 Qiao Zhang1
Qiao Zhang1 Jianli Zhou
Jianli Zhou Lin Han
Lin Han Shaoming Zhou
Shaoming Zhou- 1Division of Gastroenterology, Shenzhen Children’s Hospital, Shenzhen, China
- 2Running Gene Inc., Beijing, China
Background: Trichohepatoenteric syndrome (THES) is a rare disease that mainly causes intractable diarrhea. It is classified into THES1 and THES2, which are associated with the tetratricopeptide repeat domain 37 (TTC37) gene and Ski2-like RNA helicase (SKIV2L) gene, respectively. THES is not very prevalent in China or worldwide, but new cases have increasingly been reported.
Methods and Results: Here, we report the clinical and genetic information of a 1.5-month-old girl who was admitted to our hospital due to diarrhea and failure to thrive. Whole-exome sequencing (WES) revealed novel compound-heterozygous variants of the SKIV2L gene, c.3602_3609delAGCGCCTG (p.Q1201Rfs*2), and c.1990A > G (p.T664A) as the causative factors, which were confirmed via Sanger sequencing. Upon continuous feeding with an amino-acid formula through a gastric tube and parenteral nutrition, the patient resumed thriving and her stool frequency decreased.
Conclusion: We report a girl carrying novel variants of the SKIV2L gene that cause THES2, thereby providing valuable information on the diagnosis of THES2 and expanding the spectrum of disease-causing SKIV2L mutations.
Introduction
Trichohepatoenteric syndrome (THES), initially called syndromic diarrhea (SD), was first described by Stankler et al., in 1982 and coined by Verloes et al., in 1997 (Stankler et al., 1982; Verloes et al., 1997). The typical symptoms of THES are intrauterine growth retardation and neonatal intractable diarrhea, leading to poor weight gain and failure to thrive. Patients may also have hair abnormalities (trichorrhexis nodosa) or facial dysmorphisms (Verloes et al., 1997). Other clinical characteristics, including immunodeficiency, skin abnormalities, hepatic involvement, intellectual disability, and congenital heart disease, may also present. In Europe, the estimated prevalence of THES is 1:1,000,000, and the 10-years mortality rate is >50% (Fabre et al., 2013; Fabre et al., 2017). The mainstream management of THES is mainly based on parenteral nutrition and immunoglobulin supplementation.
THES is classified into THES1 (OMIM #222470) (69% of the cases) and THES2 (OMIM #614602) (31%). THES1 is caused by homozygous or compound-heterozygous mutations in the tetratricopeptide repeat domain 37 (TTC37) gene, whereas THES2 is associated with homozygous or compound-heterozygous mutations in the Ski2-like RNA helicase (SKIV2L) gene (Hartley et al., 2010; Fabre et al., 2012; Bourgeois et al., 2018). The clinical manifestations of these two types are identical. Two-thirds of reported THES cases were caused by pathogenic variants in TTC37, and the others were associated with mutations in the SKIV2L gene (Bourgeois et al., 2018). Both genes encode proteins that are components of the human super killer (Ski) complex, a cofactor of the RNA exosome, playing roles in the degradation of aberrant mRNAs (Schneider and Tollervey, 2013).
The SKIV2L gene (OMIM #600478) is located on chromosome 6p21.33 and has 28 exons. It encodes an RNA helicase (helicase SKI2W) of 137 kDa comprising 1,246 amino acids (Dangel et al., 1995; Lee et al., 1995; Yang et al., 1998) and contains a helicase superfamily ATP-binding domain that is involved in exosome-mediated RNA decay (Bourgeois et al., 2018). Previous reports have shown the associations of SKIV2L with several diseases, including age-related macular degeneration, inflammatory bowel disease, immunodeficiency (primary or common variable), and mitochondrial diseases (McKay et al., 2009; Kammermeier et al., 2014; van Schouwenburg et al., 2015; Stray-Pedersen et al., 2017; Riley et al., 2020). However, most mutations in SKIV2L have been reported to be responsible for THES2. To date, 45 SKIV2L mutations have been reported to the Human Gene Mutation Database (HGMD v2021.8) (Stenson et al., 2017), including 35 mutations associated with THES2. Of these THES2-causing mutations, only three were detected in the Chinese population (Zheng et al., 2016; Fung et al., 2020). Herein, we report a THES2 patient with two novel compound-heterozygous variants of the SKIV2L gene.
Methods
Clinical Examination
The medical history of this patient was provided by her parents and included the birth history, clinical manifestations, and diagnostic and therapeutic actions. Physical examination and laboratory tests were performed, including blood and fecal routine tests, fecal culture, and hepatic, renal, and immunological function tests. Imagological examinations, including abdominal ultrasound, cardiac Doppler ultrasonography, and brain magnetic resonance imaging (MRI), were also applied.
Whole-Exome Sequencing
Whole-exome sequencing (WES) and Sanger sequencing were performed to identify the causative genes. Peripheral blood samples were collected from the patient and her parents and sent to Running Gene Inc. (Beijing, China). The whole sequencing process was described in a previous study (Chen et al., 2020). DNA samples were extracted from the blood samples, qualified, and fragmented into 200–300 bp fragments for library preparation. Probes were hybridized with the prepared libraries to capture exomes, according to the protocol of the Agilent Sure Select XT2 Target Enrichment System (Agilent, Santa Clara, CA). Captured DNA samples were sequenced on Novaseq 6,000 platform (Illumina, San Diego, CA). Raw data in FASTQ format were trimmed and filtered for quality control by using fastp (Chen et al., 2018). The qualified reads were aligned to human reference sequence GRCh37/hg19 by using BWA (Li and Durbin, 2009). Single-nucleotide variants (SNVs) and insertions/deletions (indels) were called out using GATK (Van der Auwera et al., 2013). All the called variants were annotated based on the genetic databases, including ExAC v1.0 (Lek et al., 2016), gnomAD v2.1.1 (Karczewski et al., 2020), ESP6500SI-V2 (Fu et al., 2013), 1kGenomes v3.7.6 (Auton et al., 2015), HGMD v2021.8 (Stenson et al., 2017), ClinVar (Landrum and Kattman, 2018), China National GeneBank (CNGB), and an in-house database (Access date: August 31, 2021). Variants with high allele frequency (>1%) were filtered out. The pathogenicity of variants was assessed based on the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015). The SKIV2L mutations were finally selected based on its clinical relevance and pathogenicity.
Results
Case Presentation
A 1.5-month-old girl was admitted to our hospital due to persistent diarrhea and failure to thrive. She was the only child of her non-consanguineous parents without any familial history of diarrhea. She had experienced no recurrent infections before. She was born at 39 gestational weeks. Her birth height was 48 cm (10th percentile), and her birth weight was 2.5 kg (below the 3rd percentile) (intrauterine growth retardation). She received mixed feeding (regular formula and breast milk) at birth but was stunted at 1.5 months of age (height, 50 cm, below the 3rd percentile; weight, 3.16 kg, below the 3rd percentile) (Figure 1A). She had non-bloody watery stools seven to eight times a day and poor weight gain since birth. Deep hydrolysis formula and amino-acid formula were administered after diagnosis of allergy to cow-milk protein. However, her condition did not improve.
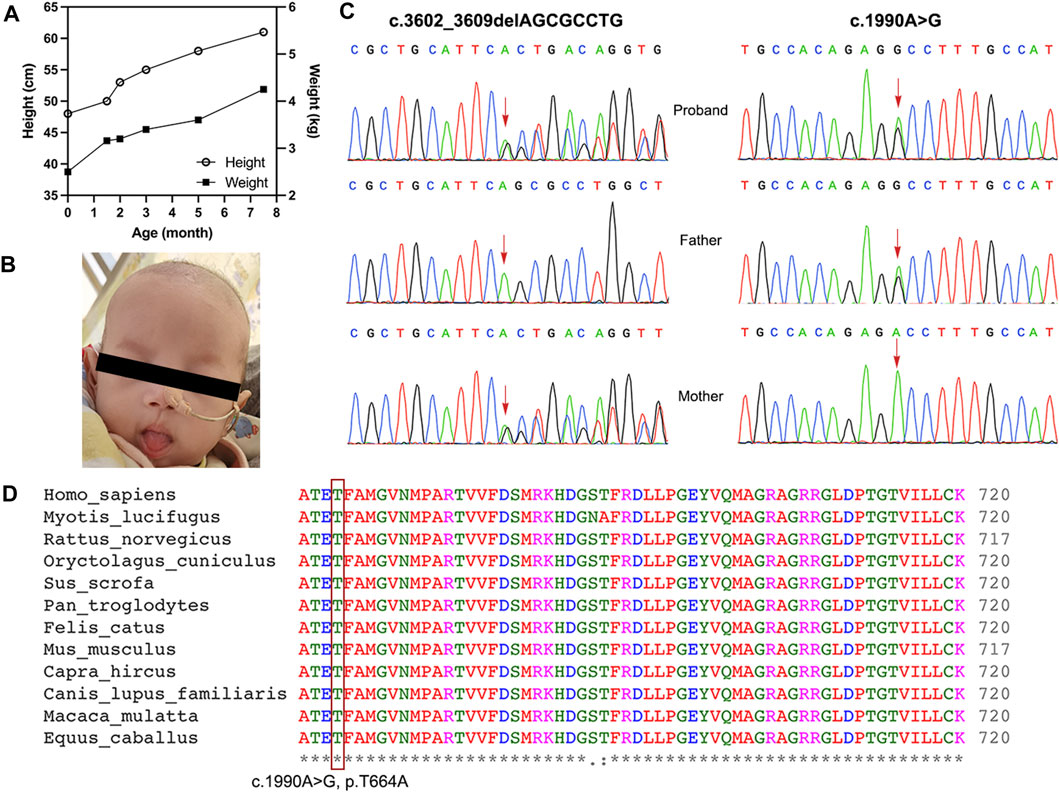
FIGURE 1. (A) The graphs show the height and weight of the patient during the treatment course. (B) The patient had woolly and brittle hair. (C) The compound-heterozygous SKIV2L mutations. The frameshift mutation with maternal origin, c.3602_3609delAGCGCCTG (p.Q1201Rfs*2), and missense mutation with paternal origin, c.1990A > G (p.T664A), were identified in the patient. (D) Threonine 664 is highly conserved across species. The level of conservation is indicated by symbols *, and from considerably conserved to relatively conserved.
Physical examination showed that the infant was weak and displayed a severe loss of subcutaneous fat. Her hair was short and sparse (woolly and brittle) (Figure 1B). No abnormalities were observed on the skin. Normal results were shown in all laboratory and imagological examinations.
Genetic Analysis
WES and Sanger sequencing were performed to identify the causative genes. A pair of novel compound-heterozygous variants, c.3602_3609delAGCGCCTG (p.Q1201Rfs*2) with the maternal origin and c.1990A > G (p.T664A) with the paternal origin, in the SKIV2L gene (NM_006,929) were identified via WES and validated via Sanger sequencing (Figure 1C). Variant p. Q1201Rfs*2 can be interpreted as “likely pathogenic” according to the ACMG standard (PVS1_strong + PM2+PP3). This variant is a null mutation which might truncate the protein, but the mutation is located at the last exon (PVS1_strong). The variant is absent from the controls (ExAC v1.0, gnomAD v2.1.1, ESP6500SI-V2, 1kGenomes v3.7.6) (PM2). Multiple lines of in silico algorithms, including MutationTaster2 (Schwarz et al., 2014) (1, disease-causing), SIFT Indel (Sim et al., 2012) (0.783, damaging), CADD v1.6 (Rentzsch et al., 2019) (23 > cutoff = 15, deleterious), and CAPIC (Li et al., 2020) (0.941 > cutoff = 0.02, pathogenic), predicted that it is deleterious, except for MutPred-LOF (Pagel et al., 2017) (0.317 < cutoff = 0.6, neutral) (PP3). The other variant c.1990A > G (p.T664A) can be classified as a “variant with uncertain clinical significance” (PM2 + PP3). This missense was not found in the controls (PM2) and also predicted to be deleterious by multiple lines of algorithms (MutationTaster2, 1.000, disease-causing; SIFT v6.2.1 (Sim et al., 2012), 0.00 < cutoff = 0.05; Provean (Choi and Chan, 2015), −4.91 < −2.5, deleterious; and Polyphen-2 (Adzhubei et al., 2013), HumDiv and HumVar, 0.991 and 0.913, probably damaging) (PP3). The missense variant is located in a highly conserved region (Figure 1D), suggesting the importance of the mutated residue. Thus, the changes in residue properties may damage the structure and function of the protein product. We considered both of these variants disease-causing.
Treatment and Prognosis
The patient was continuously fed with an amino-acid formula through a gastric tube, in addition to parenteral nutrition. Unfortunately, she developed a fever due to an infection from the central venous catheter of the parenteral nutriton. The infection was controlled by administrating cefoperazone sodium and sulbactam sodium. Additionally, as the volume of the fed amino-acid formula was increased, the amount of the intravenous nutrient fluid was gradually decreased until the parenteral nutrition was finally discontinued. Finally, she was discharged at 3 months of age and thereafter fed with 20 ml of an amino-acid formula per hour through a gastric tube. The recent follow-up at the age of 7.5 months revealed that the child still grew slowly (height, 61 cm, below the 3rd percentile; weight, 4.25 kg, below the 3rd percentile) and had intermittent diarrhea.
Discussion and Conclusion
THES2 is a rare and severe genetic disorder known to be associated with pathogenic variants of SKIV2L. Based on previous reports (Fabre et al., 2012; Fabre et al., 2013; Morgan et al., 2013; Monies et al., 2015; Lee et al., 2016; Zheng et al., 2016; Bick et al., 2017; Hiejima et al., 2017; Bourgeois et al., 2018; Vardi et al., 2018; Rudilla et al., 2019; Fung et al., 2020; Taher et al., 2020; Dyment et al., 2021; Klee et al., 2021), the incidence of this disease is not significantly different between genders (M:F = 17:20) (Table 1). Patients have their onset mostly in the neonatal period, ranging from birth to 0.8 years of age (mean, 29.5 days; median, 17 days). The symptoms of the current patient were noticed at birth. All THES2 patients have intractable diarrhea (34/34, 100%). Moreover, THES2 is also associated with intrauterine growth restriction (25/28, 89.3%), hair abnormalities (31/35, 88.6%), facial dysmorphisms (24/31, 77.4%), hepatic involvement (19/27, 70.4%), immunodeficiency (16/30, 53.3%), and cardiac abnormalities (7/19, 36.8%). This disease has been described in Middle-Eastern (11/37, 29.7%), European (10/37, 27.0%), Asian (7/37, 18.9%), American (5/37, 13.5%), and African (4/37, 10.8%) populations, with the majority seen in the Middle-Eastern background.
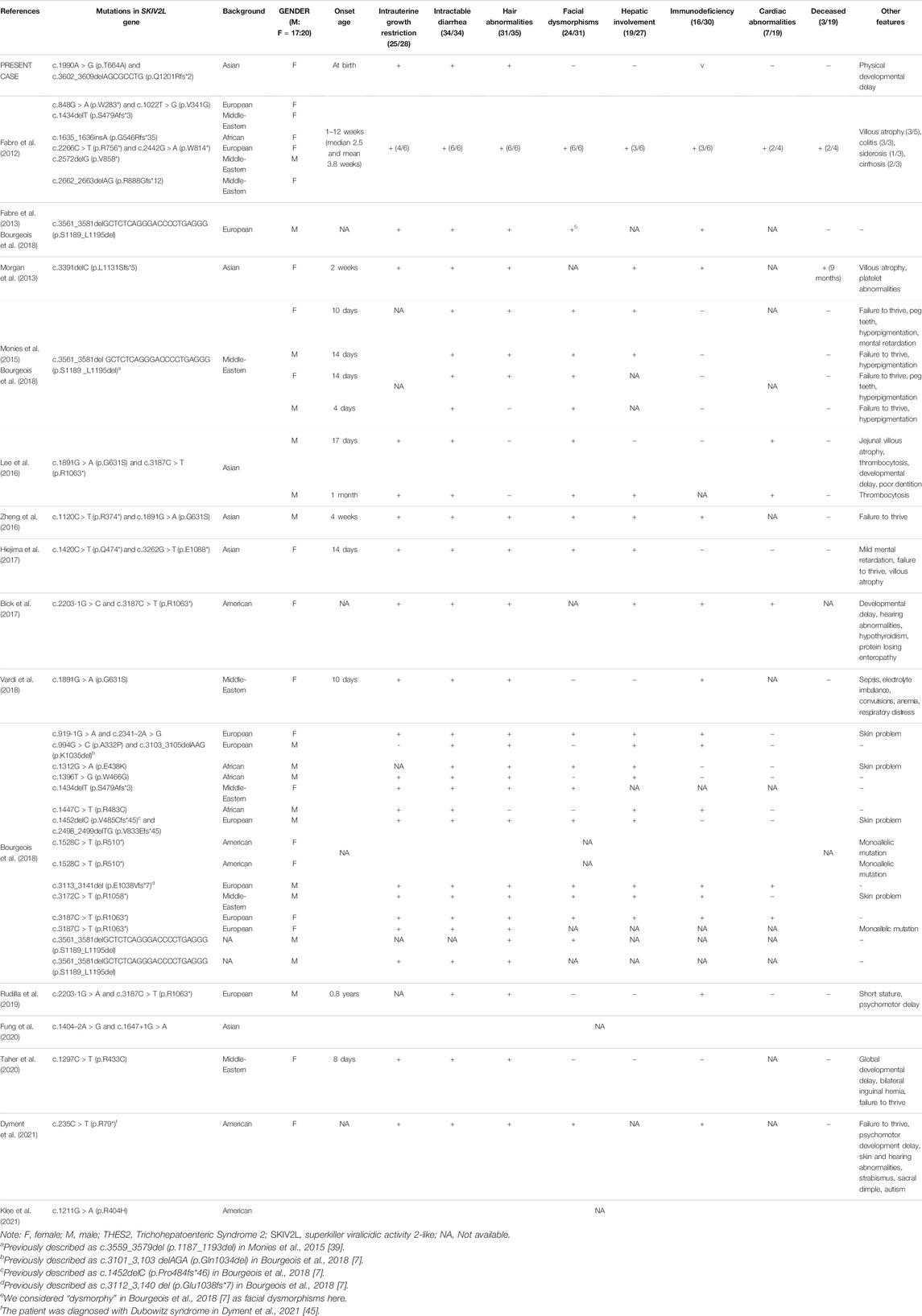
TABLE 1. Genotypic and phenotypic features of THES2 and the corresponding SKIV2L mutations.
According to the HGMD v2021.8, only 35 mutations in SKIV2L, including 32 disease-causing mutations (DM) and 3 possibly disease-causing mutations (DM?), have been reported to be associated with THES2. Nonsense mutations (10/35, 28.6%) are the most prevalent type of mutations, followed by missense (8/35, 22.9%), frameshift (7/35, 20.0%), canonical splice site (7/35, 20.0%), large-size (>20 bp) deletion (2/35, 5.7%), and inframe deletion (1/35, 2.9%) mutations. Although THES2 is considered an autosomal recessive disorder, three patients have been reported to carry a heterozygous SKIV2L mutation (Bourgeois et al., 2018). In-depth genetic analyses focusing on deep-intronic mutations may explain this discrepancy.
THES2 is usually diagnosed based on both the common THES symptoms and biallelic pathogenic variants of the SKIV2L gene. However, no phenotype–genotype correlation has been established to date. How SKIV2L mutations or defects in the Ski complex lead to the observed symptoms is still unclear. It has been revealed that SKIV2L RNA exosome limits the activation of RIG-I-like receptors (RLR), thereby negatively regulating RLR-mediated antiviral response. Human patients with SKIV2L mutations also have a potent type I interferon signature in the blood cells (Eckard et al., 2014). Such uncontrolled regulations and overwhelming responses could probably drive immune disease, leading to chronic intestinal inflammation, i.e., intractable diarrhea. Given that SKI2W is a helicase with ATPase activity, SKI2W defects may affect the normal function of the Ski complex, and consequently, the mRNA targets of this mRNA surveillance are not degraded, whereby the observed phenotypes ensue. Additional research is required to determine the pathogenesis of THES2 and the relationships between mutations and the symptoms.
Most patients with THES may need lifelong total parenteral nutrition (TPN) to survive, but some of them develop complications associated with TPN. To date, only a few infants with THES have attained normal oral nutrition (Fabre et al., 2017). In the current study, the patient was fed with an amino-acid formula and parenteral nutrition but subsequently developed complications similar to those associated with TPN. It seems that a small supplement of amino acids can be therapeutic for THES2 patients, but extreme cases might require extraoral administration of an extensively hydrolyzed formula (Taher et al., 2020). THES2 is a severe and potentially fatal disorder; however, only a few patients have been reported to die from THES2 (Fabre et al., 2012; Taher et al., 2020). Nevertheless, most cases lacked long-term follow-up. Therefore, a long follow-up period is required for patients with THES2 to acquire information on the prognosis and effectiveness of the clinical interventions.
In summary, this study reported a pair of novel compound-heterozygous variants of the SKIV2L gene in a patient with THES2, thereby expanding the spectrum of the causative mutations. Early genetic diagnosis of this syndrome could enable early and proper treatment. An amino-acid formula continuously fed through a gastric tube, alongside parenteral nutrition, could also delay the complications and improve the outcome of such cases.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by The study was approved by the Ethics Committee of Shenzhen Children’s Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
Conceptualization: QZ and ZW. Data collection: XQ and JZ. Data analysis: LH and SZ. Funding acquisition: ZW. Investigation: QZ. Project administration: ZW. Writing original draft: QZ, LH, and ZW. Writing-review and editing: QZ, XQ, JZ, LH, ZW, and SZ. All authors have read and approved the manuscript.
Funding
This work was supported by the Science Technology and Innovation Committee of Shenzhen (2021N062-JCYJ20210324115408023) and the Shenzhen Fund for Guangdong Provincial High level Clinical Key Specialties (No. SZGSP012).
Conflict of Interest
LH is employed by Running Gene Inc. (Beijing, China).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are sincerely grateful for the patient and her parents. We also appreciate Running Gene Inc. (Beijing, China) for their professional genetic sequencing and analysis, especially Liuqiang Li whose warm support helped in this study.
References
Adzhubei, I., Jordan, D. M., and Sunyaev, S. R. (2013). Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. Chapter 7 (1), Unit7–20. doi:10.1002/0471142905.hg0720s76
1000 Genomes Project Consortium Auton, A., Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., et al. (2015). A Global Reference for Human Genetic Variation. Nature 526 (7571), 68–74. doi:10.1038/nature15393
Bick, D., Fraser, P. C., Gutzeit, M. F., Harris, J. M., Hambuch, T. M., Helbling, D. C., et al. (2017). Successful Application of Whole Genome Sequencing in a Medical Genetics Clinic. J. Pediatr. Genet. 6 (2), 61–76. doi:10.1055/s-0036-1593968
Bourgeois, P., Esteve, C., Chaix, C., Béroud, C., Lévy, N., Fabre, A., et al. (2018). Tricho-Hepato-Enteric Syndrome Mutation Update: Mutations Spectrum of TTC37 and SKIV2L , Clinical Analysis and Future Prospects. Hum. Mutat. 39 (6), 774–789. doi:10.1002/humu.23418
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an Ultra-fast All-In-One FASTQ Preprocessor. Bioinformatics 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560
Chen, X., Han, L., and Yao, H. (2020). Novel Compound Heterozygous Variants of ETHE1 Causing Ethylmalonic Encephalopathy in a Chinese Patient: A Case Report. Front. Genet. 11, 341. doi:10.3389/fgene.2020.00341
Choi, Y., and Chan, A. P. (2015). PROVEAN Web Server: a Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 31 (16), 2745–2747. doi:10.1093/bioinformatics/btv195
Dangel, A. W., Shen, L., Mendoza, A. R., Wu, L.-c., and Yu, C. Y. (1995). Human Helicase geneSKI2Win the HLA Class III Region Exhibits Striking Structural Similarities to the Yeast Antiviral geneSKI2and to the Human geneKIAA0052:emergence of a New Gene Family. Nucl. Acids Res. 23 (12), 2120–2126. doi:10.1093/nar/23.12.2120
Dyment, D. A., O'Donnell‐Luria, A., Agrawal, P. B., Coban Akdemir, Z., Aleck, K. A., Antaki, D., et al. (2021). Alternative Genomic Diagnoses for Individuals with a Clinical Diagnosis of Dubowitz Syndrome. Am. J. Med. Genet. 185 (1), 119–133. doi:10.1002/ajmg.a.61926
Eckard, S. C., Rice, G. I., Fabre, A., Badens, Catherine., Gray, Elizabeth. E., Hartley, Jane. L., et al. (2014). The SKIV2L RNA Exosome Limits Activation of the RIG-I-like Receptors. Nat. Immunol. 15 (9), 839–845. doi:10.1038/ni.2948
Fabre, A., Bourgeois, P., Coste, M.-E., Roman, C., Barlogis, V., and Badens, C. (2017). Management of Syndromic Diarrhea/tricho-Hepato-Enteric Syndrome: A Review of the Literature. Irdr 6 (3), 152–157. doi:10.5582/irdr.2017.01040
Fabre, A., Charroux, B., Martinez-Vinson, C., Roquelaure, B., Odul, E., Sayar, E., et al. (2012). SKIV2L Mutations Cause Syndromic Diarrhea, or Trichohepatoenteric Syndrome. Am. J. Hum. Genet. 90 (4), 689–692. doi:10.1016/j.ajhg.2012.02.009
Fabre, A., Martinez-Vinson, C., Goulet, O., and Badens, C. (2013). Syndromic diarrhea/Tricho-Hepato-Enteric Syndrome. Orphanet J. Rare Dis. 8, 5. doi:10.1186/1750-1172-8-5
Fu, W., O’Connor, T. D., Jun, G., Kang, H. M., Abecasis, G., Leal, S. M., et al. (2013). Analysis of 6,515 Exomes Reveals the Recent Origin of Most Human Protein-Coding Variants. Nature 493 (7431), 216–220. doi:10.1038/nature11690
Fung, J. L. F., Yu, M. H. C., Huang, S., Chung, C. C. Y., Chan, M. C. Y., Pajusalu, S., et al. (2020). A Three-Year Follow-Up Study Evaluating Clinical Utility of Exome Sequencing and Diagnostic Potential of Reanalysis. Npj Genom. Med. 5, 37. doi:10.1038/s41525-020-00144-x
Hartley, J. L., Zachos, N. C., Dawood, B., Donowitz, M., Forman, J., Pollitt, R. J., et al. (2010). Mutations in TTC37 Cause Trichohepatoenteric Syndrome (Phenotypic Diarrhea of Infancy). Gastroenterology 138 (7), 2388–2398e1-2. doi:10.1053/j.gastro.2010.02.010
Hiejima, E., Yasumi, T., Nakase, H., Matsuura, M., Honzawa, Y., Higuchi, H., et al. (2017). Tricho-hepato-enteric Syndrome with Novel SKIV2L Gene Mutations. Medicine (Baltimore) 96 (46), e8601. doi:10.1097/md.0000000000008601
Kammermeier, J., Drury, S., James, C. T., Dziubak, R., Ocaka, L., Elawad, M., et al. (2014). Targeted Gene Panel Sequencing in Children with Very Early Onset Inflammatory Bowel Disease-Evaluation and Prospective Analysis. J. Med. Genet. 51 (11), 748–755. doi:10.1136/jmedgenet-2014-102624
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 581 (7809), 434–443. doi:10.1038/s41586-020-2308-7
Klee, E. W., Cousin, M. A., Pinto e Vairo, F., Rosado, Joel. A. Morales., Macke, Erica. L., Jenkinson, W. Garrett., et al. (2021). Impact of Integrated Translational Research on Clinical Exome Sequencing. Genet. Med. 23 (3), 498–507. doi:10.1038/s41436-020-01005-9
Landrum, M. J., and Kattman, B. L. (2018). ClinVar at Five Years: Delivering on the Promise. Hum. Mutat. 39 (11), 1623–1630. doi:10.1002/humu.23641
Lee, S.-G., Lee, I., Park, S. H., Kang, C., and Song, K. (1995). Identification and Characterization of a Human cDNA Homologous to Yeast SKI2. Genomics 25 (3), 660–666. doi:10.1016/0888-7543(95)80008-a
Lee, W. S., Teo, K. M., Ng, R. T., Chong, S. Y., Kee, B. P., and Chua, K. H. (2016). Novel Mutations in SKIV2L and TTC37 Genes in Malaysian Children with Trichohepatoenteric Syndrome. Gene 586 (1), 1–6. doi:10.1016/j.gene.2016.03.049
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature 536 (7616), 285–291. doi:10.1038/nature19057
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, S., van der Velde, K. J., de Ridder, D., van Dijk, A. D. J., Soudis, D., Zwerwer, L. R., et al. (2020). CAPICE: a Computational Method for Consequence-Agnostic Pathogenicity Interpretation of Clinical Exome Variations. Genome Med. 12 (1), 75. doi:10.1186/s13073-020-00775-w
McKay, G. J., Silvestri, G., Patterson, C. C., Hogg, R. E., Chakravarthy, U., and Hughes, A. E. (2009). Further Assessment of the Complement Component 2 and Factor B Region Associated with Age-Related Macular Degeneration. Invest. Ophthalmol. Vis. Sci. 50 (2), 533–539. doi:10.1167/iovs.08-2275
Monies, D. M., Rahbeeni, Z., Abouelhoda, M., Naim, E. A., Al-Younes, B., Meyer, B. F., et al. (2015). Expanding Phenotypic and Allelic Heterogeneity of Tricho-Hepato-Enteric Syndrome. J. Pediatr. Gastroenterol. Nutr. 60 (3), 352–356. doi:10.1097/mpg.0000000000000627
Morgan, N. V., Hartley, J. L., Setchell, K. D., Simpson, M. A., Brown, R., Tee, L., et al. (2013). A Combination of Mutations in AKR1D1 and SKIV2L in a Family with Severe Infantile Liver Disease. Orphanet J. Rare Dis. 8, 74. doi:10.1186/1750-1172-8-74
Pagel, K. A., Pejaver, V., Lin, G. N., Nam, H.-J., Mort, M., Cooper, D. N., et al. (2017). When Loss-Of-Function Is Loss of Function: Assessing Mutational Signatures and Impact of Loss-Of-Function Genetic Variants. Bioinformatics 33 (14), i389–i398. doi:10.1093/bioinformatics/btx272
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 47 (D1), D886–D894. doi:10.1093/nar/gky1016
Richards, S., Aziz, N., Aziz, N., Bale, S., Bick, D., Das, S., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–423. doi:10.1038/gim.2015.30
Riley, L. G., Cowley, M. J., Gayevskiy, V., Minoche, A. E., Puttick, C., Thorburn, D. R., et al. (2020). The Diagnostic Utility of Genome Sequencing in a Pediatric Cohort with Suspected Mitochondrial Disease. Genet. Med. 22 (7), 1254–1261. doi:10.1038/s41436-020-0793-6
Rudilla, F., Franco-Jarava, C., Martínez-Gallo, M., Garcia-Prat, M., Martín-Nalda, A., Rivière, J., et al. (2019). Expanding the Clinical and Genetic Spectra of Primary Immunodeficiency-Related Disorders with Clinical Exome Sequencing: Expected and Unexpected Findings. Front. Immunol. 10, 2325. doi:10.3389/fimmu.2019.02325
Schneider, C., and Tollervey, D. (2013). Threading the Barrel of the RNA Exosome. Trends Biochem. Sci. 38 (10), 485–493. doi:10.1016/j.tibs.2013.06.013
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 11 (4), 361–362. doi:10.1038/nmeth.2890
Sim, N.-L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., and Ng, P. C. (2012). SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 40, W452–W457. (Web Server issue). doi:10.1093/nar/gks539
Stankler, L., Lloyd, D., Pollitt, R. J., Gray, E. S., Thom, H., and Russell, G. (1982). Unexplained Diarrhoea and Failure to Thrive in 2 Siblings with Unusual Facies and Abnormal Scalp Hair Shafts: a New Syndrome. Arch. Dis. Child. 57 (3), 212–216. doi:10.1136/adc.57.3.212
Stenson, P. D., Mort, M., Ball, E. V., Evans, K., Hayden, M., Heywood, S., et al. (2017). The Human Gene Mutation Database: towards a Comprehensive Repository of Inherited Mutation Data for Medical Research, Genetic Diagnosis and Next-Generation Sequencing Studies. Hum. Genet. 136 (6), 665–677. doi:10.1007/s00439-017-1779-6
Stray-Pedersen, A., Sorte, H. S., Samarakoon, P., Gambin, T., Chinn, I. K., Coban Akdemir, Z. H., et al. (2017). Primary Immunodeficiency Diseases: Genomic Approaches Delineate Heterogeneous Mendelian Disorders. J. Allergy Clin. Immunol. 139 (1), 232–245. doi:10.1016/j.jaci.2016.05.042
Taher, Z. A., Alzahrani, S., Alsaghir, A., Nouh, F., and Alshumrani, M. (2020). A New Variant Mutation in SKIV2L Gene in Case of Trichohepatoenteric Syndrome. Pediatr. Rep. 12 (3), 93–97. doi:10.3390/pediatric12030021
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ Data to High Confidence Variant Calls: the Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinformatics 43 (1110), 11–33. doi:10.1-11.10.3310.1002/0471250953.bi1110s43
van Schouwenburg, P. A., Davenport, E. E., Kienzler, A.-K., Marwah, I., Wright, B., Lucas, M., et al. (2015). Application of Whole Genome and RNA Sequencing to Investigate the Genomic Landscape of Common Variable Immunodeficiency Disorders. Clin. Immunol. 160 (2), 301–314. doi:10.1016/j.clim.2015.05.020
Vardi, I., Barel, O., Sperber, M., Schvimer, M., Nunberg, M., Field, M., et al. (2018). Genetic and Structural Analysis of a SKIV2L Mutation Causing Tricho-Hepato-Enteric Syndrome. Dig. Dis. Sci. 63 (5), 1192–1199. doi:10.1007/s10620-018-4983-x
Verloes, A., Lombet, J., Lambert, Y., Hubert, A.-F., Deprez, M., Fridman, V., et al. (1997). Tricho-hepato-enteric Syndrome: Further Delineation of a Distinct Syndrome with Neonatal Hemochromatosis Phenotype, Intractable Diarrhea, and Hair Anomalies. Am. J. Med. Genet. 68 (4), 391–395. doi:10.1002/(sici)1096-8628(19970211)68:4<391:aid-ajmg3>3.0.co;2-p
Yang, Z., Shen, L., Dangel, A. W., Wu, L.-C., and Yu, C. Y. (1998). Four Ubiquitously Expressed Genes,RD(D6S45)-SKI2W(SKIV2L)-DOM3Z-RP1(D6S60E), Are Present between Complement Component Genes FactorBandC4in the Class III Region of the HLA. Genomics 53 (3), 338–347. doi:10.1006/geno.1998.5499
Keywords: trichohepatoenteric syndrome (THES) 2, SKIV2L gene, intractable diarrhea, whole-exome sequencing (WES), pediatrics
Citation: Zhang Q, Qian X, Zhou J, Han L, Zhou S and Wang Z (2021) Case Report: Novel Compound-Heterozygous Variants of SKIV2L Gene that Cause Trichohepatoenteric Syndrome 2. Front. Genet. 12:756451. doi: 10.3389/fgene.2021.756451
Received: 10 August 2021; Accepted: 17 September 2021;
Published: 06 October 2021.
Edited by:
Fan Jin, Zhejiang University, ChinaReviewed by:
Liang-Liang Fan, Central South University, ChinaRong Xiang, Central South University, China
Copyright © 2021 Zhang, Qian, Zhou, Han, Zhou and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaoxia Wang, d2FuZ3poYW94aWExOTY5QDE2My5jb20=