 Suran Liu
Suran Liu Yujie You
Yujie You Zhaoqi Tong
Zhaoqi Tong Le Zhang
Le Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 26 October 2021
Sec. Statistical Genetics and Methodology
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.761629
This article is part of the Research Topic Data Mining and Statistical Methods for Knowledge Discovery in Diseases Based on Multimodal Omics View all 15 articles
It is very important for systems biologists to predict the state of the multi-omics time series for disease occurrence and health detection. However, it is difficult to make the prediction due to the high-dimensional, nonlinear and noisy characteristics of the multi-omics time series data. For this reason, this study innovatively proposes an Embedding, Koopman and Autoencoder technologies-based multi-omics time series predictive model (EKATP) to predict the future state of a high-dimensional nonlinear multi-omics time series. We evaluate this EKATP by using a genomics time series with chaotic behavior, a proteomics time series with oscillating behavior and a metabolomics time series with flow behavior. The computational experiments demonstrate that our proposed EKATP can substantially improve the accuracy, robustness and generalizability to predict the future state of a time series for multi-omics data.
Currently, the prediction of multi-omics time series states is one of the trending areas in systems biology research (Zhang et al., 2019a). In particular, the development of high-throughput technology (Soon et al., 2013) has produced a large-scale time series multi-omics state (Liang and Kelemen 2017a), including genomics (Lockhart and Winzeler 2000), proteomics (Tyers and Mann, 2003), metabolomics (Weckwerth 2003) and more. Previous studies usually employed differential equation (Eisenhammer et al., 1991; Zhang et al., 2016; Zhang and Zhang 2017; Liu G.-D. et al., 2020) based models to abstract and formalise multi-omics time series data (Bianconi et al., 2020). Then, it became possible to explore the time-varying connections and predict their future state (Ji et al., 2017) by solving these differential equations. In particular, predicting multi-omics time series states can not only discover dynamic information for biological entities, such as genes, proteins and metabolites, but also explore complicated biological interactions and the pathogenesis of diseases (Liang and Kelemen, 2017b).
However, a multi-omics time series usually has high dimensions (Perez-Riverol et al., 2017), complicated interaction relationships (Fischer 2008) and inevitable noise (Fischer 2008; Tsimring 2014). Thus, when we employ differential equations to model the multi-omics time series state, it is hard for us to solve these equations due to their high dimensionality and nonlinear characteristics (Bianconi et al., 2020). For these reasons, the way to predict the future state of a multi-omics time series by solving these complicated nonlinear differential equations has already become challenging work.
Recently, future state prediction for a multi-omics time series has been widely studied by computational biologists. For genomic studies, we usually use a gene expression time series to develop gene regulatory networks (Davidson and Levin 2005; Zhang et al., 2018; Xiao et al., 2020; Zhang et al., 2020; Xiao et al., 2021; Zhang et al., 2021a). However, since the gene regulatory network is a complex high-dimensional nonlinear system (Zhang et al., 2012a), it often produces chaotic phenomena (Levnajić and Tadić 2010), which not only play an important role in maintaining stable gene expression patterns (Sevim and Rikvold 2008) but also are closely related to the occurrence of diseases (Suzuki et al., 2016). Usually, we employ the Lorentz system (Lorenz 1963) to describe the chaotic phenomenon. However, it is inaccurate to predict the future state of genomics time series with nonlinear complicated interactions because the Lorentz system is not good at processing nonlinear complicated interactions (Lai et al., 2018). Currently, delay embedding theory (Sauer et al., 1991; Holmes et al., 2012) is commonly used to transform the spatial information (complicated interactions) into temporal information (the future state of the time series (Chen et al., 2020)) for dimensional reduction (Gao et al., 2017; Li et al., 2017; Xia et al., 2017; Zhang et al., 2019b; Zhang et al., 2019c; Wu et al., 2020; You et al., 2020; Zhang et al., 2021b), whereas Koopman theory (Koopman, 1931) can switch the nonlinear system into a linear system to reduce computing cost. Therefore, our first research question asks if we can develop such a time series predictive model that integrates the Lorentz system with delay embedding and Koopman theory to accurately predict the future state of genomics time series with chaotic behavior.
For proteomics studies, we usually use proteomic time series data to infer protein–protein interactions (PPIs) (Wu et al., 2009). Currently, we employ mass spectrometry technology (Mann et al., 2001) to obtain proteomics time series data. However, since it is unstable to have time-course experimental data by mass spectrometry technology, proteomics time series data are prone to oscillating behavior (Iuchi et al., 2018). Previously, we employed a nonlinear pendulum system (Hirsch 1974) to describe the oscillation behavior, though it was subjected to overfitting under a strong noise environment. Since the conjugate form of delay embedding (Sauer et al., 1991; Holmes et al., 2012) can ensure the reversibility of the time series predictive model (Chen et al., 2020) and reduce the impact of noise on prediction to a certain extent, our second research question asks if we can develop such a time series predictive model that can integrate a nonlinear pendulum system with delay embedding to accurately predict the state of proteomics time series with oscillating behavior.
For metabolomics studies, we usually use metabolic time series data that represent the flow behavior of biological fluids (serum, cerebrospinal fluid, etc.) to discover key metabolites in biological fluids (Zhang et al., 2012b). A previous study (Noack et al., 2003) always employed a nonlinear biological fluid system to describe metabolic time series data. However, because most nonlinear fluid flow systems have high dimensions (Lusch et al., 2018), we not only have difficulty selecting features from high-dimensional metabolic time series data but also impede progress because of time-consuming computing (Wang et al., 2021). Currently, since neural networks (Wang et al., 2014) can decrease the computing cost (Song et al., 2017) by dimensional reduction for time series data (Hinton and Salakhutdinov, 2006), our third research question asks if we can develop such a time series predictive model that integrates a nonlinear fluid flow system with a neural network to predict the future state of the metabolomics time series accurately and quickly with flow behaviour.
To answer the above three research questions, this study innovatively develops an Embedding, Koopman and Autoencoder technologies-based multi-omics time series predictive model (EKATP) to predict the future state of the time series for the corresponding genomics, proteomics and metabolomics datasets. Compared with previous approaches (Lusch et al., 2018; Azencot et al., 2020), the contributions of the study are summarised as follows. First, we select key features from a high-dimensional nonlinear state by integrating a neural network with the delay embedding theory. Second, we switch the nonlinear system with a linear system to reduce the computing cost by the Koopman theory. Finally, we develop a neural network and delay embedding theory-based model for reversible mapping between a high-dimensional nonlinear system and a low-dimensional linear system, thereby improving the accuracy and robustness of prediction.
The rest of the manuscript is organised as follows. Related Works mainly describes the related work for Autoencoder, delay embedding theory and Koopman theory. Materials and Methods introduces the architecture of the EKATP and the related procedure. Experiments describes the computational experiments. Finally, we conclude the study and discuss the future work.
Supplementary Presentation S1 details the related theory and existing research of the Autoencoder, delay embedding theory and Koopman theory.
Figure 1 describes the workflow of the EKATP.
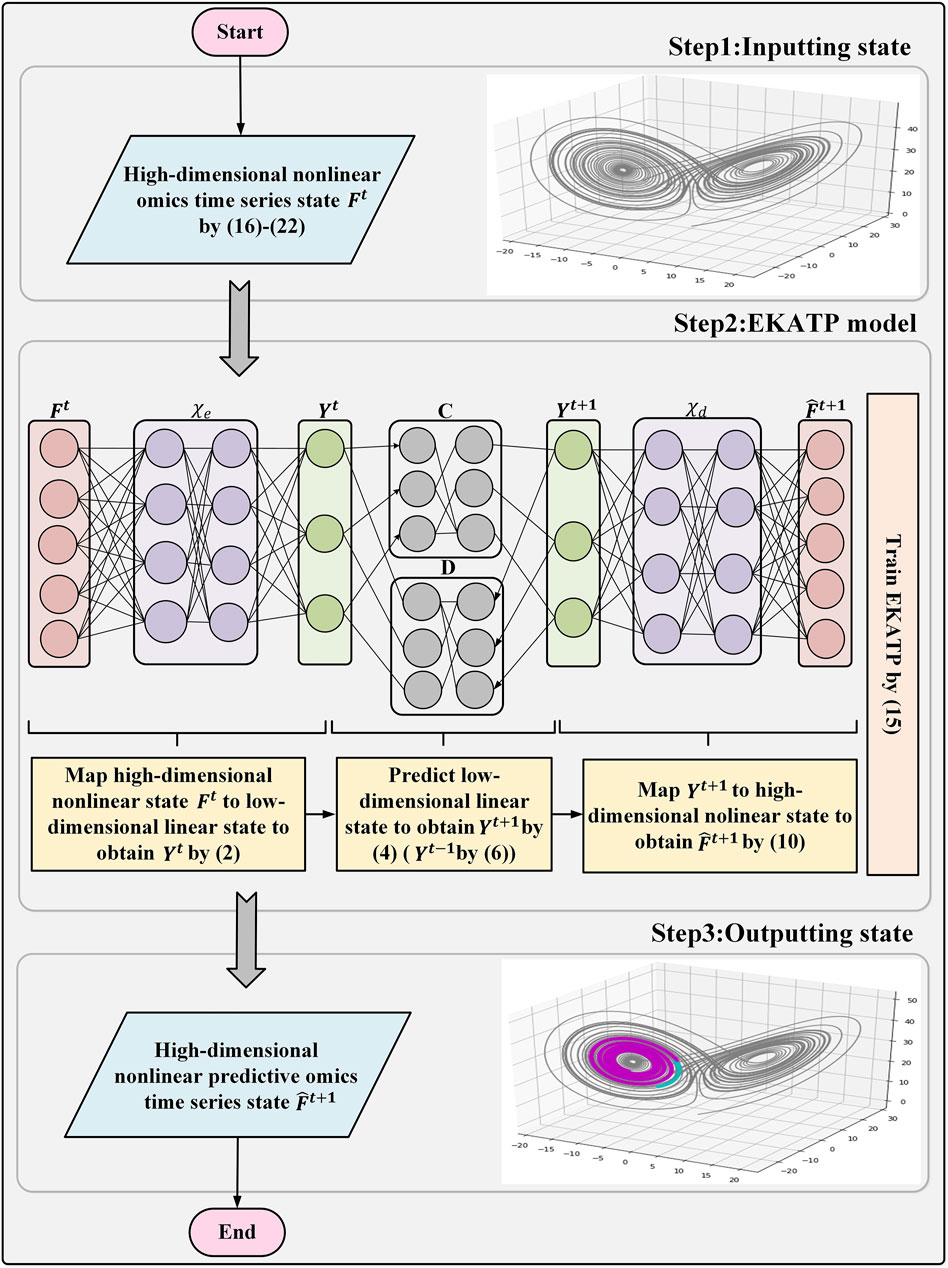
FIGURE 1. EKATP workflow.
Given a set of high-dimensional nonlinear multi-omics time series states
Since an EKATP is based on the Autoencoder framework, we employ Eq. 1 to define the objective function for Autoencoder (
Here,
According to the description in the delay embedding theory (Supplementary Presentation S1), we employ
where
Based on the Koopman theory discussed by Supplementary Presentation S1, we construct the finite dimensional linear matrix
Equation 4 can be expanded by Eq. 5.
Here,
Equation 6 can be expanded by Eq. 7.
Here,
We make the
where
Equations 11, 12 define the loss function of forward prediction (
Equation 13 defines the loss function (
Additionally, we employ loss function (
Equation 15 optimizes the key parameters for the EKATP by minimizing
Here,
This section evaluates the predictability of the proposed EKATP for high-dimensional nonlinear multi-omics datasets by comparing it with recurrent neural networks (RNNs) (Jiang and Lai, 2019), long short-term memory (LSTM) (Hochreiter and Schmidhuber, 1997), dynamic Autoencoder (DAE) (Lusch et al., 2018) and Koopman Autoencoder (KAE) (Azencot et al., 2020). The detailed experimental setup is listed in Supplementary Presentation S2. In addition, we detail the workflow chart and list the related pseudocode in Supplementary Figure S1; Supplementary Presentation S3.
We usually employ the chaotic Lorentz system (Lorenz, 1963) to describe a gene expression time series with a low-dimensional manifold (Sauer et al., 1991) by Eq. 16,
where
Since gene expression time series contains considerable noise, we employ white Gaussian noise (Li et al., 2017) to simulate the noise by Eq. 17,
where
Here, we describe how to obtain a high-dimensional gene expression time series with a low-dimensional manifold as follows. First, we generate the three-dimensional time series
To prove the accuracy and robustness of the EKATP, we generate a small-scale system containing
Figure 2 shows the predictive error in the range of 50 steps under different initial conditions and environments. Detailed information is listed in Supplementary Tables S1.7, S1.8; Supplementary Presentation S4.
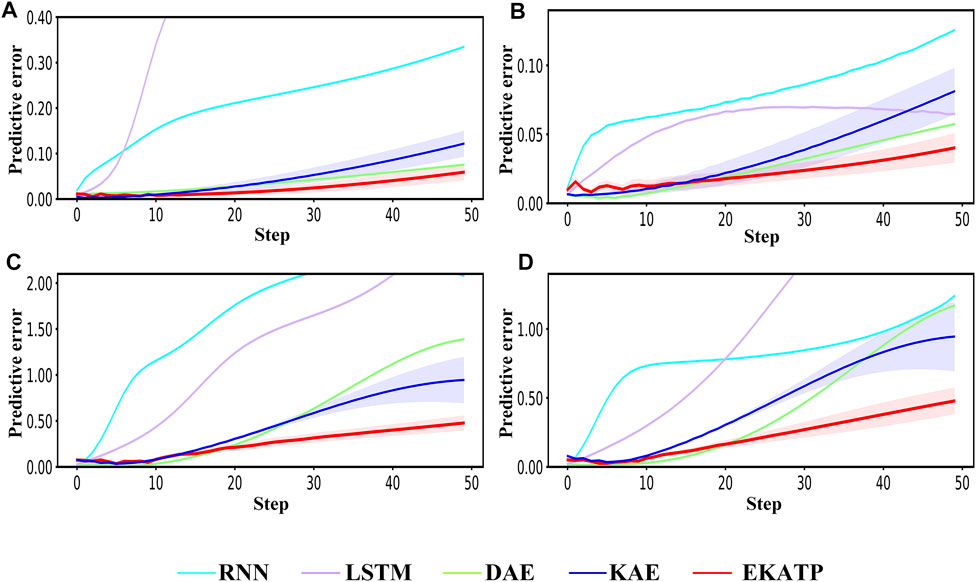
FIGURE 2. Comparison among the RNN, LSTM, DAE, KAE and EKATP. The abscissa represents the step, and the ordinate represents the predictive error. (A) The initial conditions are
Figures 2A,C demonstrates that the EKATP not only has less of a predictive error than the existing methods under a clean environment (
Figures 2B,D shows that the EKATP not only has less of a predictive error than previous methods under a noisy environment (
Figure 2 indicates that the EKATP has greater predictive accuracy and robustness than excitation methods in clean and noisy environments.
To further prove the generalizability of the EKATP, we generate a large-scale system containing
First, since the 3-dimensional time series state and 96-dimensional time series state are diffeomorphic (Sauer et al., 1991), which is indicated by the data preprocessing procedure, it implies that the mapping between these two time series is reversible. Here, we map the 96-dimensional gene expression predictive results onto a 3-dimensional space by orthogonal inverse transformation (Anderson et al., 1987) to visualize the predictive result of the EKATP.
Figures 3A,B,C demonstrates that the predictive results of the EKATP are close to the true value for different periods of a time series. Figure 3 shows that the EKATP can accurately predict the gene expression time series at different periods, implying that it has a strong generalizability, even in a very complicated nonlinear environment.

FIGURE 3. The 50-step predictive trajectories of the EKATP are under initial conditions
We always use a nonlinear pendulum model (Hirsch, 1974) with oscillatory behaviour to describe a proteomics time series with a low-dimensional manifold (Sauer et al., 1991) by Eq. 19,
where
Since a considerable amount of noise exists in a protein time series, we employ white Gaussian noise (Li et al., 2017) to describe it by Eq. 20,
where
Here, we describe how to obtain a high-dimensional proteomics time series with a low-dimensional manifold. First, we generate the 2-dimensional time series
To prove the accuracy and robustness of the EKATP, we generate a system containing
Figure 4 shows that the EKATP can effectively predict a proteomic time series under clean and noisy environments within 1,000 steps, the details of which are listed in Supplementary Tables S2.5, S2.6; Supplementary Presentation S4.
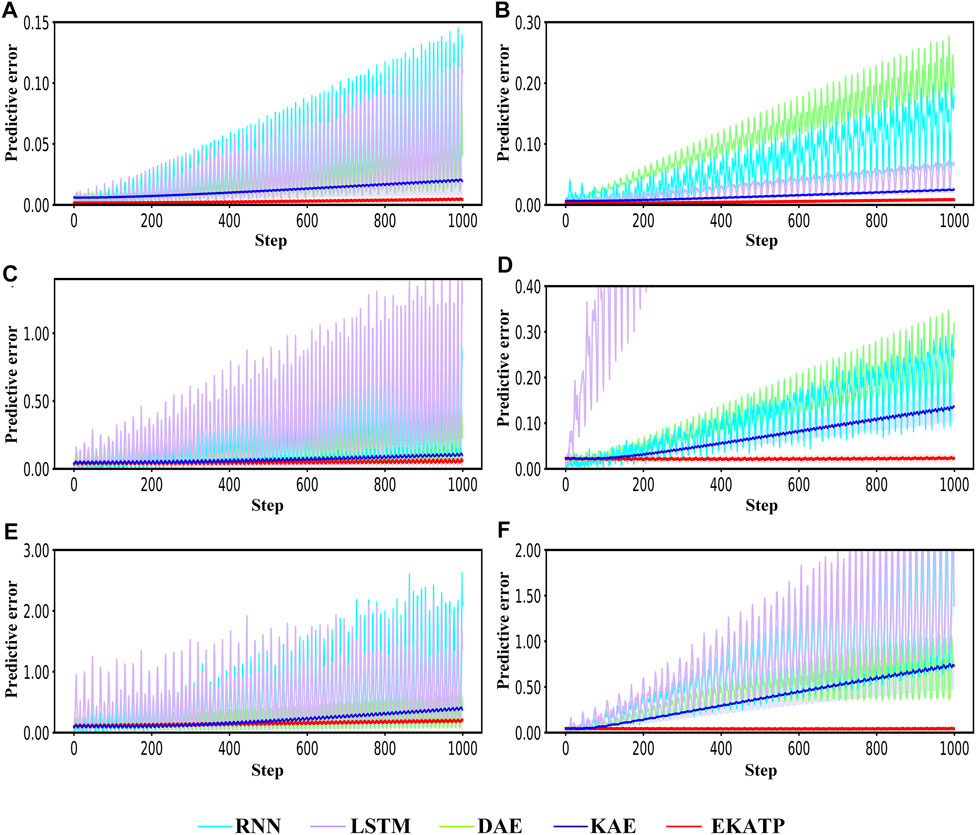
FIGURE 4. Comparison with the RNN, LSTM, DAE, KAE and EKATP. The abscissa represents the step, and the ordinate represents the predictive error. (A) The initial conditions are
Figures 4A,B shows that the EKATP not only has less of a predictive error under a clean environment (
Figures 4C,D demonstrates that the EKATP has less of a predictive error under a noise environment (
Figures 4E,F indicates that the EKATP not only has less of a predictive error under a noise environment (
Figures 4A,C,E shows that the predictive error of the EKATP remains stable when the noise intensity
Figure 4 demonstrates that the predictive accuracy and robustness of the EKATP outperforms the existing methods under clean and noisy environments.
Since Figure 4 shows that KAE has a better predictive effect than the other existing methods, we use it to compare the predictive performance with the EKATP by visualizing the predictive trajectory.
Indicated by our data preprocessing procedure, since the 2-dimensional time series state and 64-dimensional time series state are diffeomorphic (Sauer et al., 1991), the mapping between these two time series is reversible. Here, we map the 64-dimensional protein time series predictive results onto a 2-dimensional space by orthogonal inverse transformation (Anderson et al., 1987) to visualize the predictive time series trajectory. Figure 5 shows the predictive trajectories of the KAE and EKATP within 1,000 steps under the initial conditions of
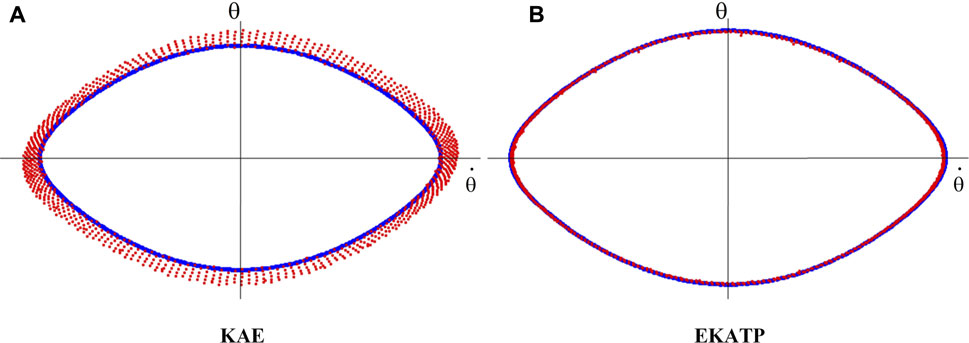
FIGURE 5. The prediction trajectories within 1,000 steps under the initial conditions of
To further prove that the EKATP has strong generalizability, we randomly selected 20 pieces of different protein time series data for model training and analysis. The details are listed in Table 1; Supplementary Table S2.7.
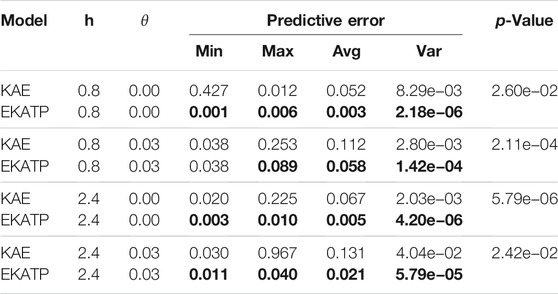
TABLE 1. Predictive error at 1,000 steps for both the KAE and EKATP.
After we employ 20 different proteomics time series datasets to test the KAE and EKATP, Table 1 shows the predictive error of the KAE and EKATP at 1,000 steps under different initial noise and complexity (
We usually employ a nonlinear biological fluid system (Noack et al., 2003) to describe the high-dimensional metabolic time series with a low-dimensional manifold (Sauer et al., 1991) for the flow behavior of biological fluids simulation by Eq. 21,
where
Since the metabolomics time series contains considerable noise, we employ white Gaussian noise (Li et al., 2017) to describe it by Eq. 22,
where
Fig. 6 Comparison of the RNN, LSTM, DAE, KAE and EKATP. The abscissa represents the time step, and the ordinate represents the predictive error. (A) The initial conditions are
Here, we show how to generate a high-dimensional metabolomics time series with a low-dimensional manifold. First, we build up the 3-dimensional time series
To demonstrate the accuracy and robustness of the EKATP, we generate a system containing
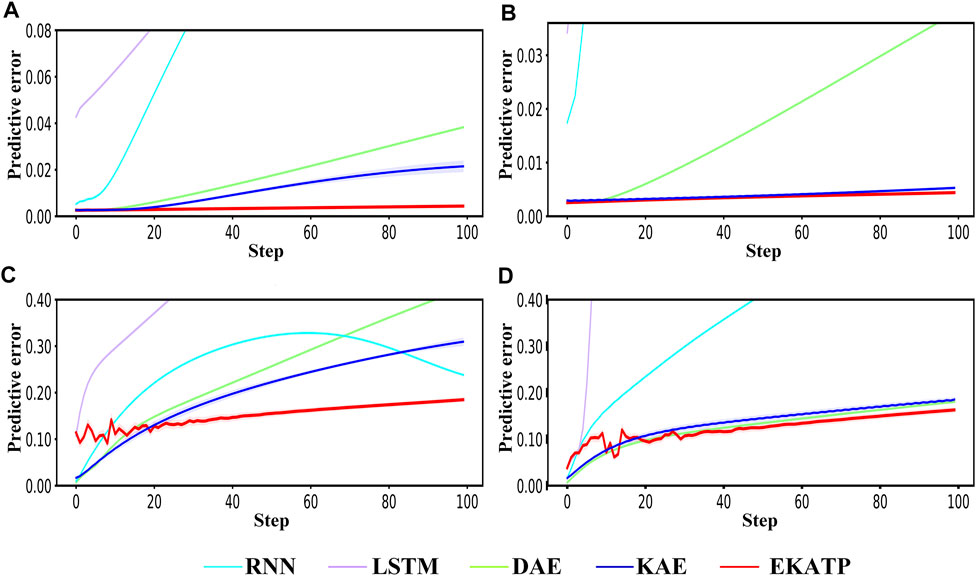
FIGURE 6. Comparison of the RNN, LSTM, DAE, KAE and EKATP. The abscissa represents the time step, and the ordinate represents the predictive error. (A) The initial conditions are
Figures 6A,C demonstrates that the EKATP has less of a predictive error under a clean environment (
Figures 6B,D suggests that the EKATP not only has less of a predictive error under a low noise intensity environment (
Figure 6 implies that the EKATP has better predictive accuracy and robustness than the existing methods under clean and weakly noisy environments.
Since a metabolomics time series usually has strong noise intensity (Mak et al., 2015), we use the EKATP to predict a high-dimensional metabolomics time series under strong noise intensities to prove its robustness. Because the 3-dimensional time series state and the 96-dimensional time series state are diffeomorphic (Sauer et al., 1991), the mapping between these two time series is reversible. Thus, after we map the 96-dimensional metabolic time series predictive results onto a 3-dimensional space by orthogonal inverse transformation (Anderson et al., 1987), Figure 7 shows the predictive time series trajectories by the EKATP under different intensities of noise. We select the last 100 steps to validate the predictive power of the EKATP as in the previous setup (Supplementary Table S3.6). The results demonstrate that although the true data become gradually messy when we increase the noise intensity
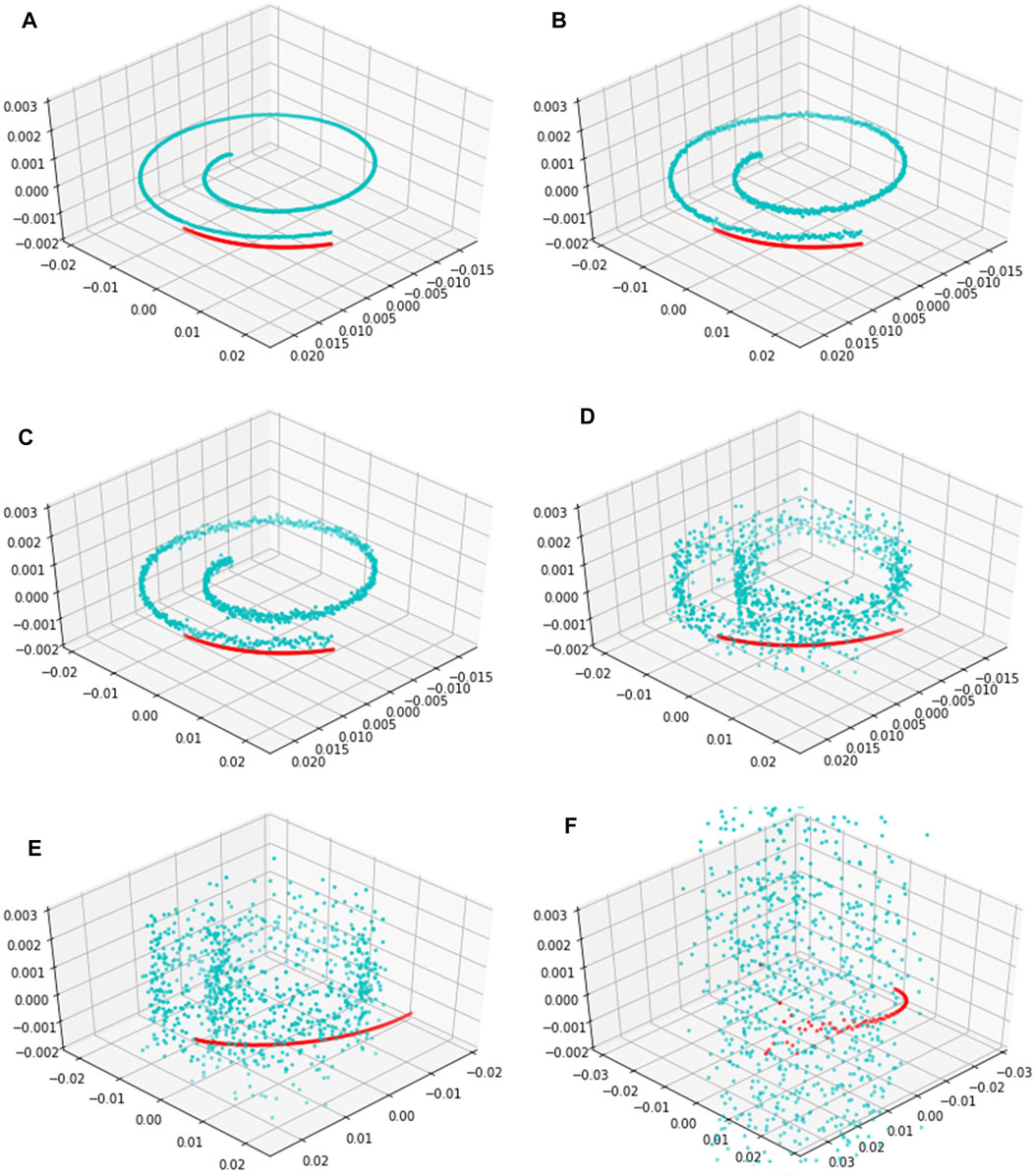
FIGURE 7. The predictive trajectories of the EKATP within 100 steps under different intensities of noise. Cyan dots represent true data with an interval of
Moreover, we use Eqs 23, 24 to calculate the Pearson correlation coefficient (PCC) (Abar et al., 2017) and the root mean squared error (RMSE) (Abar et al., 2017) between predictive and true data under different noise intensities.
Here,
Figure 8A shows that the PCC value of the EKATP gradually decreases when we increase the noise intensity

FIGURE 8. PCC and RMSE values between predictive and true data under different noise intensities. The details are listed in Supplementary Table S3.7. (A) PCC value between predictive and true data, where the abscissa represents the noise intensity and the ordinate represents the PCC value. (B) RMSE value between predictive and true data, where the abscissa represents the noise intensity and the ordinate represents the RMSE value.
To answer the three proposed questions, this study developed an EKATP to predict the future state of a high-dimensional nonlinear multi-omics time series. First, we select key features from high-dimensional nonlinear multi-omics time series data. After that, we map these key features to the low-dimensional linear space. Next, we obtain the future state of the multi-omics time series by learning the evolutionary relationship between the adjacent states of the time series in the low-dimensional linear space. Finally, we predict the future state of the high-dimensional nonlinear multi-omics time series by mapping the low-dimensional linear predictive state back to the high-dimensional nonlinear space. The experimental results demonstrate that the EKATP can greatly improve the accuracy, robustness and generalisability to predict the future state of a time series for genomics (Figures 2, 3), proteomics (Figures 4, 5; Table 1) and metabolomics (Figures 6–8) datasets.
However, there are still several shortcomings to the current study. For example, we are still unclear on the impact of embedding dimensions from high-dimensional nonlinear space to low-dimensional linear space on predictive accuracy and the way to use high-performance computing to increase the efficiency of the EKATP. Applying the EKATP to network biological datasets (Liu X. et al., 2020) is also the direction we need to continue the study. Thus, we will improve the EKATP from these perspectives in the distant future.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
YY, LZ and SL conceived the project. SL, YY and ZT carried out experiments. SL and YY visualized experiment results. SL drafted the manuscript. YY and LZ revised the article. All the authors read and approved the final article.
This work was supported by the National Science and Technology Major Project (2018ZX10201002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.761629/full#supplementary-material
Abar, T., El Asmi, A. S., and Asmi, S. E. (2017). “Machine Learning Based QoE Prediction in SDN Networks,” in Proceedings of the 2017 13th International Wireless Communications and Mobile Computing Conference (IWCMC), Valencia, Spain, June 2017. doi:10.1109/IWCMC.2017.7986488
Anderson, T. W., Olkin, I., and Underhill, L. G. (1987). Generation of Random Orthogonal Matrices. SIAM J. Sci. Stat. Comput. 8 (4), 625–629. doi:10.1137/0908055
Azencot, O., Erichson, N. B., Lin, V., and Mahoney, M. (2020). “Forecasting Sequential Data Using Consistent Koopman Autoencoders,” in Proceedings of the 37th International Conference on Machine Learning, 475–485.
Bianconi, F., Antonini, C., Tomassoni, L., and Valigi, P. (2020). Robust Calibration of High Dimension Nonlinear Dynamical Models for Omics Data: An Application in Cancer Systems Biology. IEEE Trans. Contr. Syst. Technol. 28 (1), 196–207. doi:10.1109/TCST.2018.2844362
Chen, P., Liu, R., Aihara, K., and Chen, L. (2020). Autoreservoir Computing for Multistep Ahead Prediction Based on the Spatiotemporal Information Transformation. Nat. Commun. 11 (1), 4568. doi:10.1038/s41467-020-18381-0
Davidson, E., and Levin, M. (2005). Gene Regulatory Networks. Proc. Natl. Acad. Sci. 102 (14), 4935. doi:10.1073/pnas.0502024102
Eisenhammer, T., Hübler, A., Packard, N., and Kelso, J. A. S. (1991). Modeling Experimental Time Series with Ordinary Differential Equations. Biol. Cybern. 65 (2), 107–112. doi:10.1007/BF00202385
Fischer, H. P. (2008). Mathematical Modeling of Complex Biological Systems: from Parts Lists to Understanding Systems Behavior. Alcohol. Res. Health 31 (1), 49–59.
Gao, H., Yin, Z., Cao, Z., and Zhang, L. (2017). Developing an Agent-Based Drug Model to Investigate the Synergistic Effects of Drug Combinations. Molecules 22 (12), 2209. doi:10.3390/molecules22122209
Gao, J., Liu, P., Liu, G.-D., and Zhang, L. (2021). Robust Needle Localization and Enhancement Algorithm for Ultrasound by Deep Learning and Beam Steering Methods. J. Comput. Sci. Technol. 36 (2), 334–346. doi:10.1007/s11390-021-0861-7
Hinton, G. E., and Salakhutdinov, R. R. (2006). Reducing the Dimensionality of Data with Neural Networks. Science 313 (5786), 504–507. doi:10.1126/science.1127647
Hirsch, M. (1974). Differential Equations, Dynamical Systems, and Linear Algebra Mathematics. America: Academic press. doi:10.1016/s0079-8169(08)x6044-1
Hochreiter, S., and Schmidhuber, J. (1997). Long Short-Term Memory. Neural Comput. 9 (8), 1735–1780. doi:10.1162/neco.1997.9.8.1735
Holmes, P., Lumley, J. L., Berkooz, G., and Rowley, C. W. (2012). Turbulence, Coherent Structures, Dynamical Systems and Symmetry. Cambridge, UK, Cambridge University Press.
Iuchi, H., Sugimoto, M., and Tomita, M. (2018). MICOP: Maximal Information Coefficient-Based Oscillation Prediction to Detect Biological Rhythms in Proteomics Data. BMC Bioinformatics 19 (1), 249. doi:10.1186/s12859-018-2257-4
Ji, Z., Yan, K., Li, W., Hu, H., and Zhu, X. (2017). Mathematical and Computational Modeling in Complex Biological Systems. Biomed. Res. Int. 2017, 1–16. doi:10.1155/2017/5958321
Jiang, J., and Lai, Y.-C. (2019). Model-free Prediction of Spatiotemporal Dynamical Systems with Recurrent Neural Networks: Role of Network Spectral Radius. Phys. Rev. Res. 1 (3), 033056. doi:10.1103/PhysRevResearch.1.033056
Koopman, B. O. (1931). Hamiltonian Systems and Transformation in Hilbert Space. Proc. Natl. Acad. Sci. 17 (5), 315–318. doi:10.1073/pnas.17.5.315
Lai, Q., Zhao, X.-W., Huang, J.-N., Pham, V.-T., and Rajagopal, K. (2018). Monostability, Bistability, Periodicity and Chaos in Gene Regulatory Network. Eur. Phys. J. Spec. Top. 227 (7), 719–730. doi:10.1140/epjst/e2018-700132-8
Levnajić, Z., and Tadić, B. (2010). Stability and Chaos in Coupled Two-Dimensional Maps on Gene Regulatory Network of Bacterium E. coli. Chaos 20 (3), 033115. doi:10.1063/1.3474906
Li, T., Cheng, Z., and Zhang, L. (2017). Developing a Novel Parameter Estimation Method for Agent-Based Model in Immune System Simulation under the Framework of History Matching: A Case Study on Influenza A Virus Infection. Ijms 18 (12), 2592. doi:10.3390/ijms18122592
Liang, Y., and Kelemen, A. (2017a). Computational Dynamic Approaches for Temporal Omics Data with Applications to Systems Medicine. BioData Mining 10 (1), 20. doi:10.1186/s13040-017-0140-x
Liang, Y., and Kelemen, A. (2017b). Dynamic Modeling and Network Approaches for Omics Time Course Data: Overview of Computational Approaches and Applications. Brief. Bioinform. 19 (5), 1051–1068. doi:10.1093/bib/bbx036
Liu, G.-D., Li, Y.-C., Zhang, W., and Zhang, L. (2020). A Brief Review of Artificial Intelligence Applications and Algorithms for Psychiatric Disorders. Engineering 6 (4), 462–467. doi:10.1016/j.eng.2019.06.008
Liu, X., Maiorino, E., Halu, A., Glass, K., Prasad, R. B., Loscalzo, J., et al. (2020). Robustness and Lethality in Multilayer Biological Molecular Networks. Nat. Commun. 11 (1), 6043. doi:10.1038/s41467-020-19841-3
Lockhart, D. J., and Winzeler, E. A. (2000). Genomics, Gene Expression and DNA Arrays. Nature 405 (6788), 827–836. doi:10.1038/35015701
Lorenz, E. N. (1963). Deterministic Nonperiodic Flow. J. Atmos. Sci. 20 (2), 130–141. doi:10.1175/1520-0469(1963)020<0130:dnf>2.0.co;2
Lusch, B., Kutz, J. N., and Brunton, S. L. (2018). Deep Learning for Universal Linear Embeddings of Nonlinear Dynamics. Nat. Commun. 9 (1), 4950. doi:10.1038/s41467-018-07210-0
Mak, T. D., Laiakis, E. C., Goudarzi, M., and Fornace, A. J. (2015). Selective Paired Ion Contrast Analysis: a Novel Algorithm for Analyzing Postprocessed LC-MS Metabolomics Data Possessing High Experimental Noise. Anal. Chem. 87 (6), 3177–3186. doi:10.1021/ac504012a
Mann, M., Hendrickson, R. C., and Pandey, A. (2001). Analysis of Proteins and Proteomes by Mass Spectrometry. Annu. Rev. Biochem. 70 (1), 437–473. doi:10.1146/annurev.biochem.70.1.437
Noack, B. R., Afanasiev, K., Morzyński, M., Tadmor, G., and Thiele, F. (2003). A Hierarchy of Low-Dimensional Models for the Transient and post-transient cylinder Wake. J. Fluid Mech. 497, 335–363. doi:10.1017/S0022112003006694
Perez-Riverol, Y., Kuhn, M., Vizcaíno, J. A., Hitz, M.-P., and Audain, E. (2017). Accurate and Fast Feature Selection Workflow for High-Dimensional Omics Data. PloS one 12 (12), e0189875. doi:10.1371/journal.pone.0189875
Sauer, T., Yorke, J. A., and Casdagli, M. (1991). Embedology. J. Stat. Phys. 65 (3), 579–616. doi:10.1007/BF01053745
Sevim, V., and Rikvold, P. A. (2008). Chaotic Gene Regulatory Networks Can Be Robust against Mutations and Noise. J. Theor. Biol. 253 (2), 323–332. doi:10.1016/j.jtbi.2008.03.003
Song, H., Jiang, Z., Men, A., and Yang, B. (2017). A Hybrid Semi-supervised Anomaly Detection Model for High-Dimensional Data. Comput. Intelligence Neurosci. 2017, 1–9. doi:10.1155/2017/8501683
Soon, W. W., Hariharan, M., and Snyder, M. P. (2013). High‐throughput Sequencing for Biology and Medicine. Mol. Syst. Biol. 9 (1), 640. doi:10.1038/msb.2012.61
Suzuki, Y., Lu, M., Ben-Jacob, E., and Onuchic, J. N. (2016). Periodic, Quasi-Periodic and Chaotic Dynamics in Simple Gene Elements with Time Delays. Sci. Rep. 6 (1), 21037. doi:10.1038/srep21037
Tsimring, L. S. (2014). Noise in Biology. Rep. Prog. Phys. 77 (2)–026601. doi:10.1088/0034-4885/77/2/026601
Tyers, M., and Mann, M. (2003). From Genomics to Proteomics. Nature 422 (6928), 193–197. doi:10.1038/nature01510
Wang, H., Li, M., and Yue, X. (2021). IncLSTM: Incremental Ensemble LSTM Model towards Time Series Data. Comput. Electr. Eng. 92, 107156. doi:10.1016/j.compeleceng.2021.107156
Wang, W., Huang, Y., Wang, Y., and Wang, L. (2014). “Generalized Autoencoder: A Neural Network Framework for Dimensionality Reduction,” in Proceedings of the 2014 IEEE Conference on Computer Vision and Pattern Recognition Workshops, Columbus, OH, USA, June 2014. doi:10.1109/CVPRW.2014.79
Weckwerth, W. (2003). Metabolomics in Systems Biology. Annu. Rev. Plant Biol. 54 (1), 669–689. doi:10.1146/annurev.arplant.54.031902.135014
Wu, J., Vallenius, T., Ovaska, K., Westermarck, J., Mäkelä, T. P., and Hautaniemi, S. (2009). Integrated Network Analysis Platform for Protein-Protein Interactions. Nat. Methods 6 (1), 75–77. doi:10.1038/nmeth.1282
Wu, W., Song, L., Yang, Y., Wang, J., Liu, H., and Zhang, L. (2020). Exploring the Dynamics and Interplay of Human Papillomavirus and Cervical Tumorigenesis by Integrating Biological Data into a Mathematical Model. BMC Bioinformatics 21 (7), 152. doi:10.1186/s12859-020-3454-5
Xia, Y., Yang, C., Hu, N., Yang, Z., He, X., Li, T., et al. (2017). Exploring the Key Genes and Signaling Transduction Pathways Related to the Survival Time of Glioblastoma Multiforme Patients by a Novel Survival Analysis Model. BMC Genomics 18 (1), 950. doi:10.1186/s12864-016-3256-3
Xiao, M., Liu, G., Xie, J., Dai, Z., Wei, Z., Ren, Z., et al. (2021). 2019nCoVAS: Developing the Web Service for Epidemic Transmission Prediction, Genome Analysis, and Psychological Stress Assessment for 2019-nCoV. Ieee/acm Trans. Comput. Biol. Bioinf. 18 (4), 1250–1261. doi:10.1109/TCBB.2021.3049617
Xiao, M., Yang, X., Yu, J., and Zhang, L. (2020). CGIDLA:Developing the Web Server for CpG Island Related Density and LAUPs (Lineage-Associated Underrepresented Permutations) Study. Ieee/acm Trans. Comput. Biol. Bioinf. 17 (6), 2148–2154. doi:10.1109/TCBB.2019.2935971
You, Y., Ru, X., Lei, W., Li, T., Xiao, M., Zheng, H., et al. (2020). Developing the Novel Bioinformatics Algorithms to Systematically Investigate the Connections Among Survival Time, Key Genes and Proteins for Glioblastoma Multiforme. BMC Bioinformatics 21 (13), 383. doi:10.1186/s12859-020-03674-4
Zhang, A., Sun, H., Wang, P., Han, Y., and Wang, X. (2012a). Recent and Potential Developments of Biofluid Analyses in Metabolomics. J. Proteomics 75 (4), 1079–1088. doi:10.1016/j.jprot.2011.10.027
Zhang, Z., Ye, W., Qian, Y., Zheng, Z., Huang, X., and Hu, G. (2012b). Chaotic Motifs in Gene Regulatory Networks. PLOS ONE 7 (7), e39355. doi:10.1371/journal.pone.0039355
Zhang, L., Dai, Z., Yu, J., and Xiao, M. (2020). CpG-island-based Annotation and Analysis of Human Housekeeping Genes. Brief. Bioinform. 22 (1), 515–525. doi:10.1093/bib/bbz134
Zhang, L., Fu, C., Li, J., Zhao, Z., Hou, Y., Zhou, W., et al. (2019a). Discovery of a Ruthenium Complex for the Theranosis of Glioma through Targeting the Mitochondrial DNA with Bioinformatic Methods. Ijms 20 (18), 4643. doi:10.3390/ijms20184643
Zhang, L., Li, J., Yin, K., Jiang, Z., Li, T., Hu, R., et al. (2019b). Computed Tomography Angiography-Based Analysis of High-Risk Intracerebral Haemorrhage Patients by Employing a Mathematical Model. BMC Bioinformatics 20 (7), 193. doi:10.1186/s12859-019-2741-5
Zhang, L., Liu, G., Kong, M., Li, T., Wu, D., Zhou, X., et al. (2019c). Revealing Dynamic Regulations and the Related Key Proteins of Myeloma-Initiating Cells by Integrating Experimental Data into a Systems Biological Model. Bioinformatics 37 (11), 1554–1561. doi:10.1093/bioinformatics/btz542
Zhang, L., Qiao, M., Gao, H., Hu, B., Tan, H., Zhou, X., et al. (2016). Investigation of Mechanism of Bone Regeneration in a Porous Biodegradable Calcium Phosphate (CaP) Scaffold by a Combination of a Multi-Scale Agent-Based Model and Experimental Optimization/validation. Nanoscale 8 (31), 14877–14887. doi:10.1039/C6NR01637E
Zhang, L., Xiao, M., Zhou, J., and Yu, J. (2018). Lineage-associated Underrepresented Permutations (LAUPs) of Mammalian Genomic Sequences Based on a Jellyfish-Based LAUPs Analysis Application (JBLA). Bioinformatics 34 (21), 3624–3630. doi:10.1093/bioinformatics/bty392
Zhang, L., Zhang, L., Guo, Y., Xiao, M., Feng, L., Yang, C., et al. (2021a). MCDB: A Comprehensive Curated Mitotic Catastrophe Database for Retrieval, Protein Sequence Alignment, and Target Prediction. Acta Pharmaceutica Sinica B. doi:10.1016/j.apsb.2021.05.032
Zhang, L., and Zhang, S. (2017). Using Game Theory to Investigate the Epigenetic Control Mechanisms of Embryo Development. Phys. Life Rev. 20, 140–142. doi:10.1016/j.plrev.2017.01.007
Keywords: multi-omics, time series, embedding, Koopman, deep learning
Citation: Liu S, You Y, Tong Z and Zhang L (2021) Developing an Embedding, Koopman and Autoencoder Technologies-Based Multi-Omics Time Series Predictive Model (EKATP) for Systems Biology research. Front. Genet. 12:761629. doi: 10.3389/fgene.2021.761629
Received: 20 August 2021; Accepted: 27 September 2021;
Published: 26 October 2021.
Edited by:
Jiajie Peng, Northwestern Polytechnical University, ChinaReviewed by:
Renchu Guan, Jilin University, ChinaCopyright © 2021 Liu, You, Tong and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Le Zhang, emhhbmdsZTA2QHNjdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.