 Xing Zhang
Xing Zhang Liu Yang1,2
Liu Yang1,2 Hua Li
Hua Li Hai Xiang
Hai Xiang Xingbo Zhao
Xingbo Zhao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 23 November 2021
Sec. Livestock Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.759827
This article is part of the Research TopicEarly Domestication and Artificial Selection of AnimalsView all 10 articles
Cattle, as an important tool for agricultural production in ancient China, have a complex history of domestication and distribution in China. Although it is generally accepted that ancient Chinese taurine cattle originated from the Near East, the explanation regarding their spread through China and whether or not this spread was associated with native aurochs during ancient times are still unclear. In this study, we obtained three nearly complete mitochondrial genomes (mitogenomes) from bovine remains dating back ca. 4,000 years at the Taosi and Guchengzhai sites in North China. For the first time at the mitogenome level, phylogenetic analyses confirmed the approximately 4,000-year-old bovines from North China as taurine cattle. All ancient cattle from both sites belonged to the T3 haplogroup, suggesting their origin from the Near East. The high affinity between ancient samples and southern Chinese taurine cattle indicated that ancient Chinese cattle had a genetic contribution to the taurine cattle of South China. A rapid decrease in the female effective population size ca. 4.65 thousand years ago (kya) and a steep increase ca. 1.99 kya occurred in Chinese taurine cattle. Overall, these results provide increasing evidence of the origin of cattle in the middle Yellow River region of China.
As one of the earliest domesticated animals (Diamond, 2002), cattle not only provide meat, dairy products, and leather but also played an important role in cultivation and other agricultural activities in the past 13,000 years (Ebersbach, 2002). According to their morphological characteristics and living habits, modern cattle are divided into humpless group (taurine, Bos taurus) and humped group (zebu, Bos indicus). Both archeological and genetic evidence have demonstrated that taurine cattle and zebu cattle were independently domesticated from aurochs in the Near East approximately 10,500 years before present (YBP) (Ajmone-Marsan et al., 2010) and in the Indus Valley approximately 8,500 YBP (Loftus et al., 1994), respectively, and then separately spread to the rest of the world following human migrations (Patel, 2009; Felius et al., 2014).
The abundant cattle resources in China, including the aurochs (Bos primigenius) existing in ancient China, as well as the zebu and taurine cattle in modern China, imply a complicated history of formation of Chinese taurine cattle (Qiu, 2006; Zhang et al., 2013; Cai et al., 2014; Brunson et al., 2016b). Analyses of partial mitochondrial DNA (mtDNA) sequences and complete mitochondrial genomes (mitogenomes) of various modern cattle breeds have revealed multiple region-specific maternal lineages [e.g., the T4 haplogroup was found mainly in East Asia (Felius et al., 2014)]. Analyses of mtDNA D-loop sequences revealed that the northern Chinese breeds originated from taurine cattle, the southern breeds originated from zebu cattle, whereas the central Chinese cattle breeds were of hybrid origin (Zhang et al., 2015). Whole-genome sequencing has revealed three different ancestors for the East Asian cattle, including East Asian taurine ancestry, Eurasian taurine ancestry, and Chinese indicine ancestry (Chen et al., 2018). Further genome analyses suggested that at least two historical migration events occurred in Chinese cattle in northern China (Chen et al., 2018).
Although the analyses of these modern specimens based on partial and complete genomes provide reasonable assumption into the study of cattle history, a direct ancient DNA (aDNA) analysis focusing on the ancient time has been valued more highly as it can provide a wealth of direct and solid evidence on the origin and dispersal of cattle species. In recent decades, advances in high throughput sequencing makes breakthroughs in aDNA research, which still performed inadequately. Specifically, analyses of ancient agricultural animal samples including pig, cattle, sheep and dog etc. especially from China, where exists numerous archeological sites, provide significant value to represent the history of animals and their companions, human society.
The earliest known taurine cattle remains in China were found in the Houtaomuga site in Jilin Province 5,500–5,300 YBP (Cai et al., 2018) and supported a significant population expansion of taurine cattle that spread from Northeast Asia into China. Through the discontinuous ancient DNA analysis on ancient cattle remains in China till now, it is widely accepted that Chinese taurine cattle were originated from the Near East (Brunson et al., 2016a; Cai et al., 2018) with two routes: Northwest entrance route and Northeast entrance route. In addition, genomic study of ancient cattle samples from the Shimao site has confirmed that the domestic cattle present at the site are pure East Asian taurine cattle (Chen et al., 2018). Although there is growing evidence that the domestication of Chinese cattle was a non-native process, the increasing excavation of auroch remains has made the origin of Chinese cattle contentious. Particularly, the exist of haplogroup C auroch 10,000 years ago in Northeast China (Zhang et al., 2013) and the evidence of haplogroup (P1a) incorporated into domestic cattle of northeastern Asia (Mannen et al., 2020) make it necessary to determine the species and phylogenetic relationships among suspected bovine remains from different sites in China, as well as to define the role that aurochs play in the development of taurine cattle in China.
Bovine bone remains were collected from the Taosi and Guchengzhai sites. Both sites are located at the lower and middle reaches of the Yellow River drainage basin (Figure 1) and date back to the Longshan Culture period (approximately 4,000 YBP) (Supplementary Table S1). Specifically, the Taosi site is a large urban site located between the Ta’er Mountain and the Fen River, approximately 7.5 km northeast of Xiangfen County, Shanxi Province. The Taosi site dates back 4,350–3,900 years (Brunson et al., 2016a). The paleoenvironmental conditions around the Taosi site were warmer and wetter than now (Wang et al., 2011). Many domestic animals remain, including cattle bones, were found at the Taosi site; these animals were kept primarily as animals of the wealthy inhabitants (Brunson et al., 2016a). The Guchengzhai site is located in Dafanzhuang Village, 35 km southeast of Xinmi City, Henan Province, 1.5 km northeast of the intersection of the Zhenshui River and the Yan River. The Guchengzhai site dates back 4,150–3,950 years. Similarly, the paleoenvironment of the Guchengzhai site was slightly warmer and more humid than now (Xu et al., 2013). Domestic animals, including dogs, pigs, cattle, and sheep, as well as wild animals such as bears, sika deer, and elks were extensively discovered at the Guchengzhai site (Guo, 2018). The detailed information on all samples were shown in Supplementary Table S1. Before aDNA analysis, all samples were morphologically determined to be cattle remains.
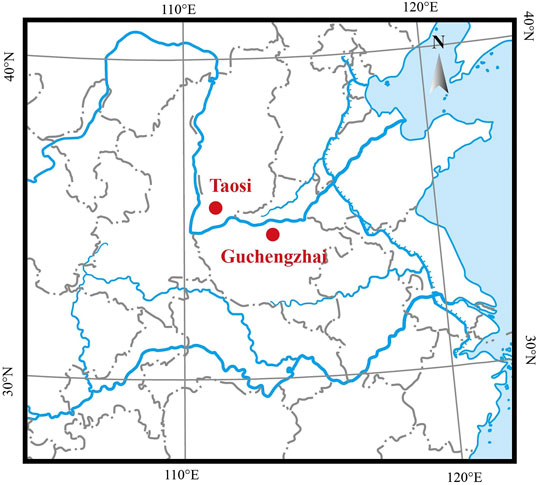
FIGURE 1. Location of the Taosi and Guchengzhai archaeological sites (red dots).
All aDNA experiments were conducted in the Ancient DNA Laboratory at China Agricultural University and repeated independently in the Animal Genetic and Evolutionary Laboratory at Foshan University, following the experimental guidelines to eliminate contamination with modern samples. First, the equipment involved in the experiment was soaked with 5% (v/v) sodium hypochlorite solution and washed with 75% (vol/vol) alcohol, followed by UV irradiation for 1 h. Thereafter, the samples were prepared by cautiously removing the adhering soils on the surface of samples with a drill, followed by washing with 5% (vol/vol) sodium hypochlorite solution and then with double-distilled water and subsequent UV irradiation. The bones were then ground to fine-grained powder using an automatic sample quick grinding machine (Jingxin Industrial Development Co., Ltd., China), and 100–200 mg of powder was subjected to DNA extraction using a silica adsorption column system. At China Agricultural University, 50 μL of extracted DNA was obtained using the QIAamp DNA Investigator Kit (QIAGEN, Germany). At Foshan University, aDNA extraction was performed following the method described by Dabney (Dabney et al., 2013) with some adaptive modifications, including setting the lytic temperature to 45°C and extending the time of incubation with double distilled water (ddH2O) to 10 min.
The mitogenomes of the genus Bos, including Holstein (Bos taurus), zebu (Bos indicus), gayal (Bos frontalis), and yak (Bos grunniens), were obtained using long-range PCR amplification. The PCR products were then used to prepare Bos hybrid baits. A total of 50 ng DNA per sample was used to construct a double-stranded DNA library using the KAPA Hyper Prep Kit for Illumina® platforms (KAPA Biosystems, San Francisco, USA) with minor modifications. The libraries were quantified using a Qubit 4.0 Fluorometer (Life Technologies Corporation, USA) and submitted for subsequent mitogenome enrichment and capture. Four libraries of Taosi samples were equally enriched to a total of 1 ng of DNA with unique adapter index sequences. Thereafter, mitogenome enrichment and hybrid capture were performed using the xGen Hybridization and Wash Kit (Integrated DNA Technologies, Inc., USA). The enriched library was then sequenced with 150 paired end reads (PE150) using the Illumina HiSeq X Ten platform. For the resequencing samples, up to 100 ng DNA per sample was used to prepare the library with the VAHTS Universal Plus DNA Library Prep Kit for MGI (Vazyme Biotech Co., Ltd, China). The qualified libraries were sequenced with PE100 using the MGI platform.
The quality of the raw data was assessed using the FastQC v0.11.9 (Andrews, 2010). Adapter and low-quality reads were filtered using clip&merge v1.7.8 from EAGER pipeline (Peltzer et al., 2016) with the following parameters: -minlength 25, --minquality 30. Before mapping, the first 250 and 500 bp of the taurine cattle reference mitogenome (GenBank accession No.: V00654) were concatenated to the end using CircularGenerate from the EAGER pipeline (Peltzer et al., 2016). The filtered reads were mapped to the circularized reference V00654 using CircularMapper v1.0 (Peltzer et al., 2016), with parameters as -n 0.01, -l 16,500. The resulting file was converted to a bam file and sorted using Samtools v1.9 (Li, 2011). Unmapped reads and reads with mapping quality <25 were removed using Samtools v1.9. Duplicate reads were removed using MarkDuplicates of Picard-tools v2.22.9 (http://broadinstitute.github.io/picard/). The endogenous DNA, coverage rate, and mapping quality distribution were calculated using Qualimap v2.2.2-dev (Okonechnikov et al., 2016). The authenticity of the retrieved sequences was evaluated according to the characteristics of the aDNA (Hofreiter et al., 2001; Willerslev and Cooper, 2005) using the mapDamage software (Jonsson et al., 2013).
Three methods, including Mapping Iterative Assembler (MIAv.1.0) (Green et al., 2008), BCFtools (Li, 2011), and UnifiedGenotyper in the Genome Analysis Tool Kit (GATK, v3.5) (DePristo et al., 2011), were comparatively deployed to generate the mitogenome sequences. For MIA, the filtered bam files were converted to Fastq format using BEDtools (Quinlan and Hall, 2010). The program ma in MIA was used to generate complete sequences. The latter two software packages generate the complete sequence based on the mutation sites relative to the reference sequence using BCFtools consensus and manual generation, respectively. Finally, the complete mitogenome sequence of the ancient samples was obtained by comparing the differences in the complete sequences from different assembly methods and the filtered bam files.
Variant calling was carried out using BCFtools v1.8 mpileup (-d 5000 -Q 20 -a DP, AD, ADF, ADR, INFO/AD, INFO/ADF, INFO/ADR, SP, DV, DP4, DPR, INFO/DPR). Single nucleotide polymorphism (SNP) sites with <3× coverage depth were filtered out. UnifiedGenotyper in GATK3.5 was also used for the variation calling, whereas SNP sites with <3× coverage were discarded. Subsequently, the filtered SNP sites were manually checked and corrected according to the bam files visualized on the Integrative Genomics Viewer (IGV) (Thorvaldsdottir et al., 2013). Eventually, common SNP loci were retained as the true mutation loci.
All the captured and re-sequenced reads for the ancient samples were aligned to the reference mitogenome (GenBank accession No.: V00654) using the online MAFFT software with default parameters (Katoh et al., 2019). Positions embracing missing data or uncertain sites were removed using MEGA 7 (Kumar et al., 2016). Then the ratio of nonsynonymous and synonymous substitutions (dN/dS) was also calculated using MEGA 7. The 3 ancient consensus sequences were aligned to 36 others extant Bovinae mitogenomes from GenBank and called Dataset A (Supplementary Table S2), which included 1 Bison, 1 Bison bonasus, 1 Bison schoetensacki, 1 Bison priscus, 1 Bubalus bubalis, 1 Bubalus depressicornis, 1 Bubalus carabanensis, 1 Bos frontalis, 1 Bos gaurus, 1 Bos grunniens, 1 Bos mutus, 1 Bos javanicus, 3 Bos primigenius, 2 Bos indicus, and 19 Bos taurus. To further identify the haplogroup of the ancient samples, 510 cattle including Bos taurus and Bos indicus were aligned with ancient samples and called Dataset B (Supplementary Table S3).
To construct phylogenetic trees and infer population structure, the nucleotide substitution model was selected using jModelTest 2.1.1 (Darriba et al., 2012), based on the model selection strategy of Bayesian information criteria. Bayesian phylogenetic trees were constructed using MrBayes 3.2.7 (Ronquist et al., 2012), with the following parameters: 20,000,000 generations of Markov Chain Monte Carlo (MCMC) chains, checking every 10,000, sampling every 2,000, and discarding the first 10% as burn in. The consensus tree was constructed using FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Principal component analysis (PCA) was used to further verify the genetic affinities of the ancient samples among the genus Bos using the R Package “adegenet” (Jombart, 2008). Part of dataset A was interrogated to infer demographics using BEAST 2.6.3 (Bouckaert et al., 2014). The ages of the ancient cattle were set at 4,000 YBP, which was the time of the culture in the respective sites of origin. We used Strict Clock as the clock model and Coalescent Bayesian Skyline as a distribution of the tree priors. The MCMC chains were set to 1000,000,000 generations. The log file was checked using Tracer v1.7.1 (Rambaut et al., 2018), and the Bayesian Skyline Plot (BSP) was obtained using Tracer v1.7.1. The optimal nucleotide substitution model was derived using MEGA 7 estimation for the maximum likelihood (ML) tree. Thereafter, an ML tree was built using MEGA 7 with 1,000 bootstrapping replicates and a timetree inferred using the Reltime method (Tamura et al., 2012). The timetree was computed using two calibration constraints from TimeTree (Kumar et al., 2017), including the time of divergence between Bos taurus, Bos primigenius, Bos taurus, and Bos indicus.
T haplogroups of Dataset B was used to calculate population pairwise FST values, the gene flow (Nm) and the net genetic distance using DNAsp 5.10 (Librado and Rozas, 2009). Meanwhile, numbers of haplotypes and variable sites, haplotype diversity (Hd), nucleotide diversity (Pi), and the average number of nucleotide differences (K) were estimated using the same data and software. Besides, T haplogroups of Dataset B was used to conduct PCoA (Principal Coordinate Analysis) among populations. The neighbor net (Bryant and Moulton, 2004) was constructed using splitstree5 (Huson and Bryant, 2006) based on Fst value. PCoA analysis was performed using the OmicStudio tools at https://www.omicstudio.cn/tool. The final result was graphed using R Package “ggplot2” (Hadley, 2016).
A total of eight ancient samples, including four bovines (4,350–3,850 YBP) from the Taosi site and four bovines (4,150–3,100 YBP) from the Guchengzhai site, were subjected to DNA extraction. All qualified aDNA extractions were selected for shotgun sequencing. However, owing to the typically low levels of endogenous DNA in these samples, only one sample from the Guchengzhai site (GCZ4C) obtained sufficient endogenous reads (0.0001%) to yield a near-complete mitogenome. Therefore, the remaining samples were subjected to mtDNA enrichment and hybridization sequencing using Bos hybrid baits. Total sequences of the near-complete mitogenome for the two samples from the Taosi site (TS1C and TS2C) were retrieved after enrichment using baits. After capture sequencing, the endogenous mtDNA read ratio increased to 0.3599% and 0.0474% for TS1C and TS2C, respectively (Table 1), resulting in the recovery of three complete ancient mitogenomes for late Neolithic archeological sites in the Middle Yellow River region. The average length of hybrid capture reads was approximately 140 bp, and the average length of resequencing reads was approximately 57 bp (Supplementary Figure S1). Damage pattern analyses showed a high C–T mutation at the 5′ end and a high G–A mutation at the 3′ end (Supplementary Figure S2), showing typical damage and fragmentation patterns characteristic of endogenous aDNA.
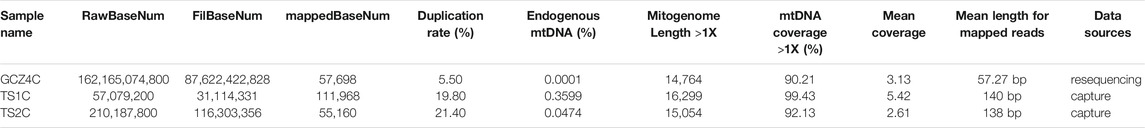
TABLE 1. Sequencing information of three ancient samples.
By considering the effect on the integrity and accuracy of ancient mitogenome assembly, we combined three methods including GATK, BCFtools and MIA to generate mitogenome sequences. The number of duplicated sequences varied from 5.50% to 21.6% (Table 1). As a result, we obtained a 16,299 bp (coverage 99.43%) mitogenome sequence for TS1C, a 15,054 bp (coverage 92.13%) sequence for TS2C, and a 14,764 bp (coverage 90.21%) for GCZ4C (Table 1). SNP sites with ≥3× coverage were used in subsequent analyses (Supplementary Table S4).
To determine the species of the ancient bovine samples, the near-complete mitogenome sequences were combined with 36 extant Bovinae mitogenomes, including the genus Bison, Bubalus, and Bos (Supplementary Table S2). The phylogenetic trees illustrated all three ancient samples belonging to the genus Bos rather than Bison or Bubalus, specifically close to the species Bos taurus (Figure 2 and Supplementary Figure S3).
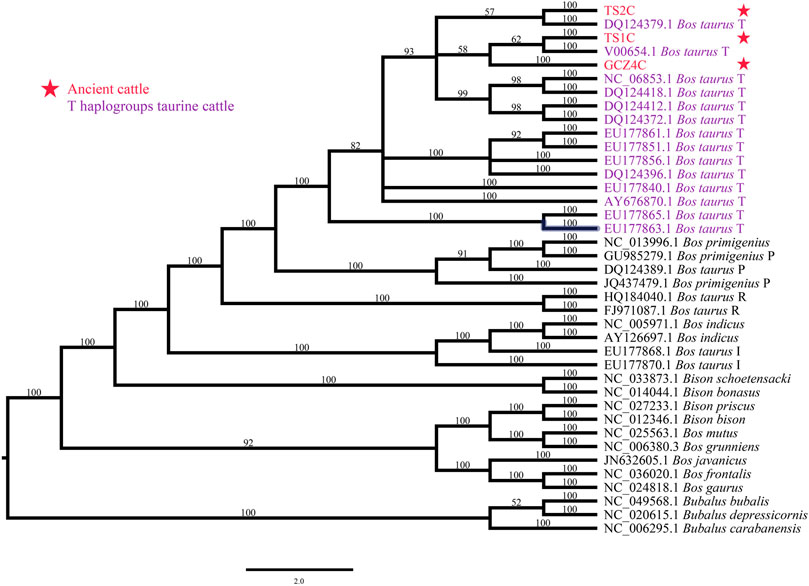
FIGURE 2. Species identification though phylogenetic analysis. ID marked with red font and star represents the ancient sample, ID with purple font represents T-haplogroup taurine cattle.
The mutation sites for each sample were discovered by alignment against the taurine cattle reference mitogenome (GenBank accession no.: V00654) (Supplementary Table S4). All ancient mitogenomes contained 13 coding genes, of which COX3 had the most variable sites and the most non-synonymous SNPs. The values of dN/dS of all genes except COX3 and ND5 were lower than 1 (Supplementary Table S5), suggesting potential purifying selection on the mitochondrial functions in the ancient bovine in North China. Based on the key SNPs in the mitogenome, all three ancient samples were classified as haplogroup T3. Specifically, TS1C from the Taosi site belonged to haplotype T3k, while another sample, TS2C, from the Taosi site and GCZ4C from the Guchengzhai site belonged to haplotype T3n (Supplementary Table S4).
To further investigate the phylogenetic relationships among the approximately 4,000-year-old cattle in North China, a Bayesian phylogenetic tree was inferred with 510 extant mitogenome sequences of Bos taurus and Bos indicus around the world (Supplementary Table S3). The Bayesian tree showed that all ancient cattle were on the T3 branch (Supplementary Figure S4). PCA (Supplementary Figure S5A and S5B) and PCoA (Supplementary Figure S6A) supported the phylogeny of the T3 haplogroup of the ancient samples. The population pairwise FST values, the gene flow, and the net genetic distance, illustrated that the ancient Chinese cattle had the closest genetic relationship to Chinese taurine cattle, followed by populations from East Asia, Europe, West Asia, America, and Africa (Table 2). Further subdivision of Chinese taurine cattle according to region showed that population in T haplogroups had high haplotype and nucleotide diversity (Table 3), and the ancient Chinese cattle were more closely related to the south Chinese taurine cattle than the north taurine Chinese cattle (Table 2 and Supplementary Table S6). Moreover, PCA (Figure 3) and PCoA (Supplementary Figure S6B) analysis of ancient samples and T3/T4 cattle suggested that TS1C was genetically close to modern Bos taurus from Southern China, while TS2C and GCZ4C were closer to cattle from Europe and East Asia.
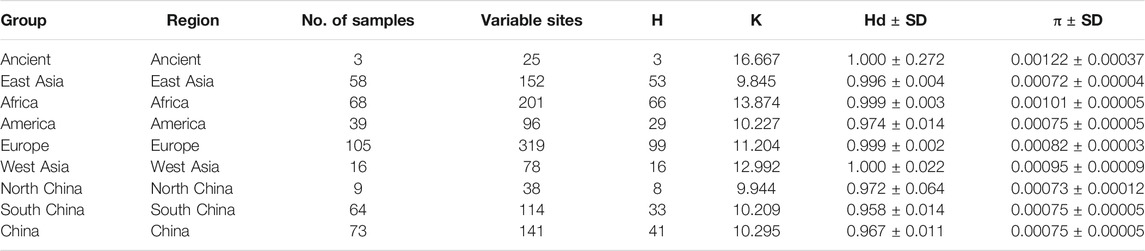
TABLE 2. The haplotype and nucleotide diversity of T haplogroup cattle in dataset B.
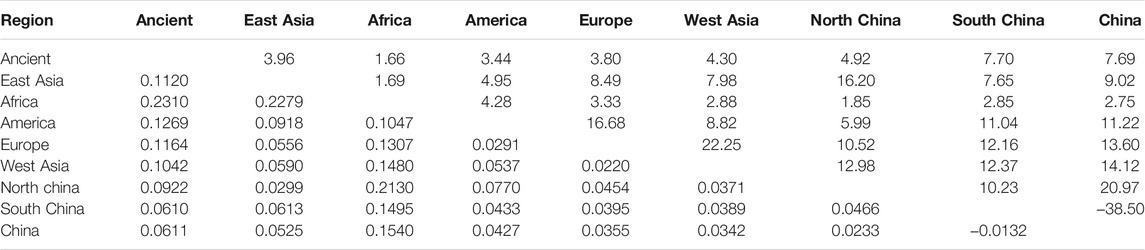
TABLE 3. Population pairwise Fst values (the below diagonal) and gene flow (Nm, the above diagonal) among T haplogroup of dataset B and ancient samples.
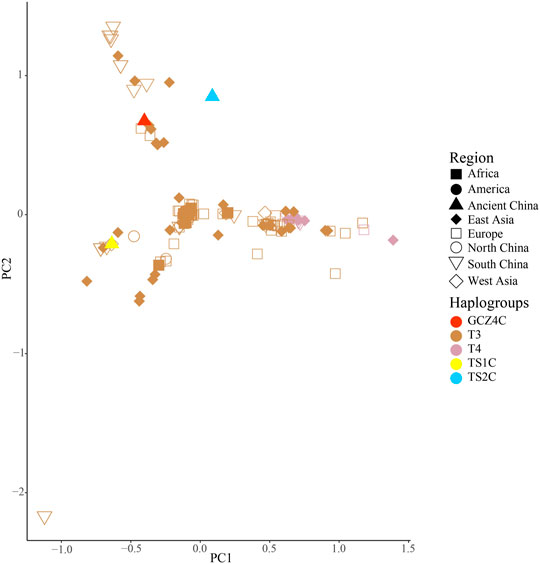
FIGURE 3. PCA analysis of ancient samples and T3/T4 species haplogroup Bos.
To assess the population dynamics of taurine cattle in China, a BSP was produced using 25 mitogenomes. The BSP points to a rapid decrease in the female effective population size approximately 4.65 kya and a steep increase in population approximately 1.99 kya (Figure 4). To infer the divergence time between ancient and modern cattle, a ML tree was constructed with 3 Bos primigenius and 25 Bos taurus cattle, using 1 Bison bonasus as the outgroup (Figure 5). Concerning the differentiation of the major branches in the phylogenetic tree, the T3 and T4 sub-lineages possibly emerged approximately 7.44 kya, and at this time, the haplotype of sample TS1C appeared among the early domesticated cattle. TS2C, another sample from the Taosi site, was divergent to modern populations approximately 4.10 kya, in accordance with the period of rapid decrease in the female effective population size. GCZ4C was divergent to the modern T3n taurine population approximately 0.86 kya, suggesting possible long-term dilution of the ancient northern Chinese cattle.
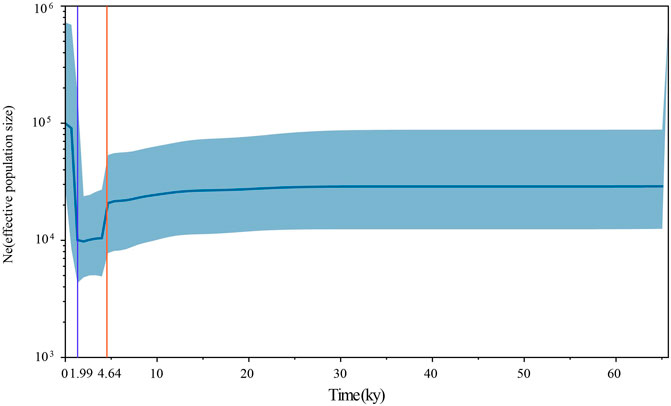
FIGURE 4. The Bayesian Skyline Plot of ancient and modern Bos. The Y-axis represents the effective number of populations, and the X-axis represents the timeline. The orange line shows the timepoint of effective population decline and the blue line shows the timepoint of the rapid increase of effective population.
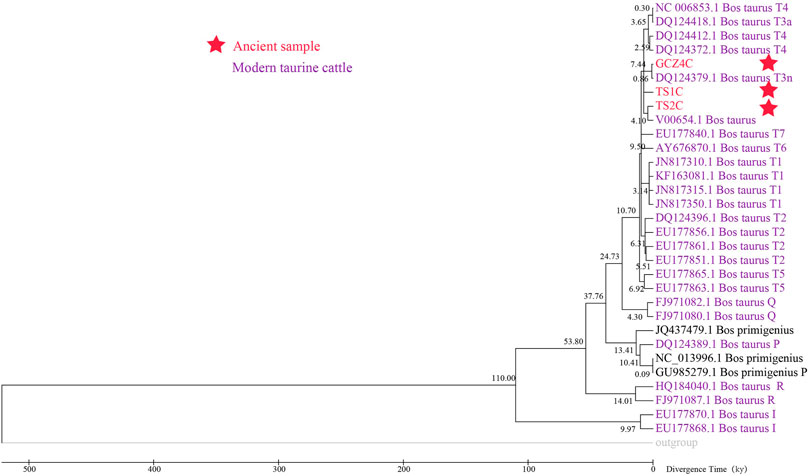
FIGURE 5. The Maximum Likelihood timetree of ancient and modern Bos.
Cattle genome research suggests that the Chinese taurine cattle originated from the Near East (Cai et al., 2014; Cai et al., 2018; Verdugo et al., 2019). According to the specific region where cattle remains were unearthed in China, it is believed that the earliest domesticated cattle in China occurred in northwestern China approximately 5,000–4,000 YBP (Flad et al., 2009). In addition, aDNA analysis of ancient Chinese cattle remains indicated that the cattle in northern China were taurine cattle since the Bronze Age (Cai et al., 2014). However, the abundant auroch remains prior to 5,000 YBP recovered from extensive regions in China suggest a potential early utilization and native domestication of Chinese bovines in the early Neolithic period (Zhang et al., 2013). Evidence from aDNA analysis of indigenous aurochs in central and northeast China during the Neolithic period adds to the uncertainty of the origins of the Chinese taurine cattle (Brunson et al., 2016b).
In this study, we obtained three nearly complete ancient mitogenomes from two ∼4,000 YBP archeological sites in North China and, for the first time, reported the complete mitogenomes from ancient bovines in this region. By determining all ancient samples as taurine cattle, no zebu population was found in these sites, providing further evidence that zebu cattle did not spread to northern China 4,000 YBP (Yue et al., 2014).
As one of the representative Longshan culture sites in China, the Taosi site is unique in that it contains numerous animal remains, such as pig mandibles, which proved that the ancestors living at the Taosi site mastered advanced animal husbandry technologies and lived a long-term, settled agricultural life (Nu, 2013). Different analyses of animal remains at the Taosi site help to understand the relationship between humans and animals under agricultural development in China. Isotopic analysis of the bones of pigs, cattle, and dogs indicated that the staple food of cattle at the Taosi site was a by-product of millet (Chen et al., 2012). Meanwhile, strontium isotope analysis indicated a mix of local and non-local animals at the Taosi site (Zhao et al., 2011). Additionally, the cattle were used for a variety of craft products at the Taosi site (Brunson et al., 2016a).
In our study, we analyzed the domestication status and genetic diversity of Chinese taurine cattle at the Taosi site at the level of the ancient mitogenome, which could be more accurate and direct than modern specimens. Both PCA, PCoA and phylogenetic analysis demonstrated that the ancient individuals of the Taosi site belonged to the T3 haplogroup, suggesting their origin from the Near East (Achilli et al., 2008), which was consistent with studies on mtDNA fragments (Cai et al., 2014; Brunson et al., 2016b). As shown, TS2C was localized to haplogroup T3n from East Asia, defined by 16119C, which probably had a founder effect (Cai et al., 2014). These data indicated that the appearance of ancient domestic cattle in northern China predated the Taosi site. A previous study based on a 294 bp mtDNA sequence showed that 15 ancient cattle in the Taosi site constituted 5 haplotypes (Cai et al., 2014). In our study, TS1C and TS2C originated from the same archeological site or even the same trench, but their haplotypes were different. This result reveals a high level of genetic diversity in the early cattle population at the Taosi site. TS1C diverged from the T3 haplogroup approximately ∼7.44 kya, while TS2C separated from the T3 haplogroup ∼4.10 kya, suggesting a rich diversity of cattle and the mixed presence of native and non-native cattle at the Taosi site.
The Guchengzhai site, dating back to the late period of the third Wangwan culture from Longshan culture in Henan Province, is one of the best-preserved ancient cities (Cai, 2002). A large number of wine vessels and animal remains, including pigs, cattle, and sheep have been found (Cai, 2008; Xu et al., 2013), indicating a locally developed agricultural level. However, genetic analysis of cattle remains at the Guchengzhai site has not been conducted. Through aDNA analysis, we generated the first complete mitogenome for Guchengzhai cattle and found that the bovine remains from this site belonged to taurine cattle. Interestingly, GCZ4C carries the same haplogroup (T3) as TS2C from the Taosi site, which has dominating founder effect in East Asian domestic cattle. Therefore, the similar genetic background shared by ancient cattle at Taosi and Guchengzhai site may indicate the early arrival of cattle at the northern China before the Longshan Culture period.
The FST values showed that ancient Chinese cattle have the closest affinity to modern Chinese cattle, indicating possible genetic contribution of ancient Chinese cattle populations to modern populations. Previously, whole-genome analysis of an ancient cattle from the Shimao site indicated that there were at least two migration events that occurred in Northern China (Chen et al., 2018). The closer genetic relationship of ancient Chinese cattle with the southern Chinese taurine than with the northwest China taurine may suggest that the ancient samples had genetic contributions to modern southern Chinese taurine cattle, and the northern taurine cattle may have been replaced by other taurine cattle. The BSP in our study also showed that the effective population size of cattle began to decline sharply approximately 4,500 YBP, in line with the arrival of taurine cattle into China. We also found that the ancient taurine cattle (TS2C) were separated approximately 4,000 YBP, which is a crucial period in the study of animal domestication in ancient China (Flad et al., 2009; Zhang et al., 2013; Brunson et al., 2016b; Cai et al., 2018). Meanwhile, the rapid increase in the effective cattle population approximately 1,900 YBP was also linked to the acceleration of communication between East and West at that time. During this period, the Han Empire in ancient China established the Silk Road to communicate with Western civilization. This information also suggests that cattle may have entered China via the Silk Road, increasing the effective population size of the Chinese cattle population.
In this study, we for the first time obtained three near-complete mitogenomes of cattle about 4,000 YBP from North China and confirmed the ∼4,000-year-old bovine from North China as taurine cattle. All ancient cattle from Taosi and Guchengzhai sites belonged to the T3 haplogroup, suggesting their origin from Near East. The high affinity between ancient samples and southern Chinese taurine cattle indicated that the ancient Chinese cattle had a genetic contribution to taurine cattle of South China. A rapid decrease in the female effective population size ca. 4.65 kya and a steep increase ca. 1.99 kya occurred in Chinese taurine cattle.
The original contributions presented in the study are publicly available in the China National GeneBank under accession number CNP0002141.
XBZ and HX designed this project; XZ, LY, and LH performed the experiments; XZ, LY, LH, HL, and HX analyzed the data; XZ, LY, HX, and XBZ interpreted the data and drafted the manuscript. All authors contributed critically to the drafts and gave final approval for publication.
This work was funded by the Foundation for Distinguished Young Talents (2019KQNCX167) and Creative Team (2019KCXTD006) in Higher Education of Guangdong, the National Natural Science Foundation of China (31961133031), the Guangdong Provincial Key Laboratory of Animal Molecular Design and Precise Breeding (2019B030301010), and the Scientific Research Fund for Postdoc of Foshan City (BKS209071). The funding bodies contributed nothing to the study design, data analyses, data interpretation, or manuscript preparation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We express sincere thanks to Prof. Jing Yuan (Institute of Archaeology, Chinese Academy of Social Science) for generously providing the ancient samples, Prof. Zhenglong Gu, Prof. Yi Zhang and Ms. Yumei Wu (China Agricultural University) for great help with experiment.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.759827/full#supplementary-material
Supplementary Figure S1 | Distribution of fragment lengths of ancient DNA from three ancient samples.
Supplementary Figure S2 | The DNA damage patterns of the ancient samples. Misincorporations frequencies are represented by the first and last 25 nucleotides of reads aligned to the bovine mitochondrial reference genome V00654.
Supplementary Figure S3 | Species identification by phylogenetic analysis of three ancient samples with 36 extant Bovinae mitogenomes, respectively.
Supplementary Figure S4 | Phylogenetic tree of three ancient mitogenomes and 510 cattle of Bos genus.
Supplementary Figure S5 | PCA of ancient samples and species from the Bos genus. (A) shows the relationship of three ancient samples to all Bos genus while (B) shows their relationship to the T haplogroup Bos genus.
Supplementary Figure S6 | PCoA of ancient samples and species from the Bos genus. (A) shows the relationship of three ancient samples to the T haplogroup Bos genus while (B) shows their relationship to the T3/T4 species haplogroup Bos.
Achilli, A., Olivieri, A., Pellecchia, M., Uboldi, C., Colli, L., Al-Zahery, N., et al. (2008). Mitochondrial Genomes of Extinct Aurochs Survive in Domestic Cattle. Curr. Biol. 18 (4), R157–R158. doi:10.1016/j.cub.2008.01.019
Ajmone-Marsan, P., Garcia, J. F., and Lenstra, J. A. (2010). On the Origin of Cattle: How Aurochs Became Cattle and Colonized the World. Evol. Anthropol. 19 (4), 148–157. doi:10.1002/evan.20267
Andrews, S. (2010). FASTQC. A Quality Control Tool for High Throughput Sequence Data. Cambridge: Babraham Institute. Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C.-H., Xie, D., et al. (2014). BEAST 2: a Software Platform for Bayesian Evolutionary Analysis. Plos Comput. Biol. 10 (4), e1003537. doi:10.1371/journal.pcbi.1003537
Brunson, K., He, N., and Dai, X. (2016a). Sheep, Cattle, and Specialization: New Zooarchaeological Perspectives on the Taosi Longshan. Int. J. Osteoarchaeol. 26 (3), 460–475. doi:10.1002/oa.2436
Brunson, K., Zhao, X., He, N., Dai, X., Rodrigues, A., and Yang, D. (2016b). New Insights into the Origins of oracle Bone Divination: Ancient DNA from Late Neolithic Chinese Bovines. J. Archaeological Sci. 74, 35–44. doi:10.1016/j.jas.2016.08.008
Bryant, D., and Moulton, V. (2004). Neighbor-Net: an Agglomerative Method for the Construction of Phylogenetic Networks. Mol. Biol. Evol. 21 (2), 255–265. doi:10.1093/molbev/msh018
Cai, D., Sun, Y., Tang, Z., Hu, S., Li, W., Zhao, X., et al. (2014). The Origins of Chinese Domestic Cattle as Revealed by Ancient DNA Analysis. J. Archaeological Sci. 41, 423–434. doi:10.1016/j.jas.2013.09.003
Cai, D., Zhang, N., Zhu, S., Chen, Q., Wang, L., Zhao, X., et al. (2018). Ancient DNA Reveals Evidence of Abundant Aurochs (Bos Primigenius) in Neolithic Northeast China. J. Archaeological Sci. 98, 72–80. doi:10.1016/j.jas.2018.08.003
Cai, Q. (2002). Guchengzhai Longshan chengzhi yu zhongyuan wenming de xingcheng. Cultural Relics of Central China 46 (6), 27–32.
Cai, Q. (2008). Investigation of the Pre-history Agriculture and the Structure of Man's Food in the Shuangji River basin. J. Huanghe S&t Uni. 10 (5), 46–49. doi:10.3969/j.issn.1008-5424.2008.05.012
Chen, N., Cai, Y., Chen, Q., Li, R., Wang, K., Huang, Y., et al. (2018). Whole-genome Resequencing Reveals World-wide Ancestry and Adaptive Introgression Events of Domesticated Cattle in East Asia. Nat. Commun. 9 (1), 2337. doi:10.1038/s41467-018-04737-0
Chen, X., Yuan, J., Hu, Y., He, N., and Wang, C. (2012). Study on Animal Husbandry in Taosi Site: Evidence Derived from Stable Isotope Analysis. Archaeology 75, 843–850.
Dabney, J., Knapp, M., Glocke, I., Gansauge, M.-T., Weihmann, A., Nickel, B., et al. (2013). Complete Mitochondrial Genome Sequence of a Middle Pleistocene Cave bear Reconstructed from Ultrashort DNA Fragments. Proc. Natl. Acad. Sci. U. S. A. 110 (39), 15758–15763. doi:10.1073/pnas.1314445110
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 9 (8), 772. doi:10.1038/nmeth.2109
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 43 (5), 491–498. doi:10.1038/ng.806
Diamond, J. (2002). Evolution, Consequences and Future of Plant and Animal Domestication. Nature 418 (6898), 700–707. doi:10.1038/nature01019
Ebersbach, R. (2002). Von Bauern und Rindern: Eine Ökosystemanalyse zur Bedeutung der Rinderhaltung in bäuerlichen Gesellschaften als Grundlage zur Modellbildung im Neolithikum. Basel: Schwabe.
Felius, M., Beerling, M.-L., Buchanan, D., Theunissen, B., Koolmees, P., and Lenstra, J. (2014). On the History of Cattle Genetic Resources. Diversity 6 (4), 705–750. doi:10.3390/d6040705
Flad, R., Yuan, J., and Li, S. (2009). On the Source and Features of the Neolithic Domestic Animals in the Gansu and Qinghai Region, China. Archaeology 5 (8), 80–86.
Green, R. E., Malaspinas, A.-S., Krause, J., Briggs, A. W., Johnson, P. L. F., Uhler, C., et al. (2008). A Complete Neandertal Mitochondrial Genome Sequence Determined by High-Throughput Sequencing. Cell 134 (3), 416–426. doi:10.1016/j.cell.2008.06.021
Guo, R. (2018). Archaeological Observations on the Food Structure of the Prehistoric Ancestors in Xinmi of Henan. Agri. Archaeol. 3, 26–32.
Hadley, W. (2016). ggplot2-Elegant Graphics for Data Analysis. New York: Springer International Publishing.
Hofreiter, M., Serre, D., Poinar, H. N., Kuch, M., and Pääbo, S. (2001). Ancient DNA. Nat. Rev. Genet. 2 (5), 353–359. doi:10.1038/35072071
Huson, D. H., and Bryant, D. (2006). Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 23 (2), 254–267. doi:10.1093/molbev/msj030
Jombart, T. (2008). Adegenet: a R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 24 (11), 1403–1405. doi:10.1093/bioinformatics/btn129
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. F., and Orlando, L. (2013). mapDamage2.0: Fast Approximate Bayesian Estimates of Ancient DNA Damage Parameters. Bioinformatics 29 (13), 1682–1684. doi:10.1093/bioinformatics/btt193
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief Bioinform. 20 (4), 1160–1166. doi:10.1093/bib/bbx108
Kumar, S., Stecher, G., Suleski, M., and Hedges, S. B. (2017). TimeTree: a Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 34 (7), 1812–1819. doi:10.1093/molbev/msx116
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi:10.1093/molbev/msw054
Li, H. (2011). A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 27 (21), 2987–2993. doi:10.1093/bioinformatics/btr509
Librado, P., and Rozas, J. (2009). DnaSP V5: a Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics 25 (11), 1451–1452. doi:10.1093/bioinformatics/btp187
Loftus, R. T., MacHugh, D. E., Bradley, D. G., Sharp, P. M., and Cunningham, P. (1994). Evidence for Two Independent Domestications of Cattle. Proc. Natl. Acad. Sci. U. S. A. 91 (7), 2757–2761. doi:10.1073/pnas.91.7.2757
Mannen, H., Yonezawa, T., Murata, K., Noda, A., Kawaguchi, F., Sasazaki, S., et al. (2020). Cattle Mitogenome Variation Reveals a post-glacial Expansion of Haplogroup P and an Early Incorporation into Northeast Asian Domestic Herds. Sci. Rep. 10 (1), 20842. doi:10.1038/s41598-020-78040-8
Nu, H. (2013). “The Longshan Period Site of Taosi in Southern Shanxi Province,” in A Companion to Chinese Archaeology. Oxford: Blackwell Publishing Ltd., 255–277. doi:10.1002/9781118325698.ch13
Okonechnikov, K., Conesa, A., and García-Alcalde, F. (2016). Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics 32 (2), 292–294. doi:10.1093/bioinformatics/btv566
Patel, A. K. (2009). Occupational Histories, Settlements, and Subsistence in Western India: What Bones and Genes Can Tell Us about the Origins and Spread of Pastoralism. Anthropozoologica 44 (1), 173–188. doi:10.5252/az2009n1a8
Peltzer, A., Jäger, G., Herbig, A., Seitz, A., Kniep, C., Krause, J., et al. (2016). EAGER: Efficient Ancient Genome Reconstruction. Genome Biol. 17, 60. doi:10.1186/s13059-016-0918-z
Qiu, Z. (2006). Quaternary Environmental Changes and Evolution of Large Mammals in North China. Vertebrata Palasiatica 44 (2), 109–132. doi:10.3969/j.issn.1000-3118.2006.02.001
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 26 (6), 841–842. doi:10.1093/bioinformatics/btq033
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 67 (5), 901–904. doi:10.1093/sysbio/syy032
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 61 (3), 539–542. doi:10.1093/sysbio/sys029
Tamura, K., Battistuzzi, F. U., Billing-Ross, P., Murillo, O., Filipski, A., and Kumar, S. (2012). Estimating Divergence Times in Large Molecular Phylogenies. Proc. Natl. Acad. Sci. U. S. A. 109 (47), 19333–19338. doi:10.1073/pnas.1213199109
Thorvaldsdottir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): High-Performance Genomics Data Visualization and Exploration. Brief. Bioinform. 14 (2), 178–192. doi:10.1093/bib/bbs017
Verdugo, M. P., Mullin, V. E., Scheu, A., Mattiangeli, V., Daly, K. G., Maisano Delser, P., et al. (2019). Ancient Cattle Genomics, Origins, and Rapid Turnover in the Fertile Crescent. Science 365 (6449), 173–176. doi:10.1126/science.aav1002
Wang, S., Wang, Z., and He, N. (2011). Study on the Charcoal Samples in the Excavations of Taosi Site. Archiaeology 3, 91–96.
Willerslev, E., and Cooper, A. (2005). Review Paper. Ancient DNA. Proc. R. Soc. B. 272 (1558), 3–16. doi:10.1098/rspb.2004.2813
Xu, J., Mo, D., Wang, H., and Zhou, K. (2013). Preliminary Research of Environment Archaeologyin Zhenshui River, Xinmi City, Henan. Quat. Sci. 33 (5), 954–964. doi:10.3969/j.issn.1001-7410.2013.05.13
Yue, X., Li, R., Liu, L., Zhang, Y., Huang, J., Chang, Z., et al. (2014). When and How Did Bos indicus Introgress into Mongolian Cattle? Gene 537 (2), 214–219. doi:10.1016/j.gene.2013.12.066
Zhang, H., Paijmans, J. L. A., Chang, F., Wu, X., Chen, G., Lei, C., et al. (2013). Morphological and Genetic Evidence for Early Holocene Cattle Management in Northeastern China. Nat. Commun. 4, 2755. doi:10.1038/ncomms3755
Zhang, L., Jia, S., Plath, M., Huang, Y., Li, C., Lei, C., et al. (2015). Impact of ParentalBos taurusandBos indicusOrigins on Copy Number Variation in Traditional Chinese Cattle Breeds. Genome Biol. Evol. 7 (8), 2352–2361. doi:10.1093/gbe/evv151
Keywords: Bos taurus, Ancient DNA, mitochondrial genome, domestication, longshan culture
Citation: Zhang X, Yang L, Hou L, Li H, Xiang H and Zhao X (2021) Ancient Mitogenomes Reveal the Domestication and Distribution of Cattle During the Longshan Culture Period in North China. Front. Genet. 12:759827. doi: 10.3389/fgene.2021.759827
Received: 17 August 2021; Accepted: 29 October 2021;
Published: 23 November 2021.
Edited by:
Ino Curik, University of Zagreb, CroatiaReviewed by:
Takahiro Yonezawa, Tokyo University of Agriculture, JapanCopyright © 2021 Zhang, Yang, Hou, Li, Xiang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xingbo Zhao, emh4YkBjYXUuZWR1LmNu; Hai Xiang, eGhAZm9zdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.