 Haochang Hu
Haochang Hu Tian Shu1†
Tian Shu1† Jian Wang
Jian Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 14 December 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.762587
This article is part of the Research TopicGenetics of Familial Hypercholesterolemia: New Insight - Volume IIView all 17 articles
As an autosomal dominant disorder, familial hypercholesterolemia (FH) is mainly caused by pathogenic mutations in lipid metabolism-related genes. The aim of this study is to investigate the genetic mutations in FH patients and verify their pathogenicity. First of all, a pedigree investigation was conducted in one family diagnosed with FH using the Dutch Lipid Clinic Network criteria. The high-throughput sequencing was performed on three family members to explore genetic mutations. The effects of low-density lipoprotein receptor (LDLR) variants on their expression levels and activity were further validated by silico analysis and functional studies. The results revealed that LDLC levels of the proband and his daughter were abnormally elevated. The whole-exome sequencing and Sanger sequencing were used to confirm that there were two LDLR missense mutations (LDLR c.226 G > C, c.1003 G > T) in this family. Bioinformatic analysis (Mutationtaster) indicated that these two mutations might be disease-causing variants. In vitro experiments suggested that LDLR c.226 G > C and c.1003 G > T could attenuate the uptake of Dil-LDL by LDLR. In conclusion, the LDLR c.226 G > C and c.1003 G > T variants might be pathogenic for FH by causing uptake dysfunction of the LDLR.
Familial hypercholesterolemia (FH) is a common autosomal genetic disorder mainly caused by pathogenic mutations in genes encoding low-density lipoprotein receptor (LDLR), apolipoprotein B (ApoB) and proprotein convertase subtilisin kexin 9 (PCSK9) (Benn et al., 2016). A meta-analysis of 11 million subjects illustrated that the prevalence of FH in the general population is 0.32%, while the prevalence of FH in ischemic heart disease and premature ischemic heart disease patients is 3.2 and 6.7%, respectively (Beheshti et al., 2020). Due to lifelong exposure to extremely high levels of low-density lipoprotein cholesterol (LDLC), FH is characterized by xanthomas, corneal arcus, and early-onset cardiovascular disease (Peterson et al., 2021). European and American guidelines suggest that FH patients should be identified as early as possible so that LDLC lowering treatment can be started early in life in order to improve the patient’s prognosis (Piepoli et al., 2016; Grundy et al., 2019).
As a membrane protein on the surface of liver cells, LDLR is the key point of LDLC metabolism. LDLR could combine with LDLC and transport it to the lysosome for metabolism. The LDLR subsequently returns to the surface of liver cells for recycling (van de Sluis et al., 2017; Chemello et al., 2021). Therefore, the pathogenic variants in LDLR could directly lead to protein dysfunction and LDLC metabolic disorders. Previous studies suggested that the disease-causing variants in LDLR might account for about 90% of FH (Gidding et al., 2015; Iacocca and Hegele, 2017). Reeskamp et al. performed targeted next-generation sequencing on 1,528 patients with LDLC greater than 5 mmol/L. The results illustrated that 227 cases (14.9%) were heterozygous carriers of pathogenic variants in LDLR (80.2%), APOB (14.5%), and PCSK9 (5.3%) (Reeskamp et al., 2021b). Furthermore, a retrospective study pointed out that LDLC gradually increased from patients with no pathogenic variants to patients with a defective variant, patients with a null variant, and patients with two variants (Di Taranto et al., 2021). Therefore, it is imperative to perform genetic diagnoses of FH patients to distinguish between different genetic states or variant types.
According to the UCL LDLR gene variant database, 3779 LDLR mutations have been reported so far; of which 77% are substitutions, 16% are deletions, and 5% are duplicates (Leigh et al., 2017). Regrettably, the pathogenicity of several LDLR variants has not been authenticated. It is crucial to research the functional verification of mutations to better clarify the molecular mechanism of FH.
In this study, a FH family was included, and high-throughput sequencing was used to explore pathogenic mutations. The effects of LDLR variants on the expression level and function of LDLR were verified in cellular experiments.
The study enrolled one FH family consisting of three members in Ningbo First Hospital. The Dutch Lipid Clinic Network criteria (DLCN) was used to diagnose FH patients (Wang et al., 2019). A detailed information collection form was designed based on the characteristics of FH patients. The content mainly included the patient’s general information, family history, personal history, past history, treatments, the results of the auxiliary examination, etc.
Whole blood samples of the three participants were collected in EDTA tubes (Gongdong Company, China) and stored in a refrigerator at -80 °C (ThermoFisher Scientific, United States). Omega Blood DNA Kit (D3392-02, Omega bio-tek, United States) was used to extract genomic DNA. High-throughput whole-exome sequencing for DNA samples was completed on the BGISEQ-500 platform (Huada Gene Technology Co. Ltd., China). First, the genomic DNA was randomly broken into fragments of about 200–300 bp, and a complete fragment library was established after PCR amplification. The quality of DNA was then inspected before sequencing, and the number of original bases (raw data) obtained by sequencing each sample should meet the standard. Clean data was subsequently obtained by removing low-quality reads from raw data. Finally, we compared the sequencing data with the human reference genome hg19 to obtain high-confidence mutations.
As one of the standard bioinformatic tools, Mutationtaster (http://www.mutationtaster.org) was applied to evaluate the disease-causing potential of DNA variants (Schwarz et al., 2014). The transcript ID of the LDLR was ENST00000558518 (NCBI Reference Sequence: NM_000,527.5) in the manuscript.
The PCR amplification method was utilized to amplify the participants’ DNA. PCR primers were designed based on the exon fragment where the mutation was located. The sequences of primers were shown as follows: LDLR (exon 3): 5′-TGACAGTTCAATCCTGTCTCTTCTG (upstream), 5′-ATAGCAAAGGCAGGGCCACACTTAC (downstream); LDLR (exon 7): 5′- AGTCTGCATCCCTGGCCCTGCGCAG (upstream), AGGGCTCAGTCCACCGGGGAATCAC (downstream) (Du and Huang, 2007). Sanger sequencing was conducted to verify the variants in each participant by the Biosystems® 3730 DNA analyzer. The sequencing results were analyzed by the Chromas software.
HEK293 cells were derived from the cell bank of the Shanghai Chinese Academy of Sciences and were cultured in a DMEM medium (Hyclone, United States) with 10% fetal bovine serum (ThermoFisher Scientific, United States). The DMEM used in the current study included 4.00 mmol/L l-glutamine, 4500 mg/L glucose, and sodium pyruvate. After the cells were grown to about 80–90% in a 37 °C incubator containing 5% CO2, they were subcultured at a ratio of 1:3.
For plasmid transfection, HEK293 cells in good growth condition were plated in a six-well plate. In this study, wild-type (WT) and mutant plasmids were constructed by chemical synthesis. Lipofectamine 2000 reagent (ThermoFisher Scientific, United States), Opti-MEM medium (Gibco, United States), and plasmid DNA were mixed and incubated at room temperature and then added to the cells. The cells were cultured in 5% CO2 at 37 °C for 6–8 h.
We divided the cells into the mutant group (HEK293 cells transfected with LDLR mutant plasmids), the WT group (HEK293 cells transfected with LDLR wild-type plasmids), and the NC group (HEK293 cells transfected with empty plasmids).
The expression level of LDLR variants was detected by Western Blot 48 h after plasmid transfection as previously described (Hu et al., 2021). The samples were lysed in RIPA buffer (Solarbio, China) containing protease and phosphatase inhibitors. Protein levels were quantified by the BCA protein assay kit (Cwbio, China). The samples containing equal amounts of protein were separated by 5x SDS-PAGE gel and transferred onto the PVDF membrane. After blocking with 5% milk, the samples were incubated with the primary antibodies: LDLR [Mouse monoclonal to LDL Receptor (Abcam, United States)] and β-actin [β-actin Rabbit mAb (Abclonal, China)] overnight at 4 °C, and secondary antibodies [peroxidase-conjugated anti-mouse IgG (Jackson Immuno Research, United States) and peroxidase-conjugated anti-rabbit IgG (Jackson Immuno Research, United States)] for 1 h at room temperature. Lastly, the signals were analyzed using the ImageJ Software.
After treatment with a serum-free medium of 0.3% bovine albumin (Solarbio, China) for 12 h, the transfected cells were incubated with 5 μg/ml Dil-LDL (Thermo Scientific, United States) for 4 h. The labeled LDL was used to study LDL uptake through endocytosis and its trafficking throughout the cell, which can be detected via fluorescence microscopy. After washing the cells with PBS, they were fixed with 4% paraformaldehyde for 10–20 min and then stained with DAPI. The fluorescence intensity of the cells was observed using a confocal laser scanning microscope (LEICA TCS SP8, Germany). The ImageJ software was used to quantitatively analyze the fluorescence intensity.
All statistical analyses were performed by the SPSS 24.0 software (SPSS Inc., Chicago, IL, United States). GraphPad Prism 8 (GraphPad Software, La Jolla, CA) was used for plots. p < 0.05 was considered statistically significant.
A 52 year-old man was diagnosed with coronary heart disease at Ningbo First Hospital. His LDLC level was abnormally elevated at 5.62 mmol/L under the treatment of atorvastatin 20 mg. Corneal arcus and xanthomas on the skin of the elbow were observed since adolescence. The results of coronary angiography illustrated 95% localized stenosis of the proximal segment of the left anterior descending branch, 90% localized stenosis of the proximal segment of the diagonal branch, and subtotal occlusion of the proximal segment of the right coronary artery. Echocardiography displayed hypertrophy of the ventricular septum, aortic valve calcification, and mild mitral regurgitation. Carotid ultrasound showed multiple plaque formation in bilateral carotid arteries.
We tracked three members in two generations of this family, including the proband’s wife and the proband’s daughter. As shown in Table 1, the blood lipid level of the proband’s wife (I-2) was normal. However, the proband’s daughter (II-1) had an increased level of LDLC (5.50 mmol/L) without corneal arcus or xanthomas. Before performing DNA analysis, the proband and his daughter were diagnosed as FH, according to the DLCN criteria.
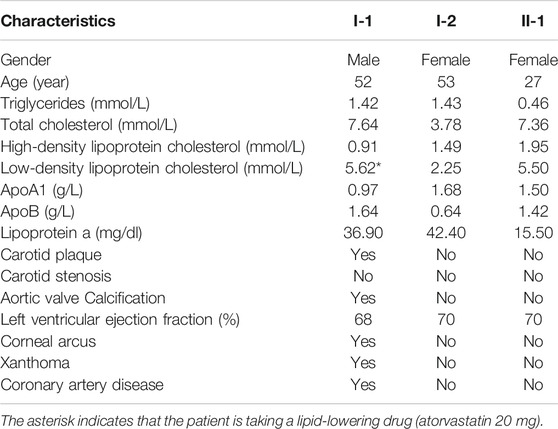
TABLE 1. Clinical data of FH patients and family members.
High-throughput sequencing was performed on all members of this FH family, and the mutations of four FH-related genes (LDLR, APOB, PCSK9, LDLRAP1) were analyzed (Tada et al., 2019). There were five LDLR variants (LDLR c.226 G > C, c.1003 G > T, c.1413A > G, c.1617 C > T, and c.2232A > G) in the proband, five LDLR variants (LDLR c.1413A > G, c.1617 C > T, c.1773 C > T, c.1959T > C, and c.2232A > G) in the proband’s wife and five LDLR variants (LDLR c.1003 G > T, c.1413A > G, c.1773 C > T, c.1959T > C, and c.2232A > G) in the proband’s daughter (Figure 1). However, no suspicious disease-causing variant was identified in the other three genes.
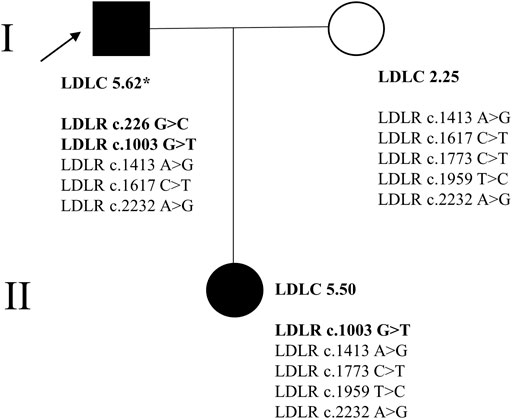
FIGURE 1. The family tree of the proband. * signified that the patient was taking the lipid-lowering drug (atorvastatin 20 mg); → represented the proband. In the current study, the background of the FH patients’ (I-1, II-1) picture was blackened. The disease-causing variants in LDLR were bolded.
The impact of LDLR variants on the receptor function was investigated by the bioinformatic tool Mutationtaster. The results revealed that LDLR c.1413A > G, c.1617 C > T, c.1773 C > T, c.1959T > C, and c.2232A > G were synonymous mutations, while LDLR c.226 G > C and c.1003 G > T were missense mutations. The amino acid sequences of these two sites were highly conserved in various species (Supplementary Figure S1).
In addition, both variants could cause changes in amino acid sequence. LDLR c.226 G > C (exon 3) caused the 76th amino acid to change from glycine to arginine, namely p. Gly76Arg. Meanwhile, LDLR c.1003 G > T (exon 7) caused the 335th amino acid to change from glycine to cysteine, namely p. Gly335Cys.
Subsequently, Sanger sequencing was performed to verify the existence of the two variants in the corresponding family members (Figure 2). The proband had two variants (LDLR c.226 G > C and c.1003 G > T), and his daughter had one variant (LDLR c.1003 G > T). Therefore, we speculate that the FH family in this study may be caused by pathogenic mutations in the LDLR.

FIGURE 2. Target sequences on LDLR by Sanger sequencing. Two disease-causing LDLR variants (LDLR c.226 G > C, c.1003 G > T) were found in the proband (I-1). The LDLR c.1003 G > T variant was found in the proband’s daughter (II-1).
The expression level of LDLR in the mutant group, WT group, and NC group was detected by Western Blot in HEK293 cells. The two bands detected in the gel map corresponded to the mature type and the precursor type LDLR, respectively. The results demonstrated that there was no significant difference between the mutant group (LDLR c.226 G > C) and the WT group (Figure 3). Compared with the WT group, less mature LDLR was detected in the LDLR c.1003 G > T group.
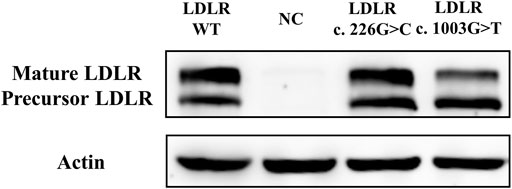
FIGURE 3. The effect of LDLR variants on LDLR expression.
The HEK293 cells carrying the LDLR c.226 G > C and c.1003 G > T variants and the WT LDLR were incubated in Dil-LDL medium for 4 h. The ability of mutant LDLR (LDLR c.226 G > C, c.1003 G > T) to take up LDL was significantly lower than that of WT LDLR (LDLR WT: 100%, LDLR c.226 G > C: 63.04%, LDLR c.1003 G > T: 42.54%, p < 0.01, Figure 4, Figure 5).
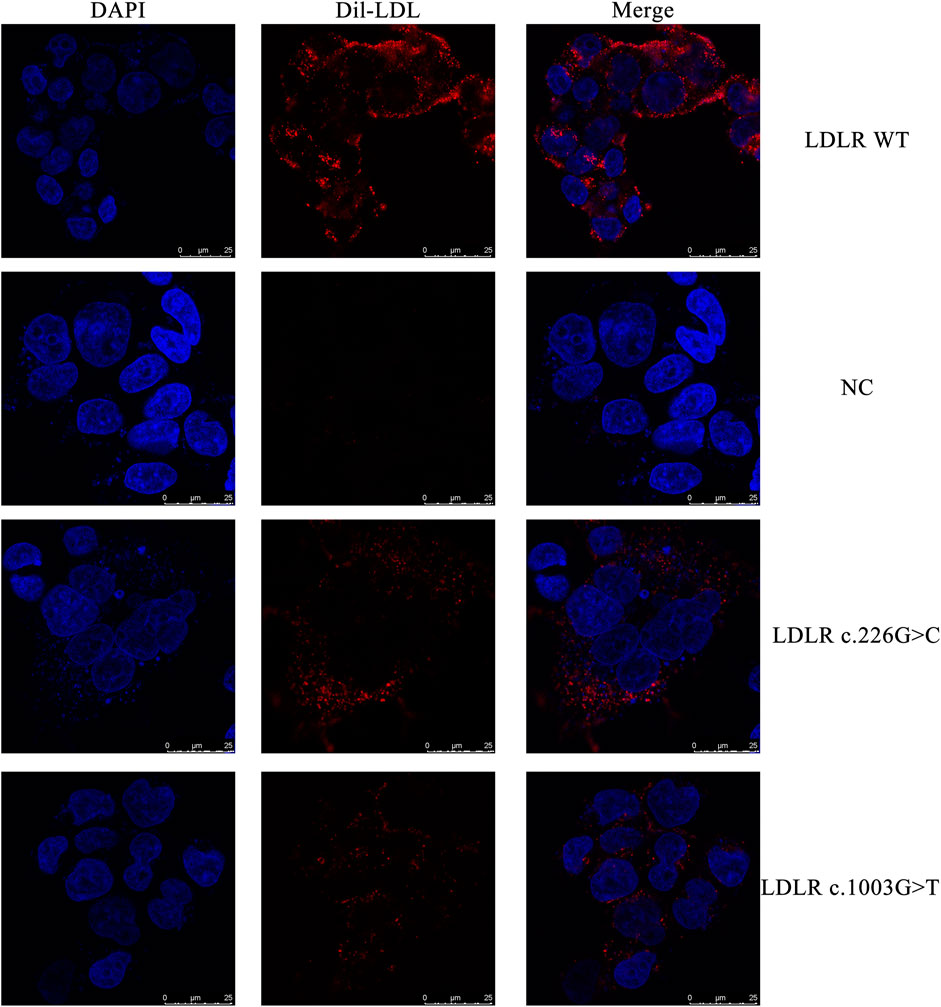
FIGURE 4. The representative confocal microscopy images. Blue fluorescence represented DAPI, while red fluorescence represented Dil-LDL.
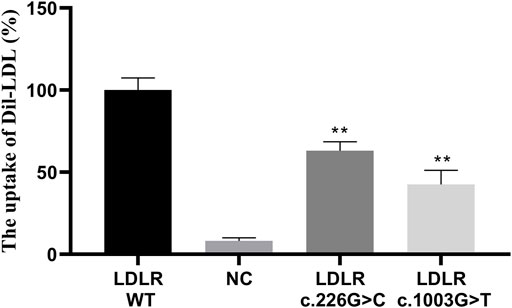
FIGURE 5. The LDL uptake in different LDLR groups. ** indicates statistical difference between mutant group and WT group (p < 0.01).
Judging from the FH patients’ clinical phenotype, the proband’s blood lipid level, corneal arcus, xanthoma, and atherosclerosis were severe. According to the sequencing results, the proband was a compound heterozygote (LDLR c.226 G > C and c.1003 G > T), and his daughter was a heterozygote (LDLR c.1003 G > T). This finding partly explained that the clinical phenotype of compound heterozygous patients was more severe than that of heterozygous.
As a typical disease of abnormal cholesterol metabolism, FH is a significant risk factor in the occurrence and development of cardiovascular disease (Hu et al., 2020). With the advancement of molecular technology, some FH patients have undergone genetic testing to elucidate the pathogenic mechanism. However, the current diagnostic rate of FH in most countries is extremely low, at < 1% even. Hence, it is crucial to carry out screening of high-risk populations in order to improve the diagnostic rate. Herein, a detailed pedigree investigation and the genogram of the proband and his family members were conducted. Two LDLR variants (LDLR c.226 G > C and c.1003 G > T) were discovered in this family by whole-exome sequencing. Functional prediction through the bioinformatic software showed that both variants might impact the expression or function of LDLR. The in vitro analysis confirmed that LDLR c.226 G > C and c.1003 G > T could diminish the ability of LDLR to uptake LDL.
By consulting the literature and the UCL database on LDLR mutations (http://www.lovd.nl/LDLR), we confirmed that both variants (LDLR c.226 G > C and c.1003 G > T) were not previously identified. The LDLR c.226 G > C variant (exon 3) is located in the coding region of the LDLR ligand-binding domain, which consists of 292 amino acids, including seven repeats (Strom et al., 2020). In the process of LDL metabolism, the variants including LDLR c.226 G > C in the LDLR ligand-binding domain might result in the LDLR being unable to reach the cell surface or in its inability to bind to LDL. This can eventually lead to hyperlipemia (Pamplona-Cunha et al., 2020). Our functional studies corroborated that LDLR c.226 G > C did not affect its expression. Instead, it caused the ability of LDLR to uptake LDL to decrease. In addition, another mutation, LDLR c.226 G > T (p. Gly76Trp), was also found at the exact location (Bourbon et al., 2008; Usifo et al., 2012). However, the expression and LDL internalization by LDLR c.226 G > T were similar to the WT, which was defined as likely benign (Benito-Vicente et al., 2015). It implies that different variants in the same position may function differently.
The LDLR c.1003 G > T variant (exon 7) is located in the homology domain of the EGF precursor, which consists of 406 amino acids, contains three EGF-like repeat units, and one β-propeller domain (Springer, 1998). It might result in the synthesized LDLR protein not being released from the endoplasmic reticulum to the cell surface, in that the receptors that reach the cell surface cannot bind to LDL, or in that the receptor cannot be recycled (Austin et al., 2004). Jeenduan et al. identified that LDLR p. D151Y and M391T located in the homology domain of EGF precursor significantly reduced the expression level of LDLR on the cell surface to 18 and 38%, respectively. Additionally, the amount of LDL uptake by LDLR was reduced to 15 and 71%, respectively (Jeenduang et al., 2010).
Our study showed that the other variant LDLR c.1003 G > T might affect the expression of the LDLR protein and impair its ability to uptake LDL (reduced to 42.54%). Previous studies have also reported the variant LDLR c.1003 G > A (p. Gly335Ser), and bioinformatics predicted that this variant was likely pathogenic (Wang et al., 2001; Laurie et al., 2004; Bertolini et al., 2013; Narang et al., 2020). Interestingly, two different variants in the translation initiation codon of LDLR (LDLR c.1A > T and c.1A > C) encoded the same amino acid (LDLR p.Met1Leu), but they cause different degrees of damage to its expression and activity (Graca et al., 2021). Therefore, functional experiments are paramount to clarify whether the mutation is pathogenic or not.
Ana Catarina Alves et al. found that a missense mutation (LDLR p. Asp601Val) might cause the loss of LDLR mature form and a complete impairment of its activity (Alves et al., 2021). The variant LDLR c. 2389 G > A might cause the erroneous cleavage of messenger RNA to retain the mutant LDLR in the Golgi apparatus (Shu et al., 2021). Existing evidence indicates that missense mutations in LDLR could affect its expression and cause dysfunction of LDLR protein through a variety of mechanisms. Regrettably, our research cannot identify the mechanism of the diminished ability of LDLR by two variants. Therefore, future research should focus on exploring the mechanism of FH caused by its variants.
Generally, the clinical phenotype of homozygous FH patients is more severe than that of heterozygous patients, whereby some patients might eventually suffer from cardiovascular events in adolescence or even childhood (Sanchez-Hernandez et al., 2016). Moreover, the double heterozygous carriers of autosomal dominant hypercholesterolemia gene mutations have an intermediate phenotype compared with heterozygous and homozygous/compound heterozygous carriers (Sjouke et al., 2016). In this study, the proband was a compound heterozygous FH, and his daughter was a heterozygous FH. Due to being exposed to high levels of LDLC at birth, FH patients often exhibit severe atherosclerosis with the time response and dose effect of LDLC (Ference et al., 2017). The LDLC level of the proband was exceptionally high, even after drug therapy, and there were severe manifestations such as corneal arcus, xanthomas, carotid artery stenosis, coronary artery stenosis, and aortic valve calcification. Comparatively, the daughter of the proband only had hypercholesterolemia without any other clinical phenotypes. The lipids*age was proposed as an indicator to predict the risk of arteriosclerotic cardiovascular disease (Ference et al., 2018). The influence of age should be considered on the FH patient’s phenotype. Furthermore, early introduction of lipid-lowering treatment and long-term medical management could reduce the occurrence of cardiovascular events for the proband’s daughter.
There are some limitations in the current study. On the one hand, only three members of one family participated in the study. On the other hand, we only detected the expression of LDLR in the whole cell lysate but did not detect the number of LDLR on the cell surface. In the future, attention should be paid to the effect of LDLR mutation on the remaining activity of LDLR to unravel the damage of LDLR mutation to its function. Finally, we mainly focused on the variants in the exon regions of LDLR in our study. It has been shown that the mutations in the intron regions of LDLR may affect the splicing of mRNA precursors and lead to the occurrence of FH (Reeskamp et al., 2021a). Further studies should be performed to explore the mechanism of LDLR intron region variation in the occurrence and development of FH.
In conclusion, two novel variants, LDLR c.226 G > C and c.1003 G > T, might be pathogenic for FH by causing LDLR uptake dysfunction.
The datasets presented in this article are not readily available because according to the requirements of the Institutional Ethics Committee, the sharing of genomic data in the public domain is not allowed. Requests to access the datasets should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Ethics Committee of Ningbo First Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
XC, SL, and HH contributed to the conception, design, and final approval of the submitted version. HH, TS, JM, SW, RC, and JW contributed to completing the experiments, writing and revising the paper. All the authors have read and approved the final manuscript.
The research was supported by grants from: Natural Science Foundation of China (81900441); Natural Science Foundation of Zhejiang Province (LY19H020003, LQ19H020010, LQ20H020001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.762587/full#supplementary-material
Alves, A. C., Azevedo, S., Benito-Vicente, A., Graça, R., Galicia-Garcia, U., Barros, P., et al. (2021). LDLR Variants Functional Characterization: Contribution to Variant Classification. Atherosclerosis 329, 14–21. doi:10.1016/j.atherosclerosis.2021.06.001
Austin, M. A., Hutter, C. M., Zimmern, R. L., and Humphries, S. E. (2004). Genetic Causes of Monogenic Heterozygous Familial Hypercholesterolemia: a HuGE Prevalence Review. Am. J. Epidemiol. 160, 407–420. doi:10.1093/aje/kwh236
Beheshti, S. O., Madsen, C. M., Varbo, A., and Nordestgaard, B. G. (2020). Worldwide Prevalence of Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 75, 2553–2566. doi:10.1016/j.jacc.2020.03.057
Benito-Vicente, A., Alves, A. C., Etxebarria, A., Medeiros, A. M., Martin, C., and Bourbon, M. (2015). The Importance of an Integrated Analysis of Clinical, Molecular, and Functional Data for the Genetic Diagnosis of Familial Hypercholesterolemia. Genet. Med. 17, 980–988. doi:10.1038/gim.2015.14
Benn, M., Watts, G. F., Tybjærg-Hansen, A., and Nordestgaard, B. G. (2016). Mutations Causative of Familial Hypercholesterolaemia: Screening of 98 098 Individuals from the Copenhagen General Population Study Estimated a Prevalence of 1 in 217. Eur. Heart J. 37, 1384–1394. doi:10.1093/eurheartj/ehw028
Bertolini, S., Pisciotta, L., Rabacchi, C., Cefalù, A. B., Noto, D., Fasano, T., et al. (2013). Spectrum of Mutations and Phenotypic Expression in Patients with Autosomal Dominant Hypercholesterolemia Identified in Italy. Atherosclerosis 227, 342–348. doi:10.1016/j.atherosclerosis.2013.01.007
Bourbon, M., Alves, A. C., Medeiros, A. M., Silva, S., and Soutar, A. K. (2008). Familial Hypercholesterolaemia in Portugal. Atherosclerosis. Investigators of Portuguese, 196, 633–642. doi:10.1016/j.atherosclerosis.2007.07.019
Chemello, K., García-Nafría, J., Gallo, A., Martín, C., Lambert, G., and Blom, D. (2021). Lipoprotein Metabolism in Familial Hypercholesterolemia. J. Lipid Res. 62, 100062. doi:10.1016/j.jlr.2021.100062
Di Taranto, M. D., Giacobbe, C., Palma, D., Iannuzzo, G., Gentile, M., Calcaterra, I., et al. (2021). Genetic Spectrum of Familial Hypercholesterolemia and Correlations with Clinical Expression: Implications for Diagnosis Improvement. Clin. Genet. 100, 529–541. doi:10.1111/cge.14036
Du, J.-K., and Huang, Q. Y. (2007). Research Progress of Lipoprotein Lipase Gene. Hereditas 29, 8–16. doi:10.1360/yc-007-0008
Ference, B. A., Ginsberg, H. N., Graham, I., Ray, K. K., Packard, C. J., Bruckert, E., et al. (2017). Low-density Lipoproteins Cause Atherosclerotic Cardiovascular Disease. 1. Evidence from Genetic, Epidemiologic, and Clinical Studies. A Consensus Statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 38, 2459–2472. doi:10.1093/eurheartj/ehx144
Ference, B. A., Graham, I., Tokgozoglu, L., and Catapano, A. L. (2018). Impact of Lipids on Cardiovascular Health. J. Am. Coll. Cardiol. 72, 1141–1156. doi:10.1016/j.jacc.2018.06.046
Gidding, S. S., Ann Champagne, M., De Ferranti, S. D., Defesche, J., Ito, M. K., Knowles, J. W., et al. (2015). The Agenda for Familial HypercholesterolemiaThe Agenda for Familial Hypercholesterolemia: A Scientific Statement from the American Heart Association. Circulation. Obesity in Young Committee of Council on Cardiovascular Disease in Young, 132, 2167–2192. doi:10.1161/cir.0000000000000297
Graça, R., Fernandes, R., Alves, A. C., Menezes, J., Romão, L., and Bourbon, M. (2021). Characterization of Two Variants at Met 1 of the Human LDLR Gene Encoding the Same Amino Acid but Causing Different Functional Phenotypes. Biomedicines 9. doi:10.3390/biomedicines9091219
Grundy, S. M., Stone, N. J., Bailey, A. L., Beam, C., Birtcher, K. K., Blumenthal, R. S., et al. (2019). 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 73, e285–e350. doi:10.1016/j.jacc.2018.11.003
Hu, H., Chen, R., Hu, Y., Wang, J., Lin, S., and Chen, X. (2021). The LDLR c.501C>A Is a Disease-Causing Variant in Familial Hypercholesterolemia. Lipids Health Dis. 20, 101. doi:10.1186/s12944-021-01536-3
Hu, H., Cheng, J., Lin, S., Wang, S., and Chen, X. (2020). Calcified Aortic Valve Disease in Patients with Familial Hypercholesterolemia. J. Cardiovasc. Pharmacol. 76, 506–513. doi:10.1097/fjc.0000000000000890
Iacocca, M. A., and Hegele, R. A. (2017). Recent Advances in Genetic Testing for Familial Hypercholesterolemia. Expert Rev. Mol. Diagn. 17, 641–651. doi:10.1080/14737159.2017.1332997
Jeenduang, N., Ruangpracha, A., Promptmas, C., Pongrapeeporn, K.-u. S., and Porntadavity, S. (2010). Two Novel D151Y and M391T LDLR Mutations Causing LDLR Transport Defects in Thai Patients with Familial Hypercholesterolemia. Clinica Chim. Acta 411, 1656–1661. doi:10.1016/j.cca.2010.06.021
Laurie, A. D., Scott, R. S., and George, P. M. (2004). Genetic Screening of Patients with Familial Hypercholesterolaemia (FH): a New Zealand Perspective. Atheroscler. Supplements 5, 13–15. doi:10.1016/j.atherosclerosissup.2004.09.001
Leigh, S., Futema, M., Whittall, R., Taylor-Beadling, A., Williams, M., Den Dunnen, J. T., et al. (2017). The UCL Low-Density Lipoprotein Receptor Gene Variant Database: Pathogenicity Update. J. Med. Genet. 54, 217–223. doi:10.1136/jmedgenet-2016-104054
Narang, A., Uppilli, B., Vivekanand, A., Naushin, S., Yadav, A., Singhal, K., et al. (2020). Frequency Spectrum of Rare and Clinically Relevant Markers in Multiethnic Indian Populations (ClinIndb): A Resource for Genomic Medicine in India. Hum. Mutat. 41, 1833–1847. doi:10.1002/humu.24102
Pamplona-Cunha, H., Medeiros, M. F., Sincero, T. C. M., Back, I. d. C., and Silva, E. L. d. (2020). Portador Heterozigoto Composto de Hipercolesterolemia Familiar Causada por Variantes no LDLR. Arq Bras Cardiol. 115, 587–589. doi:10.36660/abc.20190582
Peterson, A. L., Bang, M., Block, R. C., Wong, N. D., and Karalis, D. G. (2021). Cascade Screening and Treatment Initiation in Young Adults with Heterozygous Familial Hypercholesterolemia. J. Clin. Med. 10. doi:10.3390/jcm10143090
Piepoli, M. F., Hoes, A. W., Agewall, S., Albus, C., Brotons, C., Catapano, A. L., et al. (2016). 2016 European Guidelines on Cardiovascular Disease Prevention in Clinical Practice. Eur. Heart J. 37, 2315–2381. doi:10.1093/eurheartj/ehw106
Reeskamp, L. F., Balvers, M., Peter, J., Van De Kerkhof, L., Klaaijsen, L. N., Motazacker, M. M., et al. (2021a). Intronic Variant Screening with Targeted Next-Generation Sequencing Reveals First Pseudoexon in LDLR in Familial Hypercholesterolemia. Atherosclerosis 321, 14–20. doi:10.1016/j.atherosclerosis.2021.02.003
Reeskamp, L. F., Tromp, T. R., Defesche, J. C., Grefhorst, A., Stroes, E. S. G., Hovingh, G. K., et al. (2021b). Next-generation Sequencing to Confirm Clinical Familial Hypercholesterolemia. Eur. J. Prev. Cardiol. 28, 875–883. doi:10.1093/eurjpc/zwaa451
Sánchez-Hernández, R. M., Civeira, F., Stef, M., Perez-Calahorra, S., Almagro, F., Plana, N., et al. (2016). Homozygous Familial Hypercholesterolemia in Spain. Circ. Cardiovasc. Genet. 9, 504–510. doi:10.1161/circgenetics.116.001545
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 11, 361–362. doi:10.1038/nmeth.2890
Shu, H.-Y., Zhang, W., Zheng, C.-C., Gao, M.-Y., Li, Y.-C., and Wang, Y.-G. (2021). Identification and Functional Characterization of a Low-Density Lipoprotein Receptor Gene Pathogenic Variant in Familial Hypercholesterolemia. Front. Genet. 12, 650077. doi:10.3389/fgene.2021.650077
Sjouke, B., Defesche, J. C., Hartgers, M. L., Wiegman, A., Roeters Van Lennep, J. E., Kastelein, J. J., et al. (2016). Double-heterozygous Autosomal Dominant Hypercholesterolemia: Clinical Characterization of an Underreported Disease. J. Clin. Lipidol. 10, 1462–1469. doi:10.1016/j.jacl.2016.09.003
Springer, T. A. (1998). An Extracellular β-propeller Module Predicted in Lipoprotein and Scavenger Receptors, Tyrosine Kinases, Epidermal Growth Factor Precursor, and Extracellular Matrix Components 1 1Edited by P. E. Wright. J. Mol. Biol. 283, 837–862. doi:10.1006/jmbi.1998.2115
Strøm, T. B., Bjune, K., and Leren, T. P. (2020). Bone Morphogenetic Protein 1 Cleaves the Linker Region between Ligand-Binding Repeats 4 and 5 of the LDL Receptor and Makes the LDL Receptor Non-functional. Hum. Mol. Genet. 29, 1229–1238. doi:10.1093/hmg/ddz238
Tada, H., Okada, H., Nomura, A., Yashiro, S., Nohara, A., Ishigaki, Y., et al. (2019). Rare and Deleterious Mutations in ABCG5/ABCG8 Genes Contribute to Mimicking and Worsening of Familial Hypercholesterolemia Phenotype. Circ. J. 83, 1917–1924. doi:10.1253/circj.cj-19-0317
Usifo, E., Leigh, S. E. A., Whittall, R. A., Lench, N., Taylor, A., Yeats, C., et al. (2012). Low-density Lipoprotein Receptor Gene Familial Hypercholesterolemia Variant Database: Update and Pathological Assessment. Ann. Hum. Genet. 76, 387–401. doi:10.1111/j.1469-1809.2012.00724.x
Van De Sluis, B., Wijers, M., and Herz, J. (2017). News on the Molecular Regulation and Function of Hepatic Low-Density Lipoprotein Receptor and LDLR-Related Protein 1. Curr. Opin. Lipidol. 28, 241–247. doi:10.1097/mol.0000000000000411
Wang, J., Huff, E., Janecka, L., and Hegele, R. A. (2001). Low Density Lipoprotein Receptor (LDLR) Gene Mutations in Canadian Subjects with Familial Hypercholesterolemia, but Not of French Descent. Hum. Mutat. 18, 359. doi:10.1002/humu.1205
Keywords: familial hypercholesterolemia, low-density lipoprotein cholesterol, low-density lipoprotein receptor, disease-causing mutations, function
Citation: Hu H, Shu T, Ma J, Chen R, Wang J, Wang S, Lin S and Chen X (2021) Two Novel Disease-Causing Mutations in the LDLR of Familial Hypercholesterolemia. Front. Genet. 12:762587. doi: 10.3389/fgene.2021.762587
Received: 22 August 2021; Accepted: 10 November 2021;
Published: 14 December 2021.
Edited by:
Alpo Juhani Vuorio, University of Helsinki, FinlandReviewed by:
Aldo Grefhorst, Amsterdam University Medical Center, NetherlandsCopyright © 2021 Hu, Shu, Ma, Chen, Wang, Wang, Lin and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaoyi Lin, c2hhb3lpX2xpbkBob3RtYWlsLmNvbQ==; Xiaomin Chen, Y3htZG9jdG9yQDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.