethical clearance for the collection and use of participant samples (approval number 1900000721), in accordance with the Declaration of Helsinki.
EE and LH: Participant recruitment, assay design, DNA methylation studies, functional experiments and analyses, and wrote the original draft and revised the manuscript. RH: Clinical data review and editing. HS, LH, and LG: Study conceptualisation and final manuscript editing. LG: Participant recruitment, funding acquisition, and final manuscript editing and submission.
Funding for this study has been generously provided by the GRC Genomics Lymphoma Project fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Ms Gabrielle Bradshaw for her prior work, Mr Taufiq Arif and Mr Ian Peall for sharing their functional studies expertise, and Mr Robert Smith for his ethics submission oversight. We acknowledge Icon Cancer Center for supporting this project. We also thank the participants and volunteers for donating valuable samples.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.768913/full#supplementary-material
Abba, M. L., Patil, N., Leupold, J. H., Moniuszko, M., Utikal, J., Niklinski, J., et al. (2017). MicroRNAs as Novel Targets and Tools in Cancer Therapy. Cancer Lett. 387, 84–94. doi:10.1016/j.canlet.2016.03.043
Abba, M., Patil, N., Leupold, J. H., and Allgayer, H. (2016). MicroRNAs-from Metastasis Prediction to Metastasis Prevention. Mol. Cell Oncol. 3 (2), e1074336. doi:10.1080/23723556.2015.1074336
Alizadeh, A. A., Eisen, M. B., Davis, R. E., Ma, C., Lossos, I. S., Rosenwald, A., et al. (2000). Distinct Types of Diffuse Large B-Cell Lymphoma Identified by Gene Expression Profiling. Nature 403 (6769), 503–511. doi:10.1038/35000501
Allemani, C., Matsuda, T., Di Carlo, V., Harewood, R., Matz, M., Nikšić, M., et al. (2018). Global Surveillance of Trends in Cancer Survival 2000-14 (CONCORD-3): Analysis of Individual Records for 37 513 025 Patients Diagnosed with One of 18 Cancers from 322 Population-Based Registries in 71 Countries. Lancet 391 (10125), 1023–1075. doi:10.1016/S0140-6736(17)33326-3
Amodio, N., Rossi, M., Raimondi, L., Pitari, M. R., Botta, C., Tagliaferri, P., et al. (2015). miR-29s: a Family of Epi-miRNAs with Therapeutic Implications in Hematologic Malignancies. Oncotarget 6 (15), 12837–12861. doi:10.18632/oncotarget.3805
Arif, K. M. T., Elliott, E. K., Haupt, L. M., and Griffiths, L. R. (2020). Regulatory Mechanisms of Epigenetic miRNA Relationships in Human Cancer and Potential as Therapeutic Targets. Cancers (Basel) 12 (10), 2922. doi:10.3390/cancers12102922
Asmar, F., Punj, V., Christensen, J., Pedersen, M. T., Pedersen, A., Nielsen, A. B., et al. (2013). Genome-wide Profiling Identifies a DNA Methylation Signature that Associates with TET2 Mutations in Diffuse Large B-Cell Lymphoma. Haematologica 98 (12), 1912–1920. doi:10.3324/haematol.2013.088740
Ayers, E. C., Li, S., Medeiros, L. J., Bond, D. A., Maddocks, K. J., Torka, P., et al. (2020). Outcomes in Patients with Aggressive B‐cell non‐Hodgkin Lymphoma after Intensive Frontline Treatment Failure. Cancer 126 (2), 293–303. doi:10.1002/cncr.32526
Blenk, S., Engelmann, J., Weniger, M., Schultz, J., Dittrich, M., Rosenwald, A., et al. (2007). Germinal center B Cell-like (GCB) and Activated B Cell-like (ABC) Type of Diffuse Large B Cell Lymphoma (DLBCL): Analysis of Molecular Predictors, Signatures, Cell Cycle State and Patient Survival. Cancer Inform. 3, 399–420. doi:10.1177/117693510700300004
Bradshaw, G., Sutherland, H. G., Haupt, L. M., and Griffiths, L. R. (2016). Dysregulated MicroRNA Expression Profiles and Potential Cellular, Circulating and Polymorphic Biomarkers in Non-hodgkin Lymphoma. Genes (Basel) 7 (12), 130. doi:10.3390/genes7120130
Bradshaw, G., Sutherland, H. G., Camilleri, E. T., Lea, R. A., Haupt, L. M., and Griffiths, L. R. (2015). Genetic and Epigenetic Variants in the MTHFR Gene Are Not Associated with Non-hodgkin Lymphoma. Meta gene 6, 91–95. doi:10.1016/j.mgene.2015.09.004
Calin, G. A., and Croce, C. M. (2006). MicroRNA Signatures in Human Cancers. Nat. Rev. Cancer. 6 (11), 857–866. doi:10.1038/nrc1997
Calin, G. A., Dumitru, C. D., Shimizu, M., Bichi, R., Zupo, S., Noch, E., et al. (2002). Nonlinear Partial Differential Equations and Applications: Frequent Deletions and Down-Regulation of Micro- RNA Genes miR15 and miR16 at 13q14 in Chronic Lymphocytic Leukemia. Proc. Natl. Acad. Sci. 99 (24), 15524–15529. doi:10.1073/pnas.242606799
Cancer Council (2020). Cancer Facts and Figures 2020. Available at: https://www.cancer.org.au/about-cancer-information/what-is-cancer/facts-and-figures (Accessed on: June 10, 2021).
Caramuta, S., Lee, L., Özata, D. M., Akçakaya, P., Georgii-Hemming, P., Xie, H., et al. (2013). Role of microRNAs and microRNA Machinery in the Pathogenesis of Diffuse Large B-Cell Lymphoma. Blood Cancer J. 3, e152. doi:10.1038/bcj.2013.49
Chacon-Cortes, D., Haupt, L. M., Lea, R. A., and Griffiths, L. R. (2012). Comparison of Genomic DNA Extraction Techniques from Whole Blood Samples: a Time, Cost and Quality Evaluation Study. Mol. Biol. Rep. 39 (5), 5961–5966. doi:10.1007/s11033-011-1408-8
Chiba, S. (2016). Significance of TET2 Mutations in Myeloid and Lymphoid Neoplasms. Rinsho Ketsueki 57 (6), 715–722. doi:10.11406/rinketsu.57.715
Chiba, S. (2017). Dysregulation of TET2 in Hematologic Malignancies. Int. J. Hematol. 105 (1), 17–22. doi:10.1007/s12185-016-2122-z
Culpin, R. E., Proctor, S. J., Angus, B., Crosier, S., Anderson, J. J., and Mainou-Fowler, T. (2010). A 9 Series microRNA Signature Differentiates Between Germinal Centre and Activated B-Cell-Like Diffuse Large B-Cell Lymphoma Cell Lines. Int. J. Oncol. 37 (2), 367–376. doi:10.3892/ijo_00000685
Dakhlallah, D., Batte, K., Wang, Y., Cantemir-Stone, C. Z., Yan, P., Nuovo, G., et al. (2013). Epigenetic Regulation ofmiR-17∼92Contributes to the Pathogenesis of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 187 (4), 397–405. doi:10.1164/rccm.201205-0888oc
Dal Bo, M., Bomben, R., Hernández, L., and Gattei, V. (2015). The MYC/miR-17-92 axis in Lymphoproliferative Disorders: A Common Pathway with Therapeutic Potential. Oncotarget 6 (23), 19381–19392. doi:10.18632/oncotarget.4574
Di Lisio, L., Sánchez-Beato, M., Gómez-López, G., Rodríguez, M. E., Montes-Moreno, S., Mollejo, M., et al. (2012). MicroRNA Signatures in B-Cell Lymphomas. Blood Cancer J. 2 (2), e57. doi:10.1038/bcj.2012.1
Esquela-Kerscher, A., and Slack, F. J. (2006). Oncomirs - microRNAs with a Role in Cancer. Nat. Rev. Cancer 6 (4), 259–269. doi:10.1038/nrc1840
Fang, C., Zhu, D.-X., Dong, H.-J., Zhou, Z.-J., Wang, Y.-H., Liu, L., et al. (2012). Serum microRNAs Are Promising Novel Biomarkers for Diffuse Large B Cell Lymphoma. Ann. Hematol. 91 (4), 553–559. doi:10.1007/s00277-011-1350-9
Gado, M. M., Mousa, N. O., Badawy, M. A., El Taweel, M. A., and Osman, A. (2019). Assessment of the Diagnostic Potential of miR-29a-3p and miR-92a-3p as Circulatory Biomarkers in Acute Myeloid Leukemia. Asian Pac. J. Cancer Prev. 20 (12), 3625–3633. doi:10.31557/apjcp.2019.20.12.3625
Getaneh, Z., Asrie, F., and Melku, M. (2019). MicroRNA Profiles in B-Cell Non-hodgkin Lymphoma. EJIFCC 30 (2), 195–214.
Ghosh, A. K., Shanafelt, T. D., Cimmino, A., Taccioli, C., Volinia, S., Liu, C.-g., et al. (2009). Aberrant Regulation of pVHL Levels by microRNA Promotes the HIF/VEGF axis in CLL B Cells. Blood 113 (22), 5568–5574. doi:10.1182/blood-2008-10-185686
Gruber, J. D., Colligan, P. B., and Wolford, J. K. (2002). Estimation of Single Nucleotide Polymorphism Allele Frequency in DNA Pools by Using Pyrosequencing. Hum. Genet. 110 (5), 395–401. doi:10.1007/s00439-002-0722-6
Harada, K., Baba, Y., Ishimoto, T., Kosumi, K., Tokunaga, R., Izumi, D., et al. (2015). Suppressor microRNA-145 Is Epigenetically Regulated by Promoter Hypermethylation in Esophageal Squamous Cell Carcinoma. Anticancer Res. 35 (9), 4617–4624.
He, L., Thomson, J. M., Hemann, M. T., Hernando-Monge, E., Mu, D., Goodson, S., et al. (2005). A microRNA Polycistron as a Potential Human Oncogene. Nature 435 (7043), 828–833. doi:10.1038/nature03552
Hernández-Sánchez, M., Rodríguez, A. E., Kohlmann, A., Benito, R., García, J. L., Risueño, A., et al. (2014). TET2 Overexpression in Chronic Lymphocytic Leukemia Is Unrelated to the Presence of TET2 Variations. Biomed. Res. Int. 2014, 814294. doi:10.1155/2014/814294
Husby, S., Geisler, C., and Grønbæk, K. (2013). MicroRNAs in Mantle Cell Lymphoma. Leuk. Lymphoma 54 (9), 1867–1875. doi:10.3109/10428194.2013.766731
Ji, M., Rao, E., Ramachandrareddy, H., Shen, Y., Jiang, C., Chen, J., et al. (2011). The miR-17-92 MicroRNA Cluster Is Regulated by Multiple Mechanisms in B-Cell Malignancies. Am. J. Pathol. 179 (4), 1645–1656. doi:10.1016/j.ajpath.2011.06.008
Khare, D., Goldschmidt, N., Bardugo, A., Gur-Wahnon, D., Ben-Dov, I. Z., and Avni, B. (2017). Plasma microRNA Profiling: Exploring Better Biomarkers for Lymphoma Surveillance. PLoS ONE 12 (11), e0187722. doi:10.1371/journal.pone.0187722
Lawrie, C. H., Chi, J., Taylor, S., Tramonti, D., Ballabio, E., Palazzo, S., et al. (2009). Expression of microRNAs in Diffuse Large B Cell Lymphoma Is Associated with Immunophenotype, Survival and Transformation from Follicular Lymphoma. J. Cel Mol Med 13 (7), 1248–1260. doi:10.1111/j.1582-4934.2008.00628.x
Lenz, G., Wright, G. W., Emre, N. C. T., Kohlhammer, H., Dave, S. S., Davis, R. E., et al. (2008). Molecular Subtypes of Diffuse Large B-Cell Lymphoma Arise by Distinct Genetic Pathways. Proc. Natl. Acad. Sci. 105 (36), 13520–13525. doi:10.1073/pnas.0804295105
Liu, P., Jiang, W., Zhao, J., and Zhang, H. (2017). Integrated Analysis of Genome-wide Gene Expression and DNA Methylation Microarray of Diffuse Large B-Cell Lymphoma with TET Mutations. Mol. Med. Rep. 16 (4), 3777–3782. doi:10.3892/mmr.2017.7058
Martin-Guerrero, I., Gutierrez-Camino, A., Lopez-Lopez, E., Bilbao-Aldaiturriaga, N., Pombar-Gomez, M., Ardanaz, M., et al. (2015). Genetic Variants in miRNA Processing Genes and pre-miRNAs Are Associated with the Risk of Chronic Lymphocytic Leukemia. PLoS One 10 (3), e0118905. doi:10.1371/journal.pone.0118905
Mazzoccoli, L., Robaina, M. C., Bacchi, C. E., Soares Lima, S. C., and Klumb, C. E. (2019). miR-29 Promoter and Enhancer Methylation Identified by Pyrosequencing in Burkitt Lymhoma Cells: Interplay between MYC and miR-29 R-egulation. Oncol. Rep. 42 (2), 775–784. doi:10.3892/or.2019.7183
Mazzoccoli, L., Robaina, M. C., Apa, A. G., Bonamino, M., Pinto, L. W., Queiroga, E., et al. (2018). MiR-29 Silencing Modulates the Expression of Target Genes Related to Proliferation, Apoptosis and Methylation in Burkitt Lymphoma Cells. J. Cancer Res. Clin. Oncol. 144 (3), 483–497. doi:10.1007/s00432-017-2575-3
Michot, J.-M., Benajiba, L., Faivre, L., Baldini, C., Haddag, L., Bonnet, C., et al. (2018). Outcomes and Prognostic Factors for Relapsed or Refractory Lymphoma Patients in Phase I Clinical Trials. Invest. New Drugs 36 (1), 62–74. doi:10.1007/s10637-017-0480-x
mimirna (2021). iR-92a 2021. Available at: http://mimirna.centenary.org.au/.
miRBase (2021). miRNA 2021. Available at: http://www.mirbase.org/.
miRTarBase (2021). miRNA Database 2021. Available at: http://mirtarbase.mbc.nctu.edu.tw/php/index.php (Accessed on: March 4, 2021)
Otoukesh, B., Abbasi, M., Gorgani, H.-o. -L., Farahini, H., Moghtadaei, M., Boddouhi, B., et al. (2020). MicroRNAs Signatures, Bioinformatics Analysis of miRNAs, miRNA Mimics and Antagonists, and miRNA Therapeutics in Osteosarcoma. Cancer Cel Int. 20, 254. doi:10.1186/s12935-020-01342-4
Rao, E., Jiang, C., Ji, M., Huang, X., Iqbal, J., Lenz, G., et al. (2012). The miRNA-17∼92 Cluster Mediates Chemoresistance and Enhances Tumor Growth in Mantle Cell Lymphoma via PI3K/AKT Pathway Activation. Leukemia 26 (5), 1064–1072. doi:10.1038/leu.2011.305
Robaina, M. C., Faccion, R. S., Mazzoccoli, L., Rezende, L. M. M., Queiroga, E., Bacchi, C. E., et al. (2016). miR-17-92 Cluster Components Analysis in Burkitt Lymphoma: Overexpression of miR-17 Is Associated with Poor Prognosis. Ann. Hematol. 95 (6), 881–891. doi:10.1007/s00277-016-2653-7
Robbins, M. E., Dakhlallah, D., Marsh, C. B., Rogers, L. K., and Tipple, T. E. (2016). Of Mice and Men: Correlations between microRNA-17∼92 Cluster Expression and Promoter Methylation in Severe Bronchopulmonary Dysplasia. Am. J. Physiology-Lung Cell Mol. Physiol. 311 (5), L981–L984. doi:10.1152/ajplung.00390.2016
Rogers, L. K., Robbins, M., Dakhlallah, D., Yang, Z., Lee, L. J., Mikhail, M., et al. (2015). Attenuation ofmiR-17∼92Cluster in Bronchopulmonary Dysplasia. Ann. ATS 12 (10), 1506–1513. doi:10.1513/annalsats.201501-058oc
Roisman, A., Huamán Garaicoa, F., Metrebian, F., Narbaitz, M., Kohan, D., García Rivello, H., et al. (2016). SOXC and MiR17-92 Gene Expression Profiling Defines Two Subgroups with Different Clinical Outcome in Mantle Cell Lymphoma. Genes Chromosomes Cancer 55 (6), 531–540. doi:10.1002/gcc.22355
Sarkozy, C., and Sehn, L. H. (2018). Management of Relapsed/refractory DLBCL. Best Pract. Res. Clin. Haematol. 31 (3), 209–216. doi:10.1016/j.beha.2018.07.014
Shankland, K. R., Armitage, J. O., and Hancock, B. W. (2012). Non-Hodgkin Lymphoma. The Lancet 380 (9844), 848–857. doi:10.1016/s0140-6736(12)60605-9
Siegel, R. L., Miller, K. D., and Jemal, A. (20192019). Cancer Statistics, 2019. CA A. Cancer J. Clin. 69 (1), 7–34. doi:10.3322/caac.21551
Siref, A., McCormack, C., Huang, Q., Lim, W., and Alkan, S. (2020). Diminished Expression of 5hmc in Reed-Sternberg Cells in Classical Hodgkin Lymphoma Is a Common Epigenetic Marker. Leuk. Res. 96, 106408. doi:10.1016/j.leukres.2020.106408
Solé, C., Larrea, E., Di Pinto, G., Tellaetxe, M., and Lawrie, C. H. (2017). miRNAs in B-Cell Lymphoma: Molecular Mechanisms and Biomarker Potential. Cancer Lett. 405, 79–89. doi:10.1016/j.canlet.2017.07.020
Stumpel, D. J. P. M., Schotte, D., Lange-Turenhout, E. A. M., Schneider, P., Seslija, L., de Menezes, R. X., et al. (2011). Hypermethylation of Specific microRNA Genes in MLL-Rearranged Infant Acute Lymphoblastic Leukemia: Major Matters at a Micro Scale. Leukemia 25 (3), 429–439. doi:10.1038/leu.2010.282
Suzuki, H., Maruyama, R., Yamamoto, E., and Kai, M. (2012). DNA Methylation and microRNA Dysregulation in Cancer. Mol. Oncol. 6 (6), 567–578. doi:10.1016/j.molonc.2012.07.007
Suzuki, H., Yamamoto, E., Nojima, M., Kai, M., Yamano, H.-o., Yoshikawa, K., et al. (2010). Methylation-associated Silencing of microRNA-34b/c in Gastric Cancer and its Involvement in an Epigenetic Field Defect. Carcinogenesis 31 (12), 2066–2073. doi:10.1093/carcin/bgq203
Szurián, K., Csala, I., Piurkó, V., Deák, L., Matolcsy, A., and Reiniger, L. (2017). Quantitative miR Analysis in Chronic Lymphocytic Leukaemia/small Lymphocytic Lymphoma - Proliferation Centres Are Characterized by High miR-92a and miR-155 and Low miR-150 Expression. Leuk. Res. 58, 39–42. doi:10.1016/j.leukres.2017.04.002
Target Scan (2021). miRNA 2021. Available at: http://www.targetscan.org/vert_72/.
Teras, L. R., DeSantis, C. E., Cerhan, J. R., Morton, L. M., Jemal, A., and Flowers, C. R. (2016). 2016 US Lymphoid Malignancy Statistics by World Health Organization Subtypes. CA: a Cancer J. clinicians 66 (6), 443–459. doi:10.3322/caac.21357
Tost, J., and Gut, I. G. (2007). DNA Methylation Analysis by Pyrosequencing. Nat. Protoc. 2 (9), 2265–2275. doi:10.1038/nprot.2007.314
Venanzi, A., Marra, A., Schiavoni, G., Milner, S. G., Limongello, R., Santi, A., et al. (2021). Dissecting Clonal Hematopoiesis in Tissues of Patients with Classic Hodgkin Lymphoma. Blood Cancer Discov. 2 (3), 216–225. doi:10.1158/2643-3230.bcd-20-0203
Wang, L., Ozark, P. A., Smith, E. R., Zhao, Z., Marshall, S. A., Rendleman, E. J., et al. (2018). TET2 Coactivates Gene Expression through Demethylation of Enhancers. Sci. Adv. 4 (11), eaau6986. doi:10.1126/sciadv.aau6986
Wang, V., and Wu, W. (2009). MicroRNA-based Therapeutics for Cancer. BioDrugs 23 (1), 15–23. doi:10.2165/00063030-200923010-00002
Weber, B., Stresemann, C., Brueckner, B., and Lyko, F. (2007). Methylation of Human microRNA Genes in normal and Neoplastic Cells. Cell Cycle 6 (9), 1001–1005. doi:10.4161/cc.6.9.4209
Xiao, C., Srinivasan, L., Calado, D. P., Patterson, H. C., Zhang, B., Wang, J., et al. (2008). Lymphoproliferative Disease and Autoimmunity in Mice with Increased miR-17-92 Expression in Lymphocytes. Nat. Immunol. 9 (4), 405–414. doi:10.1038/ni1575
Yan, S., Jia, C., Quan, L., Zhao, L., Tian, Y., and Liu, A. (2019). Significance of the microRNA-17-92 G-ene C-luster E-xpressed in B-cell non-Hodgkin's L-ymphoma. Mol. Med. Rep. 20 (3), 2459–2467. doi:10.3892/mmr.2019.10448
Keywords: miR-17∼92, Burkitt lymphoma, mantle cell lymphoma, diffuse large B-cell lymphoma, micro-RNA (miRNA/miR), DNA methylation, epigenetic regulation
Citation: Elliott EK, Hopkins LN, Hensen R, Sutherland HG, Haupt LM and Griffiths LR (2021) Epigenetic Regulation of miR-92a and TET2 and Their Association in Non-Hodgkin Lymphoma. Front. Genet. 12:768913. doi: 10.3389/fgene.2021.768913
Received: 01 September 2021; Accepted: 26 October 2021;
Published: 26 November 2021.
Edited by:
Mounia S. Braza, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
David D. Eisenstat, Murdoch Childrens Research Institute, AustraliaCopyright © 2021 Elliott, Hopkins, Hensen, Sutherland, Haupt and Griffiths. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lyn R. Griffiths, bHluLmdyaWZmaXRoc0BxdXQuZWR1LmF1
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.