 Cong Zhou
Cong Zhou Yuanyuan Xiao1,2
Yuanyuan Xiao1,2- 1Department of Obstetrics and Gynecology, West China Second University Hospital, Sichuan University, Chengdu, China
- 2Key Laboratory of Birth Defects and Related Diseases of Women and Children, Ministry of Education, Sichuan University, Chengdu, China
Autosomal recessive non-syndromic deafness-28 (DFNB28) is characterized by prelingual, profound sensorineural hearing loss (HL). The disease is related to variants of the TRIOBP gene. TRIO and F-actin binding protein (TRIOBP) plays crucial roles in modulating the assembly of the actin cytoskeleton and are responsible for the proper structure and function of stereocilia in the inner ear. This study aimed to identify pathogenic variants in a patient with HL. Genomic DNA obtained from a 33-year-old woman with HL was evaluated using a disease-targeted gene panel. Using next generation sequencing and bioinformatics analysis, we identified two novel TRIOBP c.1170delC (p.S391Pfs*488) and c.3764C > G (p.S1255*) variants. Both parents of the patient were heterozygous carriers of the gene. The two variants have not been reported in general population databases or published literature. The findings of this study will broaden the spectrum of pathogenic variants in the TRIOBP gene.
Introduction
Hearing loss (HL) is one of the most common sensory disorders in humans, affecting 466 million people worldwide. The prevalence of bilateral permanent HL is estimated at one in 900–2,500 newborns, with genetic causes accounting for more than half of the cases (Cynthia and Walter, 2006). Genetic HL can be divided into syndromic and non-syndromic sensorineural HL (SNHL) (Nicolas and Anil, 2002). Hereditary hearing impairment without any other relevant clinical features is referred to as “non-syndromes” and is a genetically heterogeneous disorder (Thomas et al., 2003). To date, 123 non-syndromic HL genes have been identified (http://hereditaryhearingloss.org/). Nevertheless, pathogenic variants in common HL genes can be identified in only one-third of patients with SNHL (Morag et al., 2018).
Non-syndromic deafness-28 (DFNB28) is related to variants in the TRIOBP gene (OMIM: 609761) on chromosome 22q13 (Agnieszka et al., 2017). Multiple isoforms of the protein have been discovered (Beti et al., 2020). Human and mouse TRIOBP isoforms are divided into long forms (TRIOBP-3, TRIOBP-5 and TRIOBP-6) and short forms (TRIOBP-1, TRIOBP-2, and TRIOBP-4) (Jian et al., 2015; Jian et al., 2013; Shin et al., 2010). TRIOBP-1 consists of a pleckstrin homology (PH) domain near the N-terminal and coiled coil (CC) domains that make up the C-terminal half of the protein. TRIOBP-1 is widely expressed and binds to F-actin. It plays an important role in many processes including cell cycle, adhesion, and neuronal differentiation (Beti et al., 2020). TRIOBP-2 may encode the N-terminal sections of TRIOBP-1 including the PH domain and parts of the CC domain. However, this has not been thoroughly described so far. TRIOBP-4 is a 1,144 amino acid protein in humans, which is highly expressed in the hair cells of the inner ear and is crucial for the bundling of actin in the stereocilia of the inner ear, with variants in it causing severe or profound hearing loss (Shin et al., 2010; Beti et al., 2020). TRIOBP-5 (also called TRIOBP-3 in some earlier articles)encode 2,193 amino acid protein. TRIOBP-5 and TRIOBP-4 are expressed in the same inner ear cell types, and can be found in static ciliated rootlets. TRIOBP-6 is slightly longer than TRIOBP-5, which encoding 2,365 amino acid protein. More additional sequences than TRIOBP-5 are predicted to be unstructured, except for a stretch of α-helix in the isoform specific N-terminus of TRIOBP-6, and another segment near the center of the long isoform. It is likely that the role of TRIOBP-6 in the stereocilia may be consistent with TRIOBP-5, but this remains unclear because lack of a known murine TRIOBP-6 species (Beti et al., 2020; Jian et al., 2015; Jian et al., 2013; Shin et al., 2010).
In the present study, we present a family with isolated, prelingual HL with a recessive inheritance pattern, using 162 targeted genes, a mitochondrial whole gene enrichment panel and Sanger sequencing to identify two novel TRIOBP pathogenic variants (c.1170delC, p.S391Pfs*488 and c.3764C > G, p.S1255*) and establish a molecular diagnosis.
Patient and Methods
Patient
A 33-year-old Chinese woman visited the West China Second University Hospital of Sichuan University (Chengdu, China) for genetic diagnosis. The woman’s relatives informed the doctor that the proband had bilateral prelingual deafness and now was trying to conceive. At present, the proband has no other abnormal clinical manifestations except hearing impairment. The proband’s parents are healthy and have no close relative marriage. The mother of the proband denied exposure to teratogenic environmental factors during pregnancy. The pedigree of this family is shown in Figure 1. The present study was approved by the ethics committee of the West China Second University Hospital of Sichuan University, and written informed consent was obtained from the proband and family members.
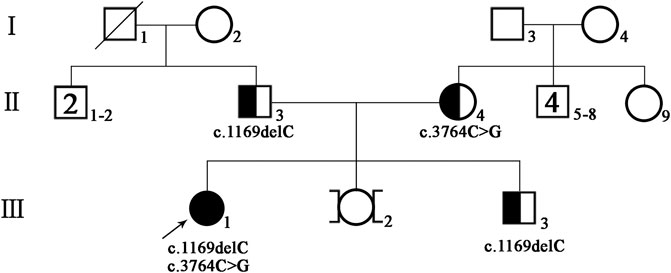
FIGURE 1. Pedigree of the patient’s family; black-filled shapes represent individuals with hearing loss, and the unfilled shapes represent unaffected ones. Males are represented by squares, females by circles.
DNA Extraction
Peripheral blood samples were drawn from four subjects (Ⅱ3, Ⅱ4, Ⅲ1, and Ⅲ3) after obtaining informed consent. Genomic DNA was extracted from leukocytes of peripheral blood samples using the QIAamp DNA Blood Mini Kit (Qiagen bioinformatics, Hilden, Germany) according to the manufacturer’s instructions. After the DNA was extracted from the samples, the concentration and purity were examined using a NanoDrop 1,000 (Thermo Fisher Scientific, Inc., Wilimington, United States).
Targeted NGS
According to the manufacturer’s protocol, we used the CM1132 and M113 Kit (MyGenostics, Inc. Beijing, China) to capture and enrich the targeted gDNA of the proband. The CM1132 kit targeted 162 genes (Supplementary Table S1) known to cause deafness, while the M113 kit contained mitochondrial whole genes and hot-spot variants that cause deafness. The double-end sequencing program (PE150) was performed on the NextSeq500 platform (Illumina, Inc. California, United States), and sequence reads of 150bp were received. The reads were mapped to the human genome reference (UCSC GRCh37/hg19) using the Burrows-Wheeler Aligner. Variants were called using the Genome Analysis Tool Kit. Annovar was used to annotate the variants. Then, all the variants were filtered based on their frequency in the 1,000 Genomes Project, ExAC, gnomad, Esp6500. Variants with Minimum allele frequency (MAF) of <0.05 were retained. We then applied several variant prediction tools to predict the functional impact of candidate variants. Finally, the pathogenicities of the variations were analyzed according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Sue et al., 2015) and Expert Specification of the ACMG/AMP variant interpretation guidelines for genetic HL (Andrea et al., 2018). To verify the variations found in the TRIOBP gene, Sanger sequencing was performed on samples obtained from the proband and her parents. The variation sites and amplification primers were as follows: c.1170 (forward 5′-CTCCTCTCCCCATCGAATCA-3′, reverse 5′-GGTTCTGGAGGCTTTGGGAT-3′) and c.3764 (forward 5′-CTCCTTCTCATCCCCACCAC-3′, reverse 5′- TGTACTCCTCCCGCTCCAA -3′). Sanger sequencing data were analyzed using the Chromas software.
Variants Detection
There were 1,529 variants identified in the targeted regions of the Deafness panel, while no variant related to HL was found in the mitochondrial whole gene panel. The summary of the next generation sequencing (NGS) was listed in Supplementary Table S2). All the 1,529 variants were filtered based on their frequency in the 1,000 Genomes Project, ExAC, gnomad, Esp6500. Variants with MAF of <0.05 were retained. We then applied several variant prediction tools including SIFT, PolyPhen, MutationTaster, GERP and SPIDEX to predict the functional impact of candidate variants. Finally, we founded two variants of the TRIOBP (c.1170delC and c.3764C > G, both in exon 7, NM_001039141.2) in the proband. Sanger sequencing of the proband and family members showed that the proband was compound heterozygous, and the parents and the healthy brother were heterozygous carriers (Figures 2A,B). The c.1170delC (p.S391Pfs*488) resulted in a frameshift, while the c.3764C > G (p.S1255*) variant directly introduces a stop codon, both of the variants may lead to TRIOBP truncation. These variants have not been reported in the general population databases, disease databases, or published literature. We analysed the location of the two novel variants in TRIOBP and identified that the exon variants were both located in the functional domains (Figure 3A). The positions of the novel variants are highly conserved across several species according to the University of California Santa Cruz Genome Browser database (UCSC) (Figure 3B).
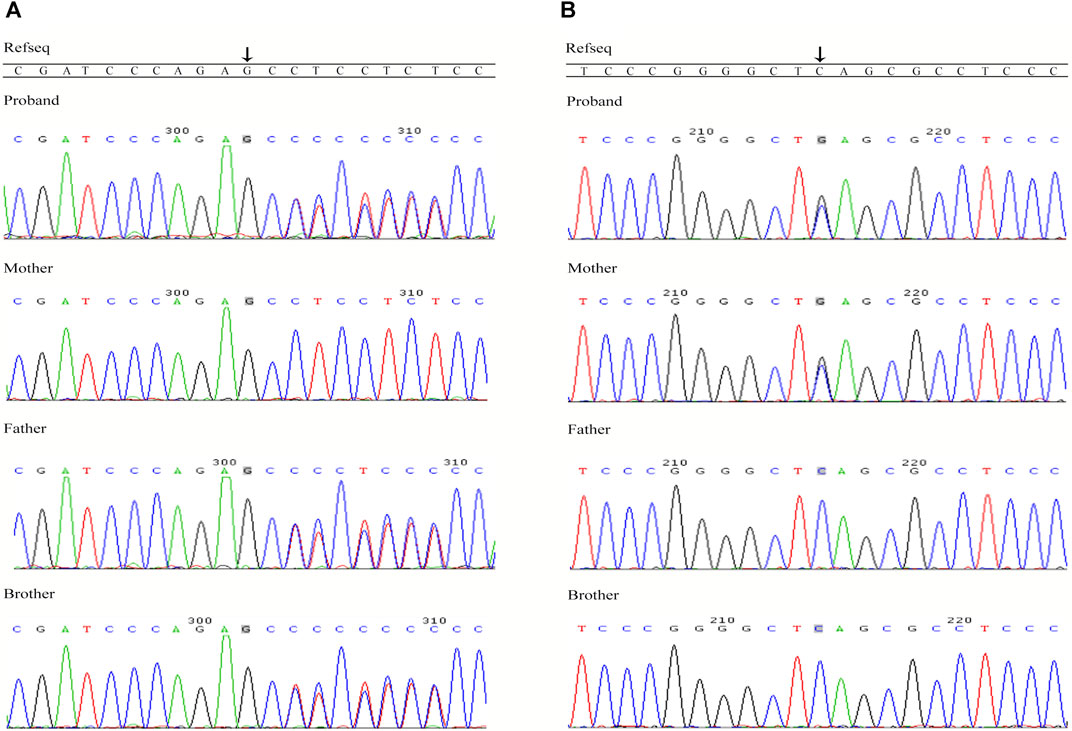
FIGURE 2. Sanger sequencing confirmation of the variants in TRIOBP identified in this study. (A) Sequences of the heterozygous frameshift variant c.1170delC (p.S391Pfs*488) and the corresponding wild-type sequence; (B) Sequences of the heterozygous Nonsense mutation c.3764C > G (p.S1255*) and the corresponding wild-type sequence.
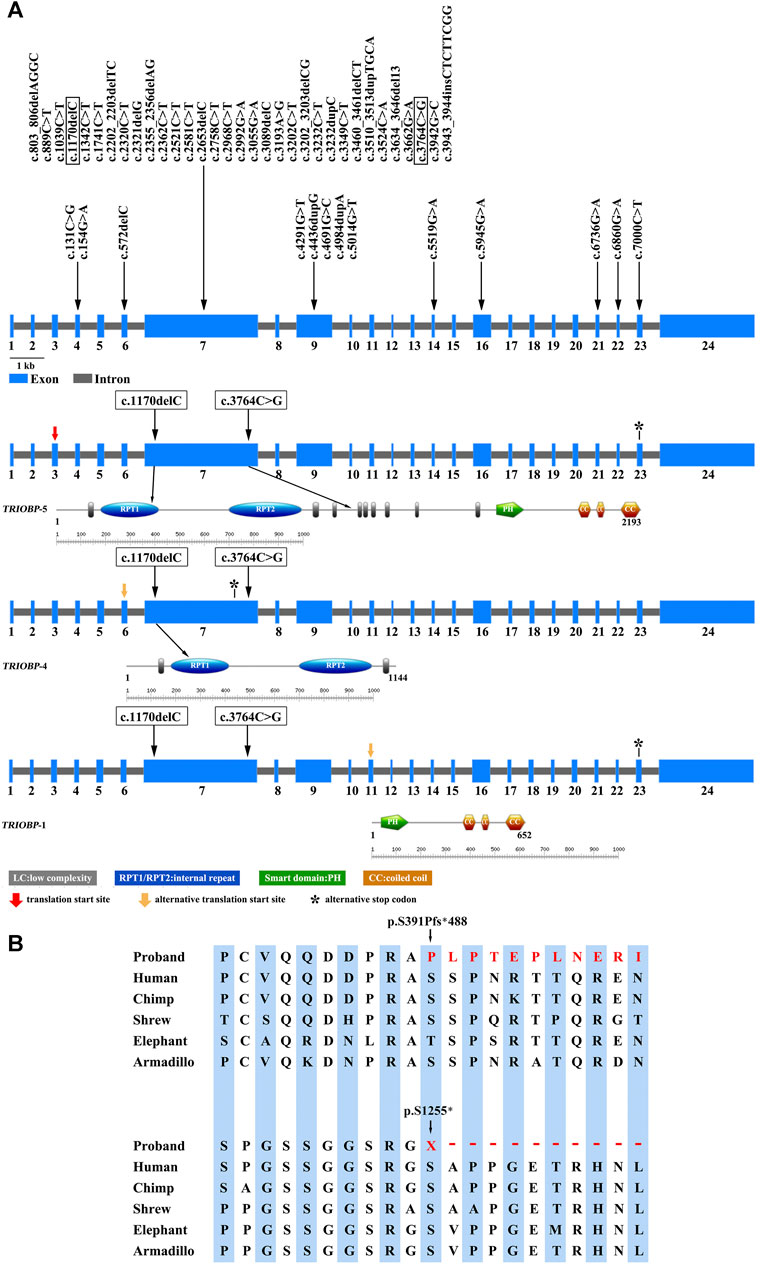
FIGURE 3. (A) Variation spectrum and domain structure of the TRIOBP gene (NM_001039141.2). The variations identified in the proband in this study are represented in box. (B) Conserved amino acid sequences of TRIOBP (amino acid 391 and 1,255) and the predicted truncated TRIOBP caused by the variants identified in this proband.
Discussion and Conclusion
Typically, the pathogenic variants of TRIOBP lead to prelingual, severe-to-profound hearing loss. According to the records and descriptions of the relatives, the patient in our study had bilateral prelingual deafness. Unfortunately, she had not received auditory tests at that time. However, not all patients with the TRIOBP pathogenic variants showed prelingual deafness, and there were some differences in severity or auditory test results among patients with different variants. According to Agnieszka et al., based on whole exome analysis, they identified two TRIOBP pathogenic variants causative of nonsyndromic, peri-to postlingual, moderate-to-severe hearing loss in three siblings from a Polish family (Agnieszka et al., 2017). Differences in age of onset and severity of hearing loss may be related to the location of TRIOBP variants and their impact on different isoforms.
DFNB28 is characterized by lingual sensorineural HL and is related to variants in the TRIOBP. The TRIOBP gene is subject to complicated alternative splicing. There were six splice variants exist, of which three transcripts are most studied, referred to as TRIOBP-1, TRIOBP-4 and TRIOBP-5. Among of them, TRIOBP-4 and/or TRIOBP-5 are required for hearing (Saima et al., 2006; Nicholas et al., 2014; Tatsuya et al., 2019; Beti et al., 2020). The sequences of TRIOBP-5/-4 amino acid contains two repeat motifs, named as the R1 repeat domain (amino acid residues 357–500) and R2 repeat domain (amino acid residues 684–896). The R1 motif is the main actin binding region of TRIOBP-4, while the binding of the R2 motif is nonspecific (Jian et al., 2013). TRIOBP-5 or TRIOBP-1 has a PH and several coiled-coil domains (CCs). There is no overlapping amino acid sequence between TRIOBP-1 and TRIOBP-4 isoform. The pathogenic variant c.1170delC, p. S391Pfs*488 may disrupts the TRIOBP-4 and TRIOBP-5 isoforms, whereas the pathogenic variant c.3764C > G, p. S1255* affects only TRIOBP-5. Both of the variants without impairing the TRIOBP-1 (Figure 3A). The c.1170delC was located in the R1 motifs, and c.3764C > G variants was located in the low complexity The two variants detected in this study were predicted to result in the premature termination of translation (after amino acids 879 and 1,255, respectively). The shortened TRIOBP protein are devoid of PH and CC, which is crucial for the actin-binding process (Figure 3A).
TRIOBP various is rare in HL patients. There are 45 disease-related variants have been reported in the TRIOBP at present (Hashem et al., 2006; Saima et al., 2006; Maiko et al., 2013; Shearer et al., 2014; Christina et al., 2016; Denise et al., 2016; Manou et al., 2016; Agnieszka et al., 2017; Celia et al., 2017; Daniel et al., 2017; Haiqiong et al., 2018; Elodie et al., 2019; Songfeng et al., 2019; Yan et al., 2019; Amal et al., 2020; Birgit et al., 2020; Yongyi et al., 2020), with HL being the only phenotypic manifestation. As presented in Table 1, although the variants cover the region from exon 4 to exon 23, most of the previously reported variations in TRIOBP are located in exon 7. The region of exon 7 is defined as a hot point and is more susceptible to variations owing to the accumulation of repeated sequences (Agnieszka et al., 2017). The TRIOBP c.1170delC (p.S391Pfs*488) and c.3764C > G (p.S1255*) variants detected in our patient were both located in exon 7. Both variants were classified as pathogenic according to the criteria of ACMG and the Expert Specification of the ACMG/AMP Variant Interpretation Guidelines for Genetic HL (Sue et al., 2015; Andrea et al., 2018).
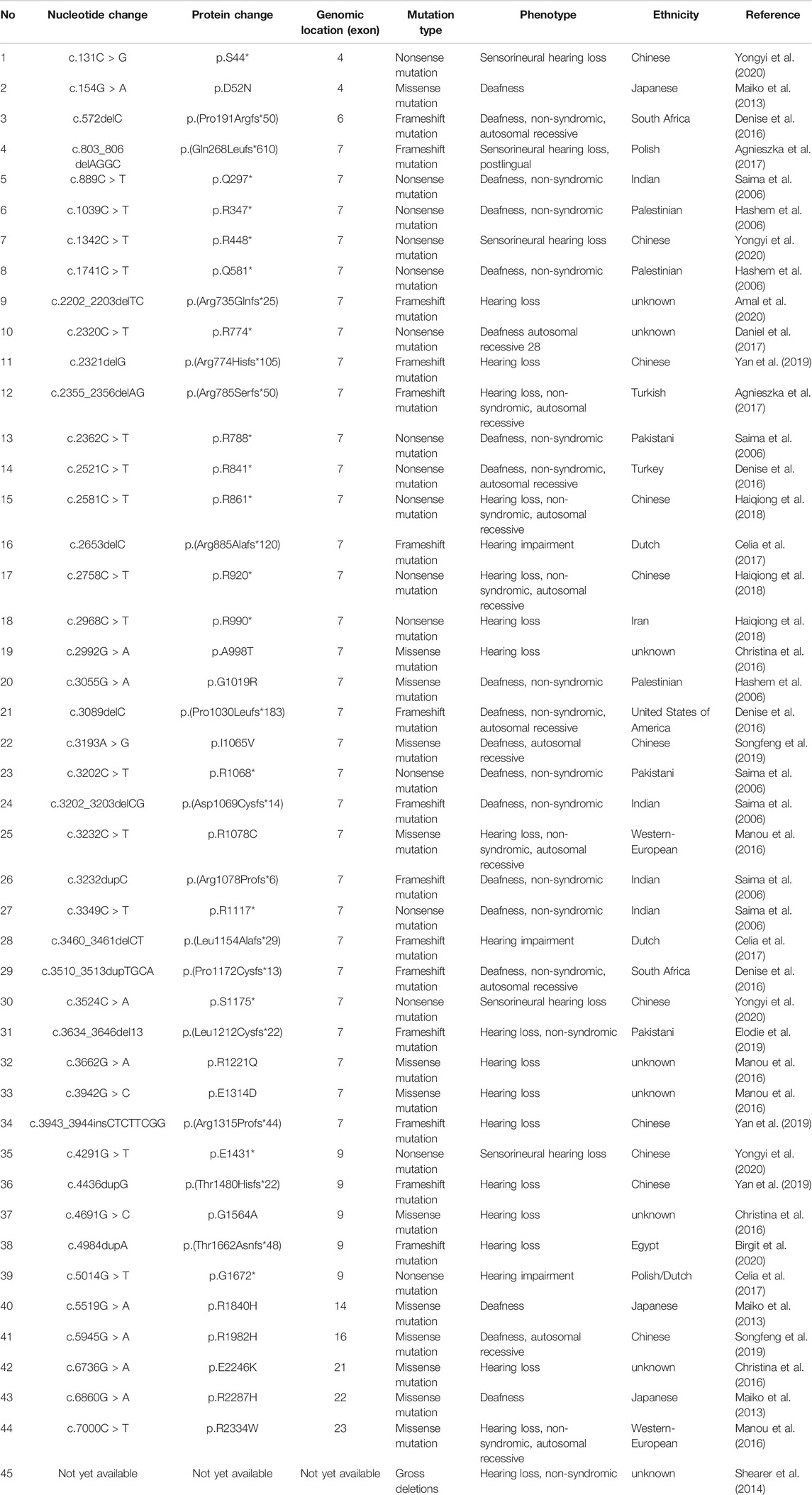
TABLE 1. Disease causing mutations in the TRIOBP gene (NM_001039141.2) according to The Human Gene Mutation database (HGMD).
This report describes a Chinese patient with a hearing impairment. Using disease-targeted gene panels, we identified two novel compound heterozygous variants in the TRIOBP gene. Both variants were predicted to lead to premature termination codons, resulting in a truncated TRIOBP protein formation. The two novel TRIOBP variants expand the spectrum of TRIOBP variants in HL. Although TRIOBP variants are not a frequent cause of HL, this gene should be thoroughly analyzed in patients with prelingua HL.
Data Availability Statement
The raw datasets analysed during the current study are not deposited in publicly available repositories because of considerations about the security of human genetic resources and patient anonymity, but are available from the corresponding author on reasonable request.
Ethics Statement
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of West China Second University Hospital of Sichuan University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
CZ, SL, and JW designed the study. CZ, YX, and HX performed the experiments. CZ and JW conducted data analysis. All authors read and approved the final manuscript.
Funding
This work was supported by The National Key R&D Program of China (2021YFC1005300, 2021YFC1005302 and 2021YFC1005303) and The Science and Technology Department of Sichuan Province (2021YFS0078).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.766973/full#supplementary-material
Abbreviations
DFNB28, The autosomal recessive non-syndromic deafness-28; HL, Hearing Loss; SNHL, sensorineural hearing loss; Genomic(g) DNA; ACMG, American college of medical genetics and genomics; NGS, next generation sequencing; UCSC, university of California santa cruz genome browser database; PH, pleckstrin homology domain; CC, coiled-coil domains.
References
Abu Rayyan, A., Kamal, L., Casadei, S., Brownstein, Z., Zahdeh, F., Shahin, H., et al. (2020). Genomic Analysis of Inherited Hearing Loss in the Palestinian Population. Proc. Natl. Acad. Sci. USA 117 (33), 20070–20076. doi:10.1073/pnas.2009628117
Bao, J., Bielski, E., Bachhawat, A., Taha, D., Gunther, L. K., Thirumurugan, K., et al. (2013). R1 Motif Is the Major Actin-Binding Domain of TRIOBP-4. Biochemistry 52 (31), 5256–5264. doi:10.1021/bi400585h
Bao, J., Wang, S., Gunther, L. K., Kitajiri, S.-i., Li, C., and Sakamoto, T. (2015). The Actin-Bundling Protein TRIOBP-4 and -5 Promotes the Motility of Pancreatic Cancer Cells. Cancer Lett. 28356 (2 Pt B), 367–373. doi:10.1016/j.canlet.2014.08.005
Birgit, S. B., Maha, A. A., Mostafa, R. M., Andreas, B., Janine, A., Susanne, M., et al. (2020). Comprehensive Molecular Analysis of 61 Egyptian Families with Hereditary Nonsyndromic Hearing Loss. Clin. Genet. 98 (1), 32–42. doi:10.1111/cge.13754
Bradshaw, N. J., Bader, V., Prikulis, I., Lueking, A., Müllner, S., and Korth, C. (2014). Aggregation of the Protein TRIOBP-1 and its Potential Relevance to Schizophrenia. PLoS One 9 (10), e111196. doi:10.1371/journal.pone.0111196
Friedman, T. B., Schultz, J. M., Ben-Yosef, T., Pryor, S. P., Lagziel, A., Fisher, R. A., et al. (2003). Recent Advances in the Understanding of Syndromic Forms of Hearing Loss. Ear Hear 24 (4), 289–302. doi:10.1097/01.AUD.0000079804.00047.CE
Katsuno, T., Belyantseva, I. A., Cartagena-Rivera, A. X., Ohta, K., Crump, S. M., Petralia, R. S., et al. (2019). TRIOBP-5 Sculpts Stereocilia Rootlets and Stiffens Supporting Cells Enabling Hearing. JCI Insight 204 (12), e128561. doi:10.1172/jci.insight.128561
Kitajiri, S.-i., Sakamoto, T., Belyantseva, I. A., Goodyear, R. J., Stepanyan, R., Fujiwara, I., et al. (2010). Actin-bundling Protein TRIOBP Forms Resilient Rootlets of Hair Cell Stereocilia Essential for Hearing. Cell 141 (5), 786–798. doi:10.1016/j.cell.2010.03.049
Lewis, M. A., Nolan, L. S., Cadge, B. A., Matthews, L. J., Schulte, B. A., Dubno, J. R., et al. (2018). Whole Exome Sequencing in Adult-Onset Hearing Loss Reveals a High Load of Predicted Pathogenic Variants in Known Deafness-Associated Genes and Identifies New Candidate Genes. BMC Med. Genomics 11 (1), 1177. doi:10.1186/s12920-018-0395-1
Miyagawa, M., Naito, T., Nishio, S.-y., Kamatani, N., and Usami, S.-i. (2013). Targeted Exon Sequencing Successfully Discovers Rare Causative Genes and Clarifies the Molecular Epidemiology of Japanese Deafness Patients. PLoS One 8 (8), e71381. doi:10.1371/journal.pone.0071381
Morton, C. C., and Nance, W. E. (2006). Newborn Hearing Screening - A Silent Revolution. N. Engl. J. Med. 354 (20), 2151–2164. doi:10.1056/NEJMra050700
Nicolas, G., and Anil, K. L. (2002). Etiology of Syndromic and Nonsyndromic Sensorineural Hearing Loss. Otolaryngol. Clin. North. Am. 35 (4), 891–908. doi:10.1016/s0030-6665(02)00053-1
Oza, A. M., DiStefano, M. T., Hemphill, S. E., Cushman, B. J., Grant, A. R., Siegert, R. K., et al. (2018). Expert Specification of the ACMG/AMP Variant Interpretation Guidelines for Genetic Hearing Loss. Hum. Mutat. 39 (11), 1593–1613. doi:10.1002/humu.23630
Pollak, A., Lechowicz, U., Murcia Pieńkowski, V. A., Stawiński, P., Kosińska, J., Skarżyński, H., et al. (2017). Whole Exome Sequencing Identifies TRIOBP Pathogenic Variants as a Cause of post-lingual Bilateral Moderate-To-Severe Sensorineural Hearing Loss. BMC Med. Genet. 18 (1), 142. doi:10.1186/s12881-017-0499-z
Riazuddin, S., Khan, S. N., Ahmed, Z. M., Ghosh, M., Caution, K., Nazli, S., et al. (2006). Mutations in TRIOBP, Which Encodes a Putative Cytoskeletal-Organizing Protein, Are Associated with Nonsyndromic Recessive Deafness. Am. J. Hum. Genet. 78 (1), 137–143. doi:10.1086/499164
Richard, E. M., Santos-Cortez, R. L. P., Faridi, R., Rehman, A. U., Lee, K., Shahzad, M., et al. (2019). Global Genetic Insight Contributed by Consanguineous Pakistani Families Segregating Hearing Loss. Hum. Mutat. 40 (1), 53–72. doi:10.1002/humu.23666
Richards, S., Nazneen, A., Aziz, N., Bale, S., Bick, D., Das, S., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–423. doi:10.1038/gim.2015.30
Shahin, H., Walsh, T., Sobe, T., Abu Sa’ed, J., Abu Rayan, A., Lynch, E. D., et al. (2006). Mutations in a Novel Isoform of TRIOBP that Encodes a Filamentous-Actin Binding Protein Are Responsible for DFNB28 Recessive Nonsyndromic Hearing Loss. Am. J. Hum. Genet. 78 (1), 144–152. doi:10.1086/499495
Shang, H., Yan, D., Tayebi, N., Saeidi, K., Sahebalzamani, A., Feng, Y., et al. (2018). Targeted Next-Generation Sequencing of a Deafness Gene Panel (MiamiOtoGenes) Analysis in Families Unsuitable for Linkage Analysis. Biomed. Res. Int. 2018, 1–7. doi:10.1155/2018/3103986
Shearer, A., Kolbe, D. L., Azaiez, H., Sloan, C. M., Frees, K. L., Weaver, A. E., et al. (2014). Copy Number Variants Are a Common Cause of Non-syndromic Hearing Loss. Genome Med. 6 (65), 37. doi:10.1186/gm554
Sloan-Heggen, C. M., Bierer, A. O., Shearer, A. E., Kolbe, D. L., Nishimura, C. J., Frees, K. L., et al. (2016). Comprehensive Genetic Testing in the Clinical Evaluation of 1119 Patients with Hearing Loss. Hum. Genet. 135, 441–450. doi:10.1007/s00439-016-1648-8
Sommen, M., Schrauwen, I., Vandeweyer, G., Boeckx, N., Corneveaux, J. J., van den Ende, J., et al. (2016). DNA Diagnostics of Hereditary Hearing Loss: A Targeted Resequencing Approach Combined with a Mutation Classification System. Hum. Mutat. 37 (8), 812–819. doi:10.1002/humu.22999
Songfeng, Z., Xueshuang, M., Weiqiang, Y., Rvfei, Z., Tao, Y., and Hongyi, H. (2019). Whole-exome Sequencing Identifies Rare Pathogenic and Candidate Variants in Sporadic Chinese Han Deaf Patients. Clin. Genet. 97 (2), 352–356. doi:10.1111/cge.13638
Sun, Y., Yuan, J., Wu, L., Li, M., Cui, X., Yan, C., et al. (2019). Panel-based NGS Reveals Disease-Causing Mutations in Hearing Loss Patients Using BGISEQ-500 Platform. Medicine (Baltimore) 98 (12), e14860. doi:10.1097/MD.0000000000014860
Trujillano, D., Bertoli-Avella, A. M., Kumar Kandaswamy, K., Weiss, M. E., Köster, J., Marais, A., et al. (2017). Clinical Exome Sequencing: Results from 2819 Samples Reflecting 1000 Families. Eur. J. Hum. Genet. 25 (2), 176–182. doi:10.1038/ejhg.2016.146
Yan, D., Tekin, D., Bademci, G., Foster, J., Cengiz, F. B., Kannan-Sundhari, A., et al. (2016). Spectrum of DNA Variants for Non-syndromic Deafness in a Large Cohort from Multiple Continents. Hum. Genet. 135 (8), 953–961. doi:10.1007/s00439-016-1697-z
Yuan, Y., Li, Q., Su, Y., Lin, Q., Gao, X., Liu, H., et al. (2020). Comprehensive Genetic Testing of Chinese SNHL Patients and Variants Interpretation Using ACMG Guidelines and Ethnically Matched normal Controls. Eur. J. Hum. Genet. 28 (2), 231–243. doi:10.1038/s41431-019-0510-6
Zaharija, B., Samardžija, B., and Bradshaw, N. J. (2020). The TRIOBP Isoforms and Their Distinct Roles in Actin Stabilization, Deafness, Mental Illness, and Cancer. Molecules 25 (21), 4967. doi:10.3390/molecules25214967
Keywords: TRIOBP, DFNB28, variation, hearing loss, next generation sequencing
Citation: Zhou C, Xiao Y, Xie H, Wang J and Liu S (2021) Case Report: Novel Compound Heterozygous Variants in TRIOBP Associated With Congenital Deafness in a Chinese Family. Front. Genet. 12:766973. doi: 10.3389/fgene.2021.766973
Received: 06 September 2021; Accepted: 25 October 2021;
Published: 17 November 2021.
Edited by:
Lingqian Wu, Central South University, ChinaReviewed by:
Li Zhuo, Central South University, ChinaZhengfeng Xu, Nanjing Medical University, China
Copyright © 2021 Zhou, Xiao, Xie, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Wang, aGh3al8xMjNAMTYzLmNvbQ==; Shanling Liu, c3Vubnk2MzBAMTI2LmNvbQ==
†These authors have contributed equally to this work