 Raghad Alharthi1,2Muhannad A. Alnahdi1,3Ahad Alharthi1†
Raghad Alharthi1,2Muhannad A. Alnahdi1,3Ahad Alharthi1† Seba Almutairi1,4Sultan Al-Khenaizan1,2
Seba Almutairi1,4Sultan Al-Khenaizan1,2 Mohammed A. AlBalwi1,5,6*
Mohammed A. AlBalwi1,5,6*- 1College of Medicine, King Saud Bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia
- 2Department of Dermatology, King Abdulaziz Medical City, Ministry of National Guard Health Affairs, Riyadh, Saudi Arabia
- 3Department of Ophthalmology, Ministry of National Guard Health Affairs, King Abdulaziz Medical City, Riyadh, Saudi Arabia
- 4Department of Dermatology, King Fahad University Hospital, Al Khobar, Saudi Arabia
- 5Department of Pathology and Laboratory Medicine, Ministry of National Guard Health Affairs, King Abdulaziz Medical City, Riyadh, Saudi Arabia
- 6Medical Genomic Research Department, Ministry of National Guard Health Affairs, King Abdullah International Medical Research Center, Riyadh, Saudi Arabia
Epidermolysis bullosa (EB) is a rare heterogeneous genetic mechanobullous skin disorder that is characterized by increased skin fragility leading to blistering following minor trauma. EB may be inherited as an autosomal dominant or an autosomal recessive disorder and can be classified into dystrophic EB (DEB), junctional EB (JEB), and EB simplex (EBS). A total of 28 Saudi patients with EB were included in this observational, retrospective chart-review study. A consecutive non-probability sampling technique was used to approach all affected patients. Molecular analysis was done to test the patients’ genomic DNA using a custom-designed AmpliSeq panel of suspected genes. All disease-causing variants were checked against available public databases. Twelve patients (42.9%) were found to have DEB, 6 patients (21.4%) with JEB, and 10 patients (35.7%) with EBS. The molecular genetic results revealed detections of 24 various homozygous genetic variations in the genes associated with EB, of which 14 were novel mutations. The most frequent variations were detected in COL7A1 in 12 cases (42.9%), followed by LAMB3 in 5 cases (17.9%), TGM5 in 4 cases (14.3%), and other genes. Furthermore, the majority (87.5%) of EB cases were confirmed to have homozygous mutations, and few were documented with positive consanguinity history. Only 3 cases (12.5%) were found to be autosomal dominant displaying heterozygous mutations. This is the first study to establish the EB genetic profile in Saudi Arabia where DEB is the most frequent type. A total of 14 novel mutations were identified that had not been previously reported. Consanguineous marriage is clearly recognized in the Saudi population; therefore, we propose a nationwide EB program that would help extend the spectrum of the genetic profile and help in the diagnosis and better understanding of this disease.
Introduction
Inherited epidermolysis bullosa (EB) is a heterogeneous group of skin disorders characterized by increased skin fragility leading to blister formation following minor trauma (Fine 2010; Mariath et al., 2020). Worldwide, it is estimated that the EB prevalence is about 19.6 per one million of live-born infants (Fine 2016). EB may be inherited as either autosomal dominant or autosomal recessive. This disorder is caused so far by more than 29 gene mutations encoding structural proteins within the skin with functional absence or loss that leads to instability of the micro-architectural connections between the dermis and epidermis, leading to blister formation (Has et al., 2020; Mariath et al., 2020). To date, there are over 30 subtypes of EB recognized, which are classified into four major groups based on clinical or molecular studies: dystrophic EB (DEB), junctional EB (JEB), EB simplex (EBS), and recently Kindler syndrome (Has et al., 2020). Kindler syndrome is a rare type of EB caused by mutations in the FERMT1 gene and is inherited in an autosomal recessive pattern. Dystrophic EB is caused by mutations in the gene encoding type VII collagen leading to the separation of the sub-basal lamina. DEB is inherited in an autosomal recessive or autosomal dominant pattern. Junctional EB results from mutations in genes encoding either laminin-332 or collagen type XVII, resulting in blister formation within the lamina lucida of the basement membrane. JEB is inherited in an autosomal recessive pattern. EBS results from intra-epidermal separation with mild systemic involvement ascribed to mutations encoding KRT5 and KRT14, resulting in a disturbance of the stability of the keratin filament network. EBS is usually inherited in an autosomal dominant pattern, but in rare cases, it is inherited as autosomal recessive.
In Saudi Arabia (SA), few EB cases were reported in the Eastern Province among dermatology clinic case reviews without detailing their genetic characteristics (Fine 2016). EB research is scarce in the region unlike in other parts of the world, so this study aims to highlight the genetic perspective in Saudi EB patients at a tertiary healthcare center.
The EB patients’ quality of life is highly impacted, as even the mildest form of the disorder leads to blisters and wounds that are quite painful (Abahussein et al., 1993; Tabolli et al., 2009). Potential complications are anemia, vocal cord stenosis, obstructive urethral lesions, and scarring and visual impairment (Abahussein et al., 1993; Fine et al., 2008; Fine and Mellerio 2009a; Fine and Mellerio 2009b). Patients have claimed suffering from physical and psychological restrictions like physical pain, lack of engagement in social activities, and embarrassment owing to their skin appearance (Horn and Tidman 2002a; Fine et al., 2009).
Materials and methods
Subjects
We performed an observational and retrospective chart-review study of 28 Saudi’ patients at King Abdulaziz Medical City, a tertiary care hospital in Riyadh, SA. The enrolled patients were diagnosed with EB and skin fragility disorders in the period between 1998 and 2020 and treated at the same center under the divisions of dermatology, general pediatrics, ophthalmology, and dentistry. A consecutive non-probability sampling technique was used to review the files of the patients. All required data were retrieved and gathered from the hospital BestCare system as well as from the database of the molecular pathology and genetics laboratory. Institutional Review Board (IRB) approval was obtained from the ethics committee of King Abdullah International Medical Research Center under RC19/250/R. Data collected from the patients’ files include sociodemographic, clinical, laboratory, and genetic data.
Genetic analysis
Molecular analysis of these cases was carried out by testing genomic DNA and checking for genetic variations of all exons and exon/intron boundaries using a custom-designed AmpliSeq panel that includes the following genes: CD151, CHST8, COL17A1, COL7A1, CSTA, DSG1, DSG2, DSG3, DSG4, DSP, DST, EXPH5, FERMT1, GRIP1, ITGA3, ITGA6, ITGB4, KRT1, KRT10, KRT14, KRT5, LAMA3, LAMB3, LAMC2, MMP1, NID1, PKP1, PLEC, and TGM5. All disease-causing variants were checked against the Human Gene Mutation Database (HGMD), ClinVar, the Genome Aggregation Database (gnomAD), and the Exome Aggregation Consortium (ExAC). The in silico tools SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (http://www.mutationtaster.org were used to predict coding variant effects on protein function. The collected data were entered into Microsoft Excel and analyzed using a simple statistical parameter through IBM Statistical Package for Social Sciences (SPSS) version 24. Numerical variables are presented as mean and standard deviation, and categorical variables are presented as frequencies and percentages.
Results
The population is represented with a 1.3:1 male-to-female ratio, as male patients were 16 (57%) and female patients were 12 (42.9%). The mean age was 8.9 ± 5.4 years old, the youngest patient was 3 years, and the oldest was 21 years old. Positive consanguinity history was documented in 9 patients, while family history was noted in 6 patients.
Dystrophic EB 12 (42.9%) was the most frequent subtype, followed by EB simplex 10 (35.7%) and junctional EB 6 (21.4%). Phenotypic presentations of each classification are shown in Figures 1–3. The mutations were detected in 7 genes: COL7A1, LAMB3, TGM5, PLEC, DST, KRT14, and COL17A1. Mutations implicated with COL7A1 were the most frequent in which they were found in 12 (42.9%) patients, followed by mutations with LAMB3 in 5 (17.9%) patients, TGM5 with 4 (14.3%) patients, PLEC with 3 (10.7%) patients, DST with 2 (7%) patients, KRT14 with 1 (3.6%) patient, and COL17A1 with 1 (3.6%) patient (Table 1).
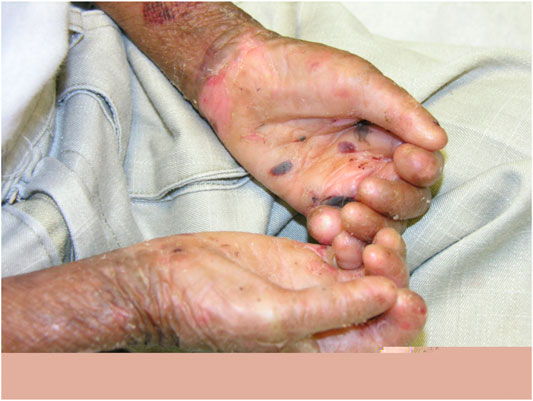
FIGURE 1. Clinical presentation of dystrophic epidermolysis bullosa.
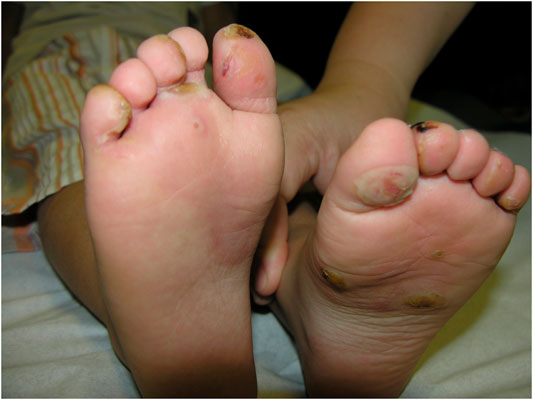
FIGURE 2. Clinical presentation of epidermolysis bullosa simplex.
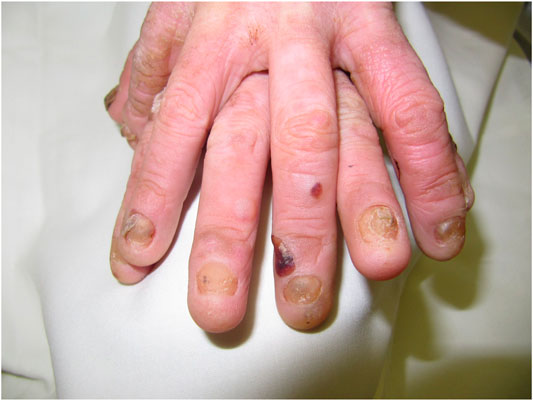
FIGURE 3. Clinical presentation of junctional epidermolysis bullosa.

TABLE 1. Frequency of the genes involved in all EB patients.
The clinical features of all patients were of the usual phenotype seen in EB patients, namely, mechanobullous fragility and blisters with a wide range of severity according to the genotype. Furthermore, nail deformities, tooth decay, lesions and erosive ulcerations in the oral cavity, and recurrent respiratory and urinary tract infections have been observed. None of patients had any gastrointestinal complications with an exception of one patient who had pyloric atresia. However, we did not detect any unusual other clinical features even in patients with novel mutations.
Genetic analysis of the implicated genes revealed 24 mutations identified among all enrolled cases. Among them, 14 mutations have not been reported to date. Autosomal recessive inheritance prevailed in 25 (89.3%) cases, and only 3 (10.7%) cases were found to be autosomal dominant (Table 2). A total of 4 different mutations were found in more than one patient, and those genes were diagnosed within another member of the same family. A total of 3 cases had the same mutation in COL7A1 (case nos. 6, 7, and 8), and another mutation of COL7A1 was found in two cases (case nos. 10, 11). Two cases had the same mutation involved in LAMB3 (case nos. 17 and 18), and two additional patients had the same gene mutations in TGM5 (case nos. 26 and 27).
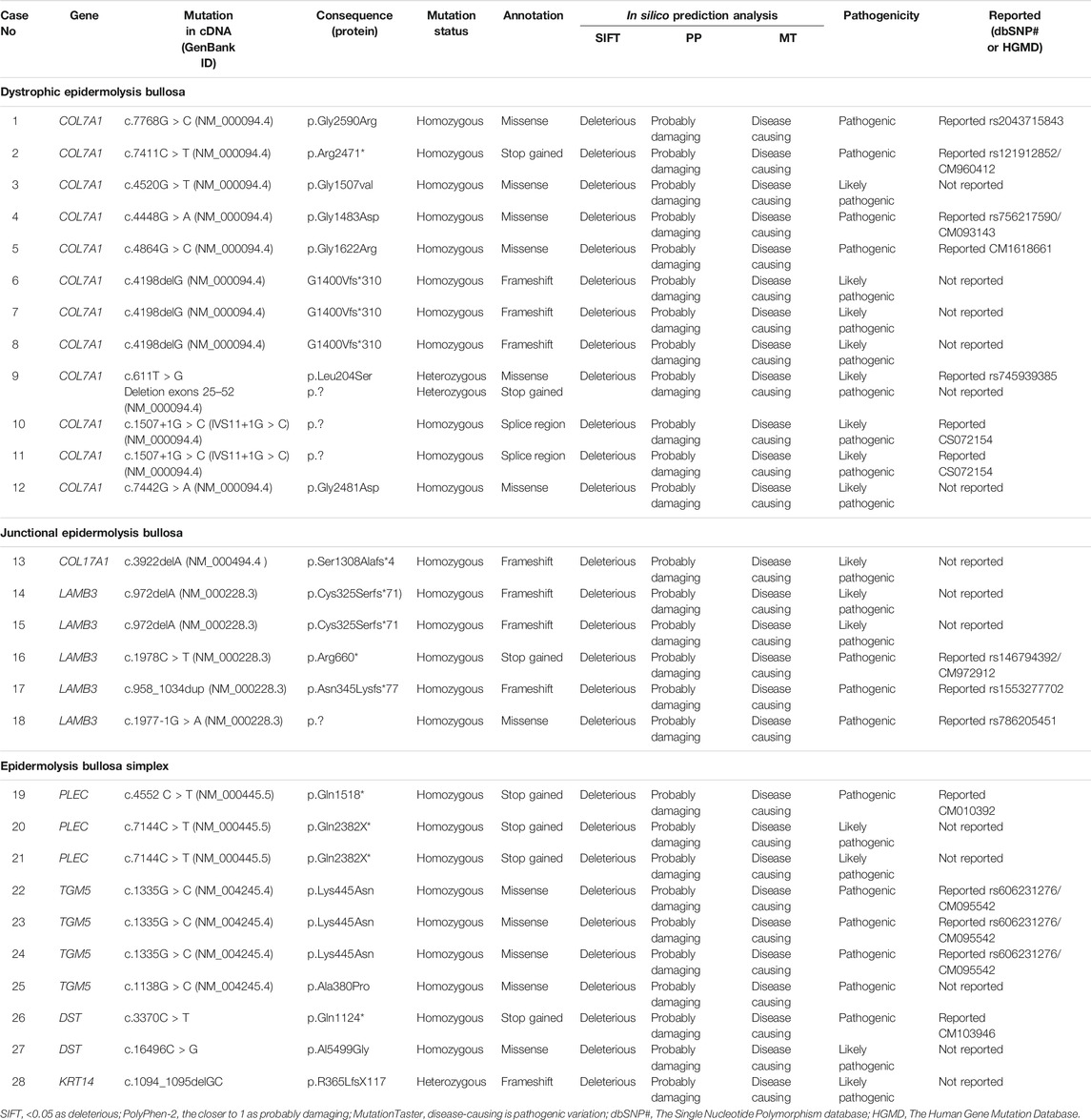
TABLE 2. Genes, variants, mutation types, and novelty status per EB classifications.
Discussion
DEB is a rare inherited EB caused by mutations involving the genes that encode type VII collagen leading to the separation of the sub-basal lamina (Brun et al., 2017). Recessive DEB has a wider array of severity and milder/localized form that has acral and nail involvement, similar to other forms of DEB. In particular, DEB patients have a significant risk of developing aggressive squamous cell carcinoma in chronic lesion sites (Mitsuhashi and Hashimoto 2003). The severe form is characterized by generalized blistering of the hands and feet, usually involving the acral surfaces, leading to pseudosyndactyly and flexural contractures that intensify with age (Bruckner-Tuderman 2010). Clinically, our cases do not differ much from what were described internationally. Although gastrointestinal complications are common in patients with EB, this was not the case in our patients since only one patient had pyloric atresia.
DEB was the most frequent subtype in the study population. Twelve patients with DEB were detected with 10 different homozygous variants in COL7A1. A total of 5 mutations are novel: 4 missense and 1 frameshift (Table 2). The other 5 reported variants are 4 missense mutations and 1 non-sense mutation that were previously reported (Horn and Tidman 2002b; Almaani et al., 2009). These results expand the spectrum of identified mutations implicated with DEB.
Six patients with JEB were among the study population. A total of 3 patients had novel mutations, and the other 3 patients had previously reported mutations. Two novel homozygous mutations were detected in 2 genes, COL17A1 and LAMB3; 1 mutation with frameshift in COL17A1; and 1 frameshift in LAMB3 gene (Table 2). In those with consanguineous history, homozygous mutations were identified. The reported mutations are 1 non-sense, 1 frameshift, and 1 missense in LAMB3 genes, which were previously reported by Christiano et al. and Pulkkinen et al. (Christiano et al., 1996; Pulkinnen and Uitto, 1999).
EBS was diagnosed in 10 patients with 8 different mutations in 4 genes: TGM5, PLEC, DST, and KRT14. Four are pathogenic mutations that are not reported to date. The 4 novel mutations of EBS in 3 different genes are 1 missense in TGM5, 2 non-sense in PLEC, and 1 missense in DST (Table 2). The other 5 reported mutations are 2 missense in TGM5, one non-sense mutation involving PLEC, and one frameshift in KRT14 gene that were previously reported (Fine and Mellerio 2009b; Fine et al., 2008; Mitsuhashi and Hashimoto 2003). TGM5 mutations may belong to other disorders with skin fragility and not to classical EB (Has et al., 2020). Almost all cases (Table 2) were inherited in an autosomal recessive pattern harboring either homozygous or compound heterozygous allele variants, although the loss-of-function mutations might have resulted from missense or frameshift mutations through the mechanism of non-sense-mediated mRNA decay. Indeed, it seems likely that the presence of homozygosity in our Saudi patients conformed to a high fertility rate and consanguinity rate, which reached more than 50% in some areas (Scott et al., 2016).
It is worth mentioning that all newly reported variants in this study were evaluated for their impact on protein function and structure using in silico prediction tools such as Sorting Intolerant From Tolerant (SIFT), Polymorphism Phenotyping v2 (PolyPhen-2), and MutationTaster. However, the final protein function pathogenicity effect of these mutant variants and the association with the EB disease may require further functional verification.
Study limitation
The study was conducted at a single tertiary care center at central SA, yet it is the first pillar to establish the Saudi EB genetic profile. Secondly, the retrospective design has made the study more subjective to missing some relevant data. However, the effects do not have any major impact on the findings. We propose therefore further collaboration between various centers from different parts of the region to have substantial effects on the sample size, including diversifying the patients’ backgrounds.
Conclusion
The study elaborates and reports on the genetic profile and prevalence of EB in Saudi patients at a single tertiary healthcare center in central SA. Dystrophic EB was the highest reported subcategory among all the other EB classifications. COL7A1 was the most common gene identified among the patients. Positive family history of consanguinity was evident as expected; further highlighting its role through education is needed among Saudi EB patients.
This study entails an impact on the future of identifying the genetic characteristics of Saudi EB patients, along with emphasizing on the need to launch an entity that would be responsible for directing the efforts of initiating a national center for EB.
Author’s Note
This manuscript is dedicated to Ahad Alharthi one of our interns who participated in this research and passed away on 27th July 2021. Despite her short professional life as a medical student and intern, she participated in many researches and was a leader in many skin awareness campaigns.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by King Abdullah International Medical Research Center (RC19/250/R). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Authors Contributions
RA, MuAA, AA, and SA were involved in collection of the patients’ clinical information, data analysis, and drafting of the manuscript. SA-K reviewed the patients’ clinical presentation, genetic data, and reviewed and revised the manuscript. MoAA designed and supervised the entire research and reviewed and revised the manuscript. All authors have read and approved the final version of manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors acknowledge the assistance of Zoe Poral Camarig for proofreading and editing the manuscript. This project is not funded.
References
Abahussein, A. A., al-Zayir, A. A., Mostafa, W. Z., and Okoro, A. N. (1993). Epidermolysis Bullosa in the Eastern Province of Saudi Arabia. Int. J. Dermatol. 32, 579–581. doi:10.1111/j.1365-4362.1993.tb05029.x
Almaani, N., Liu, L., Harrison, N., Tanaka, A., Lai-Cheong, J., and McGrath, J. (2009). New glycine Substitution Mutations in Type VII Collagen Underlying Epidermolysis Bullosa Pruriginosa but the Phenotype Is Not Explained by a Common Polymorphism in the Matrix Metalloproteinase-1 Gene Promoter. Acta Derm Venerol 89, 6–11. doi:10.2340/00015555-0605
Bruckner-Tuderman, L. (2010). Dystrophic Epidermolysis Bullosa: Pathogenesis and Clinical Features. Dermatol. Clin. 28, 107–114. doi:10.1016/j.det.2009.10.020
Brun, J., Chiaverini, C., Chiaverini, C., Devos, C., Leclerc-Mercier, S., Mazereeuw, J., et al. (2017). Pain and Quality of Life Evaluation in Patients with Localized Epidermolysis Bullosa Simplex. Orphanet J. Rare Dis. 12, 119. doi:10.1186/s13023-017-0666-5
Christiano, A. M., McGrath, J. A., Tan, K. C., and Uitto, J. (1996). Glycine Substitutions in the Triple-Helical Region of Type VII Collagen Result in a Spectrum of Dystrophic Epidermolysis Bullosa Phenotypes and Patterns of Inheritance. Am. J. Hum. Genet. 58, 671–681.
Fine, J.-D. (2016). Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates from the National Epidermolysis Bullosa Registry. JAMA Dermatol. 152, 1231–1238. doi:10.1001/jamadermatol.2016.2473
Fine, J.-D. (2010). Inherited Epidermolysis Bullosa: Past, Present, and Future. Ann. N. Y Acad. Sci. 1194, 213–222. doi:10.1111/j.1749-6632.2010.05463.x
Fine, J.-D., Johnson, L. B., Weiner, M., Li, K.-P., and Suchindran, C. (2009). Epidermolysis Bullosa and the Risk of Life-Threatening Cancers: The National EB Registry Experience, 1986-2006. J. Am. Acad. Dermatol. 60, 203–211. doi:10.1016/j.jaad.2008.09.035
Fine, J.-D., Johnson, L. B., Weiner, M., and Suchindran, C. (2008). Gastrointestinal Complications of Inherited Epidermolysis Bullosa: Cumulative Experience of the National Epidermolysis Bullosa Registry. J. Pediatr. Gastroenterol. Nutr. 46, 147–158. doi:10.1097/mpg.0b013e31812f5667
Fine, J.-D., and Mellerio, J. E. (2009). Extracutaneous Manifestations and Complications of Inherited Epidermolysis Bullosa. J. Am. Acad. Dermatol. 61, 367–384. doi:10.1016/j.jaad.2009.03.052
Fine, J.-D., and Mellerio, J. E. (2009). Extracutaneous Manifestations and Complications of Inherited Epidermolysis Bullosa. J. Am. Acad. Dermatol. 61, 387–402. doi:10.1016/j.jaad.2009.03.053
Has, C., Bauer, J. W., Bodemer, C., Bolling, M. C., Bruckner‐Tuderman, L., Diem, A., et al. (2020). Consensus Reclassification of Inherited Epidermolysis Bullosa and Other Disorders with Skin Fragility. Br. J. Dermatol. 183, 614–627. doi:10.1111/bjd.18921
Horn, H. M., and Tidman, M. J. (2002). Quality of Life in Epidermolysis Bullosa. Clin. Exp. Dermatol. 27, 707–710. doi:10.1046/j.1365-2230.2002.01121.x
Horn, H. M., and Tidman, M. J. (2002). The Clinical Spectrum of Dystrophic Epidermolysis Bullosa. Br. J. Dermatol. 146, 267–274. doi:10.1046/j.1365-2133.2002.04607.x
Mariath, L. M., Santin, J. T., Santin, L., and Kiszewski, A. E. (2020). Inherited Epidermolysis Bullosa: Update on the Clinical and Genetic Aspects. Anais Brasileiros de Dermatologia 95, 551–569. doi:10.1016/j.abd.2020.05.001
Mitsuhashi, Y., and Hashimoto, I. (2003). Genetic Abnormalities and Clinical Classification of Epidermolysis Bullosa. Arch. Dermatol. Res. 295, S29–S33. doi:10.1007/s00403-002-0369-0
Pulkinnen, L., and Uitto, J. (1999). Mutation Analysis and Molecular Genetics of Epidermolysis Bullosa. Matrix Biol. 18, 29–42.
Scott, E. M., Halees, A., Halees, A., Itan, Y., Spencer, E. G., He, Y., et al. (2016). Characterization of Greater Middle Eastern Genetic Variation for Enhanced Disease Gene Discovery. Nat. Genet. 48, 1071–1076. doi:10.1038/ng.3592
Keywords: epidermolysis bullosa, dystrophic epidermolysis bullosa, junctional epidermolysis bullosa, epidermolysis bullosa simplex, Saudi Arabia
Citation: Alharthi R, Alnahdi MA, Alharthi A, Almutairi S, Al-Khenaizan S and AlBalwi MA (2022) Genetic Profile of Epidermolysis Bullosa Cases in King Abdulaziz Medical City, Riyadh, Saudi Arabia. Front. Genet. 12:753229. doi: 10.3389/fgene.2021.753229
Received: 04 August 2021; Accepted: 24 December 2021;
Published: 10 February 2022.
Edited by:
Ming Li, Shanghai Jiaotong University, ChinaReviewed by:
Hiroyuki Wakiguchi, Yamaguchi University, JapanHiram Almeida Jr, Federal University of Pelotas, Brazil
Copyright © 2022 Alharthi, Alnahdi, Alharthi, Almutairi, Al-Khenaizan and AlBalwi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammed A. AlBalwi, YmFsd2ltQG5naGEubWVkLnNh
†Ahad Alharthi passed away in 2021