 Areerat Hnoonual1
Areerat Hnoonual1 Phawin Kor-anantakul2,3
Phawin Kor-anantakul2,3 Chariyawan Charalsawadi1
Chariyawan Charalsawadi1 Juthamas Worachotekamjorn4
Juthamas Worachotekamjorn4 Pornprot Limprasert5*
Pornprot Limprasert5*- 1Department of Pathology, Faculty of Medicine, Prince of Songkla University, Songkhla, Thailand
- 2Center of Excellence for Medical Genomics, Medical Genomics Cluster, Department of Pediatrics, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand
- 3Excellence Center for Genomics and Precision Medicine, King Chulalongkorn Memorial Hospital, Thai Red Cross Society, Bangkok, Thailand
- 4Department of Pediatrics, Faculty of Medicine, Prince of Songkla University, Songkhla, Thailand
- 5Faculty of Medicine, Siam University, Bangkok, Thailand
Autism spectrum disorder (ASD) is a group of neurodevelopmental disorders which are etiologically heterogeneous. Chromosomal microarray is now recommended as the first-tier clinical diagnostic test for ASD. We performed chromosomal microarray in 16 Thai patients with ASD using an Illumina HumanCytoSNP-12 v2.1 array and found one case with uniparental disomy (UPD) of chromosome 15. Methylation-specific PCR showed abnormal methylation of the maternal SNRPN allele. Haplotype analysis revealed that the patient had received both chromosomes 15 from his father. These results were consistent with Angelman syndrome. However, his clinical features had no clinical significance for classic Angelman syndrome. He had first presented at the pediatric clinic with no speech, poor social interaction skills and repetitive behaviors consistent with ASD based on the DSM-IV criteria at 2 years of age and later confirmed by ADOS at 5 years of age. He was strikingly overweight but had no dysmorphic facies, seizures nor ataxia and was diagnosed as non-syndromic ASD, a diagnosis which was believed until at 10 years of age, his DNA was included for analysis in this current cohort study. Our findings suggest that ASD patients with unknown etiology should be considered for methylation-specific PCR testing for Angelman syndrome where chromosomal microarray is not available. In the study, we also review the clinical features of Angelman syndrome caused by UPD and the frequency of ASD in individuals with Angelman syndrome.
Introduction
Autism spectrum disorder (ASD) is a heterogeneous group of neurodevelopmental disorders characterized by impaired social interaction and communication, and stereotyped behaviors and interests. Currently, a number of genetic syndromes are known to be related to ASD. Abnormalities of chromosome 15 are the most frequently reported in ASD, especially 15q11-q13 duplications which have been reported to occur in 0.25–3% of ASD cases depending on sample ascertainment (Cook et al., 1997; Schroer et al., 1998; Depienne et al., 2009; Sanders et al., 2015). Chromosome 15q11-q13, the Prader-Willi/Angelman syndrome (PWS/AS) critical region, contains a number of imprinted and non-imprinted genes including the UBE3A and GABAA receptor subunit genes that may be associated with the presence of ASD. Several association studies of these genes have found significant associations with autism (Cook et al., 1997; Cook et al., 1998; Buiting et al., 2016; Khatri and Man, 2019).
Angelman syndrome (AS) is a neurogenetic disorder characterized by severe developmental delay usually noticeable at 6–12 months of age. The notable clinical features include developmental delay, severe speech delay with minimal to no use of word, ataxia, hand flapping, happy demeanor and inappropriate laughter or smiling. Other clinical features associated with AS include microcephaly and seizure with a characteristic abnormal electroencephalogram (EEG) pattern with large amplitude slow-spike waves. Irregular sleep-wake cycles, wide-spaced teeth, frequent drooling and hypopigmentation are also frequently observed (Williams et al., 2006). AS is caused by four known genetic mechanisms including deletion of chromosome 15q11-q13, mutations of UBE3A, uniparental disomy (UPD) of chromosome 15 and imprinting center defect (Buiting et al., 2016; Beygo et al., 2019). While the loss of function of the maternally inherited UBE3A gene causes AS, abnormalities of chromosome 15q (deletions/duplications) that contain the UBE3A gene have been reported in association with ASD (Cook et al., 1997; Depienne et al., 2009; Vatsa and Jana, 2018; Khatri and Man, 2019), suggesting that the UBE3A gene may also be implicated in autism. AS has a high comorbidity with autism and shows many overlapping clinical features of autism (Steffenburg et al., 1996; Peters et al., 2004; Trillingsgaard and ØStergaard, 2004; Bonati et al., 2007). AS is also considered as a syndromic form of ASD. However, previous studies have shown a wide range of ASD prevalence in patients with AS (Chan et al., 1993; Steffenburg et al., 1996; Peters et al., 2004; Trillingsgaard and ØStergaard, 2004; Sahoo et al., 2006; Bonati et al., 2007; Peters et al., 2012; Moss et al., 2013; Mertz et al., 2014; Wink et al., 2015) but there are very few studies reporting on the prevalence of AS in patients with ASD (Schroer et al., 1998; Depienne et al., 2009). Several recent studies have suggested that ASD is strongly associated with genetic variations, including copy number variations (CNVs). Chromosomal microarray (CMA) has enabled identification of CNVs with a much higher resolution than standard chromosome analysis. In addition, single nucleotide polymorphism (SNP) arrays based on CMA can also detect copy number neutral abnormalities such as regions of long contiguous stretches of homozygosity (LCSH) and UPD which are associated with an increased risk of genetic diseases including PWS and AS. CMA is now recommended as the first-tier clinical diagnostic test for individuals with ASD, developmental delay/intellectual disability, and multiple congenital anomalies of unknown causes (Miller et al., 2010; Battaglia et al., 2013; Schaefer et al., 2013).
In this study, we aimed to identify novel or pathogenic CNVs in selected Thai children with ASD, and we found a patient with atypical AS who had originally been diagnosed as ASD. SNP arrays identified the absence of heterozygosity (AOH) along the long (q) arm of chromosome 15. Subsequently, methylation-specific PCR (MS-PCR) analysis confirmed the AS diagnosis in this patient. A haplotype analysis of chromosome 15 showed that the patient had inherited both chromosomes 15 from his father, indicating paternal UPD consistent with a diagnosis of AS. In addition, we studied 100 patients with ASD for PWS/AS methylation-specific PCR screening, and no other patients with AS or PWS were detected in our ASD cohort.
Case Presentation
A 2-year-old boy initially presented with no speech, poor social interaction skills and repetitive behaviors consistent with ASD based on the DSM-IV criteria, which also fulfilled the DSM-V criteria. The patient was the second child born to healthy and unrelated Thai parents. He had one older sister with normal development. His birth weight was 3,600 g with no perinatal complications. He first stood with support at 10 months, but did not walk without support until the age of 27 months. He was unable to hold a spoon and feed himself until 45 months of age. He never developed verbal language skills. His growth curves were within normal ranges, including head circumference, except he had been overweight since 2 years of age (Supplementary Figure S1). His BMI at 10 years was 29.7 kg/m2 (>97th centile, Z score = 3.79). He had no hypopigmentation, and had never shown signs of seizure, ataxia or hypotonia. He usually walked on tip toes. He had poor eye contact and had a repetitive behavior in constantly spinning the wheels of a toy car. He engaged in hand-flapping whenever he was happy. He had no self-injury nor aggressive behaviors. He liked to bite things (i.e., clothes, drinking straws). At the age of 5 years, he was assessed using the Autism Diagnosis Observation Schedule (ADOS)-Module 1 by a developmental pediatrician. His scores for communication were 4 (cutoff = 4), 8 for reciprocal social interaction (cutoff = 7), and 1 for repetitive behaviors (Supplementary Table S1). The scores were consistent with a diagnosis of autistic disorder. At 10 years of age, he had a non-verbal IQ rating of 42 (Stanford Binet Intelligence Scales-Fifth edition) and the Vineland Adaptive Behavior Scales-II showed moderate deficits in all three domains, communications, daily living skills and socialization. We also observed that his autistic features evolved as his social interaction skills improved over time. For example, at 11 years of age, he liked to play with his peers and parents, although he still could not speak understandable words.
Initially, he was tested for common genetic causes of ASD, which revealed normal karyotyping and normal CGG repeats on the FMR1 gene. Electroencephalogram (EEG) was also normal. Following the recommendations from the International Standard Cytogenomic Array (ISCA) and the American College of Medical Genetics (ACMG) for the use of CMA, he was screened with the CMA at 10 years of age to look for pathogenic CNVs. Written informed consent was obtained from the patient’s parents for publication of this case report.
Materials and Methods
Patients
A total of 16 patients with ASD of unknown cause were recruited from a large cohort of Thai children with non-syndromic ASD reported in a previous study (Charalsawadi et al., 2014). The patients fulfilled the criteria for ASD diagnosis according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) (Hansakunachai et al., 2014). Karyotyping and Fragile X DNA testing had been done in all patients and also MECP2 sequencing testing in females as initial tests and all tests had shown normal results. In addition, we performed PWS/AS methylation specific PCR on 100 patients with ASD to screen for AS and PWS to find the frequency of PWS/AS in ASD patients.
Chromosomal Microarray Analysis
SNP arrays were performed for the 16 patients with unexplained ASD using a HumanCytoSNP-12 DNA Analysis BeadChip v2.1 kit (Illumina, San Diego, California, United States), which contains approximately 3,00,000 SNP markers. The results were analyzed by BlueFuse Multi software and GenomeStudio Data Analysis Software v. 2011.1 based on the reference human genome (hg19/GRCh37). The data were then interpreted based on guidelines from the ISCA and ACMG (Miller et al., 2010; Kearney et al., 2011; Riggs et al., 2020).
Methylation-specific Polymerase Chain Reaction (MS-PCR) Analysis
The DNA samples were treated with bisulfate using a process described previously (Kubota et al., 1997) with modifications (Supplementary Material). The modified DNA sample was then used as a template in the following MS-PCR. MS-PCR analysis of the SNRPN gene was performed using original and alternative primers (Kubota et al., 1997; Hussain Askree et al., 2011) (Supplementary Table S2). Multiplex PCR was performed in a total volume of 10 µL containing 50 ng modified DNA template, 1X PCR buffer, 1.5 mM MgCl2, 0.2 mM dNTPs and 0.6 units of FastStart Taq DNA polymerase (Roche Applied Science). PCRs were carried out by denaturation at 96°C for 10 min, followed by 35 cycles at 94°C for 30 s, 64°C for 30 s, 72°C for 1 min, and final extension at 72°C for 10 min. The PCR products were run on 2.5% agarose gel electrophoresis and visualized by staining with ethidium bromide.
Fluorescence in situ Hybridization (FISH)
FISH was performed to check specifically for chromosome 15 deletions, according to the manufacturer’s instructions with minor modifications. The DNA probes used in the FISH analysis included a triple probe mix of Vysis Prader-Willi/Angelman Region Probe - LSI D15S10 SpectrumOrange at 15q11.2, CEP 15 (D15Z1) SpectrumAqua at 15p11.2, and PML SpectrumGreen Probe at 15q22 (Abbott Molecular Inc.). The FISH images were captured and analyzed using the Isis program (MetaSystems).
Haplotype Analysis of UPD
To investigate either paternal or maternal UPD in the proband with autism, six microsatellite markers (D15S1012, D15S643, D15S983, D15S979, D15S657, and D15S966) located along the q arm of chromosome 15 were selected from the GenBank database (Supplementary Table S3). The details of haplotype analysis using capillary electrophoresis are shown in Supplementary Material.
Results
CMA, MS-PCR Analysis and FISH
We screened 16 patients with unexplained ASD for genomic imbalances and SNP array analysis in a 10-year-old boy with ASD detected an absence of heterozygosity (AOH) region of 82.2 Mb (15q11.1–15q26.3), possibly indicative of UPD (Figure 1). No pathogenic CNVs were detected in the other 15 patients. To validate the results of the microarray and to investigate the possible alterations of the SNRPN methylation status, MS-PCR analyses with both original and alternative primer sets were further performed, which found the absence of a maternal methylation of the SNRPN gene in the proband, which was consistent with a diagnosis of AS (Figure 2A). FISH testing confirmed that the patient did not carry deletions on chromosome 15 (Supplementary Figure S2) and chromosome analysis using G-banding in this patient showed a normal karyotype, and thus he was suspected of having UPD.
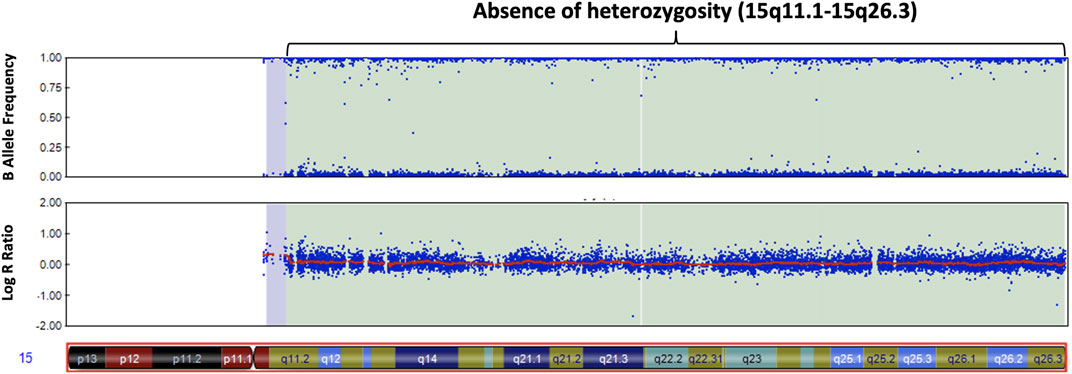
FIGURE 1. Chromosomal microarray analysis of our patient with autism. The SNP microarray shows an absence of heterozygosity along the q arm of chromosome 15 in the B allele frequency plot (upper panel). The Log R ratio plot (lower panel) shows no change in copy number in the q arm of chromosome 15.
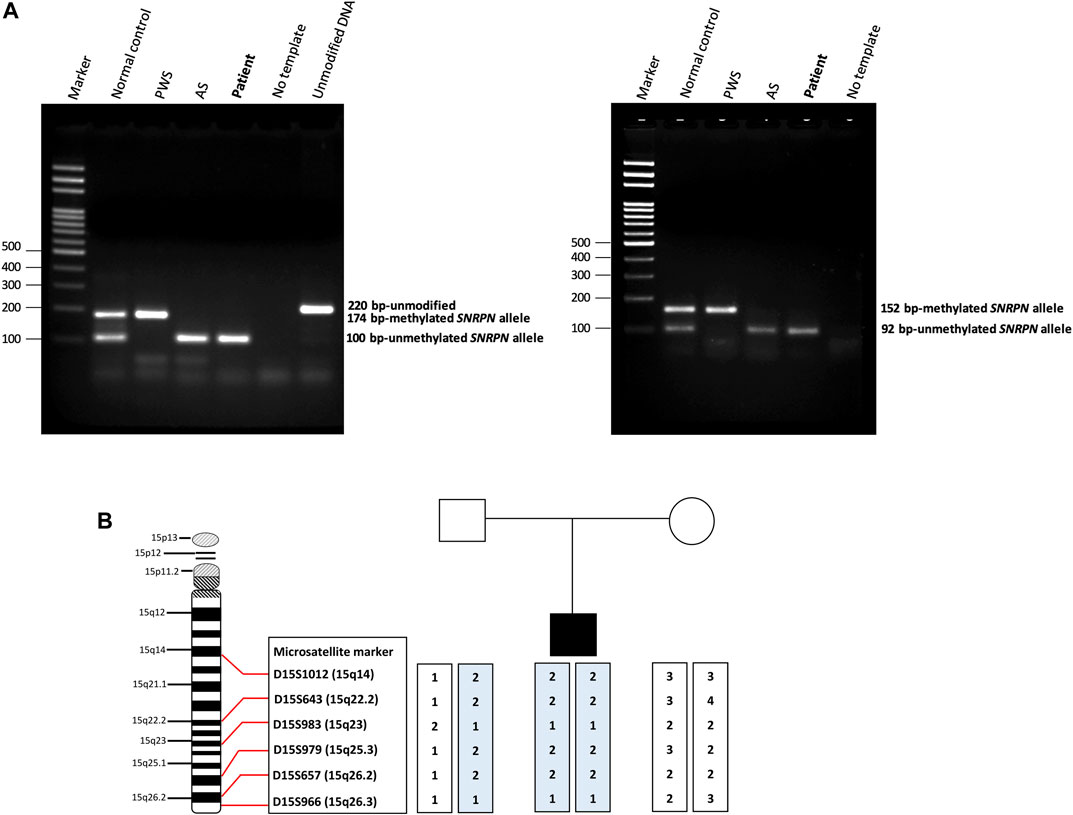
FIGURE 2. Methylation-specific PCR (MS-PCR) results and haplotype analysis. (A) The (left panel) shows the results of MS-PCR analysis of a patient using an original primer set designed by Kubota et al. The MS-PCR results from a normal control show a 100-bp PCR product representing the unmethylated paternal allele and a 174-bp PCR product representing the methylated maternal allele. Consistent with the clinical diagnosis of AS, our patient showed only a 100-bp PCR product derived from the unmethylated paternal allele. The (right panel) shows the results of MS-PCR analysis of our patient using an alternative primer set designed by Hussain Askree et al. A normal control shows a 92-bp PCR product of the unmethylated paternal allele and a 152-bp PCR product of the methylated maternal allele. Our patient showed only a 92-bp PCR product derived from the unmethylated paternal allele, consistent with a diagnosis of AS. AS, Angelman syndrome; PWS, Prader-Willi syndrome. (B) Haplotype analysis of the patient and his parents. The results of a haplotype analysis using six microsatellite markers across the q arm of chromosome 15 revealed that the patient had inherited both chromosomes 15 from his father. It also showed isodisomy which indicated the most likely cause to be a meiosis II nondisjunction event.
Haplotype Analysis of UPD
Haplotype analysis with six microsatellite markers of the q arm of chromosome 15 was then used to investigate the genetic mechanisms of AS in the patient. The results confirmed that the patient had received both chromosomes 15 from his father, indicating that paternal UPD was the likely cause of AS in this patient. In addition, both paternally-derived alleles showed the same haplotypes, indicating paternal isodisomy of chromosome 15. The results of the haplotype analysis are shown in Figure 2B.
Screening of PWS and AS in Patients With ASD
Because of the unexpected result of detecting AS in the patient with ASD, we further screened a total of 100 ASD patients to look for PWS/AS in our ASD patient’s cohort using MS-PCR analysis. In this group, all of the patients had normal paternal and maternal methylation of the SNRPN gene, indicating that none of them had PWS or AS, suggesting that PWS/AS is not a common genetic cause of patients with ASD. The workflow and summary of the results of this study are shown in Supplementary Figure S3.
Discussion
We identified an AS patient with ASD caused by paternal UPD of chromosome 15. The patient was first diagnosed with ASD of unknown cause and a chromosomal microarray was performed following the ACMG and ISCA recommendations. The microarray results revealed UPD of chromosome 15. The MS-PCR analysis of this patient showed abnormal methylation of the maternal SNRPN allele and haplotype analysis showed that the patient had paternal uniparental isodisomy, resulting from meiosis II nondisjunction. Therefore, the results of all molecular genetic testing of this patient were consistent with AS. In general, in approximately 75% of individuals with AS, the AS results from maternal 15q11-q13 deletion, which results in a more severe clinical phenotype than other genetic types of AS. AS is also caused by the UBE3A mutations (5–10%), paternal UPD of chromosome 15 (3–7%), or an imprinting center defect (2–3%). The remaining approximately 10% of patients with AS have no detectable genetic abnormality (Buiting et al., 2016; Beygo et al., 2019). Different genetic types in AS patients may show different phenotypes in performance (Yang et al., 2021).
Many aspects of the case presented here are consistent with previous reports of AS patients with the underlying cause of paternal UPD (Lossie et al., 2001; Poyatos et al., 2002; Thompson and Bolton, 2003; Tsai et al., 2004; Varela et al., 2004; Saitoh et al., 2005; Bonati et al., 2007; Depienne et al., 2009; Horváth et al., 2013; Luk and Lo, 2016), who generally had a lower prevalence of seizure, normal head circumference and absence of hypopigmentation (Lossie et al., 2001; Depienne et al., 2009; Horváth et al., 2013; Samanta, 2021). AS patients with UPD also have a higher risk of obesity than deletion type (Lossie et al., 2001; Poyatos et al., 2002; Varela et al., 2004; Saitoh et al., 2005; Brennan et al., 2015; Luk and Lo, 2016). A previous study found that children with UPD showed hyperphagic behavior and their weight increased significantly after the age of 2 years compared with children in the other genetic groups. These UPD children also had significantly higher birth weight than children with deletions or UBE3A mutations (Mertz et al., 2014). AS patients caused by UPD with no ataxia (Poyatos et al., 2002; Saitoh et al., 2005; Horváth et al., 2013; Luk and Lo, 2016) or no happy demeanor (Fridman et al., 2002; Varela et al., 2004; Bonati et al., 2007; Depienne et al., 2009; Luk and Lo, 2016) have also been reported. Moreover, it is notable that, despite one earlier study suggesting a decreased risk of ASD in AS in the absence of seizure (Thompson and Bolton, 2003), our case presented with autistic features without any history of seizure. This case adds to the pool of milder phenotypes in AS patients with UPD compared to a typical AS patients with deletions. A summary of the clinical features of our case and other AS cases with UPD is shown in Table 1. Of note, ataxic gait and frequent laughter/happy demeanor as consistent (100%) clinical features of patients with AS as claimed in a previous report (Williams et al., 2006) were not present in all AS patients with UPD in our literature review (Fridman et al., 2002; Poyatos et al., 2002; Thompson and Bolton, 2003; Varela et al., 2004; Saitoh et al., 2005; Bonati et al., 2007; Depienne et al., 2009; Horváth et al., 2013; Luk and Lo, 2016), supporting our hypothesis that AS resulting from UPD can manifest as a milder AS phenotype.
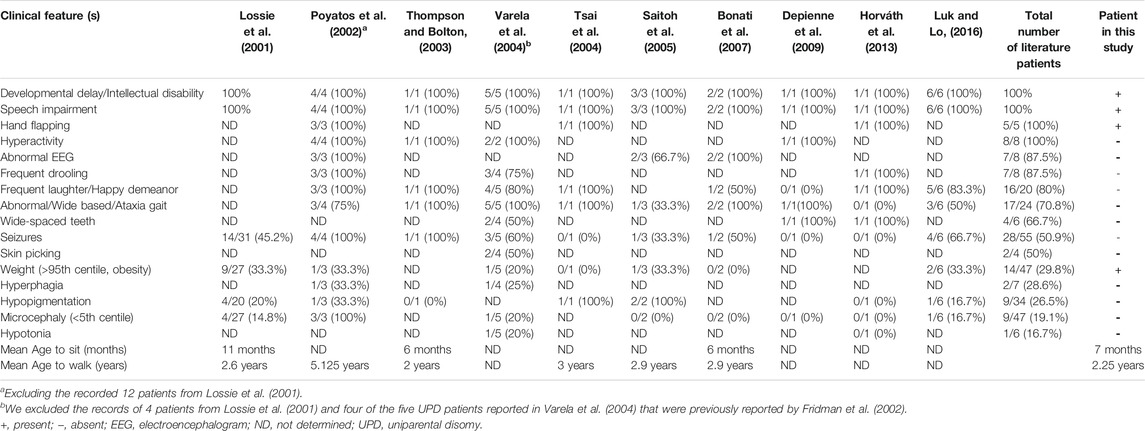
TABLE 1. Clinical features of Angelman syndrome patients with UPD of the patient in this study and patients from previous reports.
Previous studies have reported a wide range (1%–100%) of prevalence of ASD in AS caused by deletion, imprinting center defect, UPD or UBE3A mutations. The wide variety of percentages of ASD incidence in AS patients may be due to the small cohorts in such studies, which ranged from 4 to 93 AS cases (Chan et al., 1993; Steffenburg et al., 1996; Peters et al., 2004; Trillingsgaard and ØStergaard, 2004; Sahoo et al., 2006; Bonati et al., 2007; Mertz et al., 2014; Wink et al., 2015). The findings of the studies of ASD in AS are summarized in Table 2. However, many of the AS cases in these studies did not meet the full diagnostic criteria of ASD and lacked genetic verification for the diagnoses of AS. These studies mostly used standardized observation schedules such as ADOS for ASD diagnosis, however the ADOS is not a valid measure for children with mental ages below 18–24 months. Thus social and communication deficits in individuals with AS may be qualitatively different from these phenotypes in individuals with ASD. Among the studies of ASD in AS, Peters et al. and Bonati et al. reported two cases of AS with UPD who met the autism criteria from a total of 4 cases of AS with UPD in their studies. Furthermore, there is little information about the prevalence of AS in patients with ASD. Schroer et al. reported one AS case (a maternal 15q11-q13 deletion) among 100 patients with ASD (1%) and Depienne et al. reported two AS cases in a study of 552 cases with ASD (0.36%), one with a deletion and the other with UPD. Another previous study reported a prevalence of ASD in PWS individuals of 26.7% (Bennett et al., 2015). A systematic review found that ASD occurred more frequently in PWS patients with maternal UPD (35.3%) than those with deletion (18.5%) (Bennett et al., 2015). Although we found no cases of PWS or AS in our 100 patients with ASD, there is sufficient evidence for the occurrence of ASD in AS (Table 2), indicating that MS-PCR screening should be considered for AS and PWS in ASD patients, especially in patients with severe speech impairment and obesity (Lossie et al., 2001; Poyatos et al., 2002; Mertz et al., 2014; Luk and Lo, 2016). MS-PCR analysis is an easy and rapid method to screen for these syndromes simultaneously. MS-PCR analysis can detect more than 99% of affected individuals with PWS and approximately 80% of affected individuals with AS, including those with deletion, imprinting center defect or UPD (Beygo et al., 2019). However, further investigations such as haplotype analysis are needed to elucidate the underlying genetic mechanisms of disease for genetic counseling. The recurrence risk of having AS to the siblings of an affected child depends on the pathogenic mechanism. The risk to siblings is less than 1% if the affected child has a deletion or UPD, but up to 50% if the affected child has an imprinting center defect or a mutation of the UBE3A gene (Beygo et al., 2019).
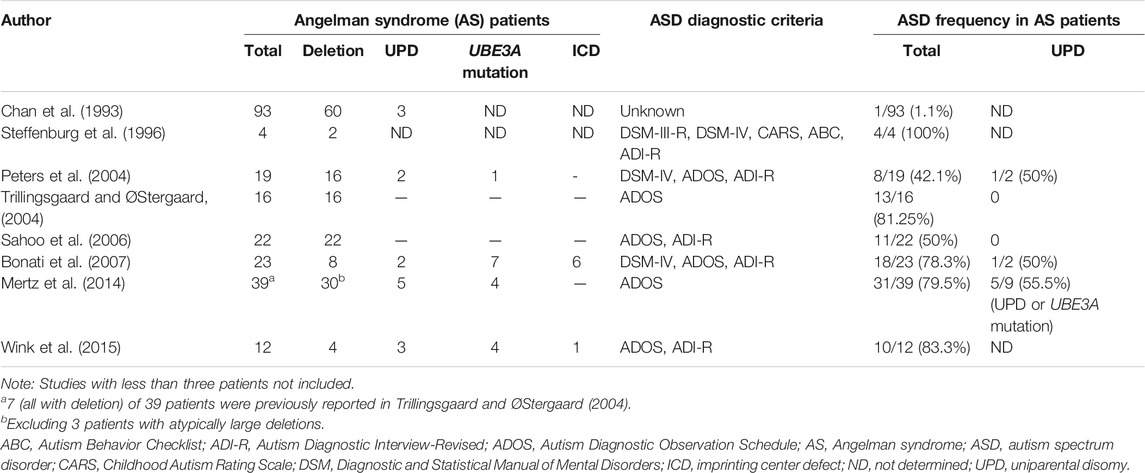
TABLE 2. Summary of the studies of ASD in Angelman syndrome patients.
In conclusion, we report an atypical AS child with autistic features and obesity caused by UPD of chromosome 15. Microarray analysis first identified the absence of heterozygosity of the q arm of chromosome 15. Subsequently, MS-PCR confirmed AS in this patient and microsatellite haplotype analysis revealed that he had received both chromosomes 15 from his father, which confirmed paternal UPD, leading to a final diagnosis of AS with UPD, with clinical features milder than a typical AS patient. Our findings suggest that a PWS/AS investigation may be useful in ASD cases with unknown etiology, as finding the underlying cause of ASD can lead to better genetic counseling for the family of the patient.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed directed to the first author or corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Ethics Committee of the participating institutes (REC 48/364–006-3). The legal guardian of each patient provided written informed consent to participate in this study. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
AH designed and performed the experiments, analyzed the data and co-wrote the manuscript. PK performed the FISH experiment, co-wrote the manuscript. CC analyzed the chromosomal microarray data. JW recruited and evaluated the patients. PL supervised the project, designed the experiments, obtained funding, and co-wrote the manuscript. All authors reviewed and approved the final manuscript.
Funding
This work was supported by a grant from the Faculty of Medicine, Prince of Songkla University (48/364-006-3).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Kantapon Dissaneewate who performed the FISH analysis and helped to collect additional clinical data on the proband in the study. We also thank Thanya Sripo and Oradawan Plong-On for performing and interpreting the MS-PCR analysis. We thank David Patterson of the International Affairs Office, Faculty of Medicine, Prince of Songkla University, for English language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.755605/full#supplementary-material
References
Battaglia, A., Doccini, V., Bernardini, L., Novelli, A., Loddo, S., Capalbo, A., et al. (2013). Confirmation of Chromosomal Microarray as a First-Tier Clinical Diagnostic Test for Individuals with Developmental Delay, Intellectual Disability, Autism Spectrum Disorders and Dysmorphic Features. Eur. J. Paediatric Neurol. 17, 589–599. doi:10.1016/j.ejpn.2013.04.010
Bennett, J. A., Germani, T., Haqq, A. M., and Zwaigenbaum, L. (2015). Autism Spectrum Disorder in Prader-Willi Syndrome: A Systematic Review. Am. J. Med. Genet. 167, 2936–2944. doi:10.1002/ajmg.a.37286
Beygo, J., Buiting, K., Ramsden, S. C., Ellis, R., Clayton-Smith, J., and Kanber, D. (2019). Update of the EMQN/ACGS Best Practice Guidelines for Molecular Analysis of Prader-Willi and Angelman Syndromes. Eur. J. Hum. Genet. 27, 1326–1340. doi:10.1038/s41431-019-0435-0
Bonati, M. T., Russo, S., Finelli, P., Valsecchi, M. R., Cogliati, F., Cavalleri, F., et al. (2007). Evaluation of Autism Traits in Angelman Syndrome: a Resource to Unfold Autism Genes. Neurogenetics 8, 169–178. doi:10.1007/s10048-007-0086-0
Brennan, M.-L., Adam, M. P., Seaver, L. H., Myers, A., Schelley, S., Zadeh, N., et al. (2015). Increased Body Mass in Infancy and Early Toddlerhood in Angelman Syndrome Patients with Uniparental Disomy and Imprinting center Defects. Am. J. Med. Genet. 167, 142–146. doi:10.1002/ajmg.a.36831
Buiting, K., Williams, C., and Horsthemke, B. (2016). Angelman Syndrome - Insights into a Rare Neurogenetic Disorder. Nat. Rev. Neurol. 12, 584–593. doi:10.1038/nrneurol.2016.133
Chan, C. T., Clayton-Smith, J., Cheng, X. J., Buxton, J., Webb, T., Pembrey, M. E., et al. (1993). Molecular Mechanisms in Angelman Syndrome: a Survey of 93 Patients. J. Med. Genet. 30, 895–902. doi:10.1136/jmg.30.11.895
Charalsawadi, C., Maisrikhaw, W., Praphanphoj, V., Wirojanan, J., Hansakunachai, T., Roongpraiwan, R., et al. (2014). A Case with a Ring Chromosome 13 in a Cohort of 203 Children with Non-syndromic Autism and Review of the Cytogenetic Literature. Cytogenet. Genome Res. 144, 1–8. doi:10.1159/000365909
Cook, E. H., Lindgren, V., Leventhal, B. L., Courchesne, R., Lincoln, A., Shulman, C., et al. (1997). Autism or Atypical Autism in Maternally but Not Paternally Derived Proximal 15q Duplication. Am. J. Hum. Genet. 60, 928–934.
Cook, E. H., Courchesne, R. Y., Cox, N. J., Lord, C., Gonen, D., Guter, S. J., et al. (1998). Linkage-disequilibrium Mapping of Autistic Disorder, with 15q11-13 Markers. Am. J. Hum. Genet. 62, 1077–1083. doi:10.1086/301832
Depienne, C., Moreno-De-Luca, D., Heron, D., Bouteiller, D., Gennetier, A., Delorme, R., et al. (2009). Screening for Genomic Rearrangements and Methylation Abnormalities of the 15q11-Q13 Region in Autism Spectrum Disorders. Biol. Psychiatry 66, 349–359. doi:10.1016/j.biopsych.2009.01.025
Fridman, C., Varela, M. C., Valente, K., Marques-Dias, M. J., and Koiffmann, C. P. (2002). Phenotypic and Behavioral Variability within Angelman Syndrome Group with UPD. Genet. Mol. Biol. 25, 127–130. doi:10.1590/S1415-47572002000200002
Hansakunachai, T., Roongpraiwan, R., Sombuntham, T., Limprasert, P., and Ruangdaraganon, N. (2014). A New Structured Interview for Children with Autism Spectrum Disorder Based on the DSM-IV. J. Med. Assoc. Thai. 97 (Suppl. 8), S7–S14.
Horváth, E., Horváth, Z., Isaszegi, D., Gergev, G., Nagy, N., Szabó, J., et al. (2013). Early Detection of Angelman Syndrome Resulting from De Novo Paternal Isodisomic 15q UPD and Review of Comparable Cases. Mol. Cytogenet. 6, 35. doi:10.1186/1755-8166-6-35
Hussain Askree, S., Hjelm, L. N., Ali Pervaiz, M., Adam, M., Bean, L. J. H., Hedge, M., et al. (2011). Allelic Dropout Can Cause False-Positive Results for Prader-Willi and Angelman Syndrome Testing. J. Mol. Diagn. 13, 108–112. doi:10.1016/j.jmoldx.2010.11.006
Kearney, H. M., Thorland, E. C., Brown, K. K., Quintero-Rivera, F., and South, S. T. (2011). Working Group of the American College of Medical Genetics Laboratory Quality Assurance CommitteeAmerican College of Medical Genetics Standards and Guidelines for Interpretation and Reporting of Postnatal Constitutional Copy Number Variants. Genet. Med. 13, 680–685. doi:10.1097/GIM.0b013e3182217a3a
Khatri, N., and Man, H.-Y. (2019). The Autism and Angelman Syndrome Protein Ube3A/E6AP: The Gene, E3 Ligase Ubiquitination Targets and Neurobiological Functions. Front. Mol. Neurosci. 12, 109. doi:10.3389/fnmol.2019.00109
Kubota, T., Das, S., Christian, S. L., Baylin, S. B., Herman, J. G., and Ledbetter, D. H. (1997). Methylation-specif Ic PCR Simplifies Imprinting Analysis. Nat. Genet. 16, 16–17. doi:10.1038/ng0597-1510.1038/ng0597-16
Lossie, A. C., Whitney, M. M., Amidon, D., Dong, H. J., Chen, P., Theriaque, D., et al. (2001). Distinct Phenotypes Distinguish the Molecular Classes of Angelman Syndrome. J. Med. Genet. 38, 834–845. doi:10.1136/jmg.38.12.834
Luk, H. M., and Lo, I. F. M. (2016). Angelman Syndrome in Hong Kong Chinese: A 20 years' Experience. Eur. J. Med. Genet. 59, 315–319. doi:10.1016/j.ejmg.2016.05.003
Mertz, L. G. B., Thaulov, P., Trillingsgaard, A., Christensen, R., Vogel, I., Hertz, J. M., et al. (2014). Neurodevelopmental Outcome in Angelman Syndrome: Genotype-Phenotype Correlations. Res. Dev. Disabilities 35, 1742–1747. doi:10.1016/j.ridd.2014.02.018
Miller, D. T., Adam, M. P., Aradhya, S., Biesecker, L. G., Brothman, A. R., Carter, N. P., et al. (2010). Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 86, 749–764. doi:10.1016/j.ajhg.2010.04.006
Moss, J., Howlin, P., Hastings, R. P., Beaumont, S., Griffith, G. M., Petty, J., et al. (2013). Social behavior and characteristics of autism spectrum disorder in Angelman, Cornelia de Lange, and Cri du Chat syndromes. Am. J. Intellect. Dev. Disabil. 118, 262–283. doi:10.1352/1944-7558-118.4.262
Peters, S., Beaudet, A., Madduri, N., and Bacino, C. (2004). Autism in Angelman Syndrome: Implications for Autism Research. Clin. Genet. 66, 530–536. doi:10.1111/j.1399-0004.2004.00362.x
Peters, S. U., Horowitz, L., Barbieri-Welge, R., Taylor, J. L., and Hundley, R. J. (2012). Longitudinal Follow-Up of Autism Spectrum Features and Sensory Behaviors in Angelman Syndrome by Deletion Class. J. Child. Psychol. Psychiatry 53, 152–159. doi:10.1111/j.1469-7610.2011.02455.x
Poyatos, D., Guitart, M., Gabau, E., Brun, C., Mila, M., Vaquerizo, J., et al. (2002). Severe Phenotype in Angelman Syndrome Resulting from Paternal Isochromosome 15. J. Med. Genet. 39, 4e–4. doi:10.1136/jmg.39.2.e4
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical Standards for the Interpretation and Reporting of Constitutional Copy-Number Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22, 245–257. doi:10.1038/s41436-019-0686-8
Sahoo, T., Peters, S. U., Madduri, N. S., Glaze, D. G., German, J. R., Bird, L. M., et al. (2006). Microarray Based Comparative Genomic Hybridization Testing in Deletion Bearing Patients with Angelman Syndrome: Genotype-Phenotype Correlations. J. Med. Genet. 43, 512–516. doi:10.1136/jmg.2005.036913
Saitoh, S., Wada, T., Okajima, M., Takano, K., Sudo, A., and Niikawa, N. (2005). Uniparental Disomy and Imprinting Defects in Japanese Patients with Angelman Syndrome. Brain Dev. 27, 389–391. doi:10.1016/j.braindev.2003.12.013
Samanta, D. (2021). Epilepsy in Angelman Syndrome: A Scoping Review. Brain Dev. 43, 32–44. doi:10.1016/j.braindev.2020.08.014
Sanders, S. J., He, X., Willsey, A. J., Ercan-Sencicek, A. G., Samocha, K. E., Cicek, A. E., et al. (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. doi:10.1016/j.neuron.2015.09.016
Schaefer, G. B., Mendelsohn, N. J., and Professional Practice and Guidelines Committee, (2013). Clinical Genetics Evaluation in Identifying the Etiology of Autism Spectrum Disorders: 2013 Guideline Revisions. Genet. Med. 15, 399–407. doi:10.1038/gim.2013.32
Schroer, R. J., Phelan, M. C., Michaelis, R. C., Crawford, E. C., Skinner, S. A., Cuccaro, M., et al. (1998). Autism and Maternally Derived Aberrations of Chromosome 15q. Am. J. Med. Genet. 76, 327–336. doi:10.1002/(sici)1096-8628(19980401)76:4<327::aid-ajmg8>3.0.co;2-m
Steffenburg, S., Gillberg, C. L., Steffenburg, U., and Kyllerman, M. (1996). Autism in Angelman Syndrome: a Population-Based Study. Pediatr. Neurol. 14, 131–136. doi:10.1016/0887-8994(96)00011-2
Thompson, R. J., and Bolton, P. F. (2003). Case Report: Angelman Syndrome in an Individual with a Small SMC(15) and Paternal Uniparental Disomy: a Case Report with Reference to the Assessment of Cognitive Functioning and Autistic Symptomatology. J. Autism Dev. Disord. 33, 171–176. doi:10.1023/a:1022991410822
Trillingsgaard, A., and Østergaard, J. R. (2004). Autism in Angelman Syndrome. Autism 8, 163–174. doi:10.1177/1362361304042720
Tsai, A. C., Gibby, T., Beischel, L., McGavran, L., and Johnson, J. P. (2004). A Child with Angelman Syndrome and Trisomy 13 Findings Due to Associated Paternal UPD 15 and Segmental UPD 13. Am. J. Med. Genet. 126A, 208–212. doi:10.1002/ajmg.a.20581
Varela, M. C., Kok, F., Otto, P. A., and Koiffmann, C. P. (2004). Phenotypic Variability in Angelman Syndrome: Comparison Among Different Deletion Classes and between Deletion and UPD Subjects. Eur. J. Hum. Genet. 12, 987–992. doi:10.1038/sj.ejhg.5201264
Vatsa, N., and Jana, N. R. (2018). UBE3A and its Link with Autism. Front. Mol. Neurosci. 11, 448. doi:10.3389/fnmol.2018.00448
Williams, C. A., Beaudet, A. L., Clayton-Smith, J., Knoll, J. H., Kyllerman, M., Laan, L. A., et al. (2006). Angelman Syndrome 2005: Updated Consensus for Diagnostic Criteria. Am. J. Med. Genet. 140A, 413–418. doi:10.1002/ajmg.a.31074
Wink, L. K., Fitzpatrick, S., Shaffer, R., Melnyk, S., Begtrup, A. H., Fox, E., et al. (2015). The Neurobehavioral and Molecular Phenotype of Angelman Syndrome. Am. J. Med. Genet. 167, 2623–2628. doi:10.1002/ajmg.a.37254
Keywords: angelman syndrome, autism, methylation-specific PCR, microarray, uniparental disomy
Citation: Hnoonual A, Kor-anantakul P, Charalsawadi C, Worachotekamjorn J and Limprasert P (2021) Case Report: An Atypical Angelman Syndrome Case With Obesity and Fulfilled Autism Spectrum Disorder Identified by Microarray. Front. Genet. 12:755605. doi: 10.3389/fgene.2021.755605
Received: 09 August 2021; Accepted: 07 September 2021;
Published: 22 September 2021.
Edited by:
Lawrence Todd Reiter, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Sarika Peters, Vanderbilt University Medical Center, United StatesJames Resnick, University of Florida, United States
Copyright © 2021 Hnoonual, Kor-anantakul, Charalsawadi, Worachotekamjorn and Limprasert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pornprot Limprasert, bHBvcm5wcm9AeWFob28uY29t