 Xiao Xiong1,2
Xiao Xiong1,2 Yogeshwar D Kelkar3Chris J Geden4
Yogeshwar D Kelkar3Chris J Geden4 Chao Zhang5
Chao Zhang5 Yidong Wang6
Yidong Wang6 Evelien Jongepier7Ellen O. Martinson3,8
Evelien Jongepier7Ellen O. Martinson3,8 Eveline C Verhulst6Jürgen Gadau9John H Werren3
Eveline C Verhulst6Jürgen Gadau9John H Werren3 Xu Wang1,10,11*†
Xu Wang1,10,11*†- 1Department of Pathobiology, College of Veterinary Medicine, Auburn University, Auburn, AL, United States
- 2School of Life Sciences and Technology, Tongji University, Shanghai, China
- 3Department of Biology, University of Rochester, Rochester, NY, United States
- 4Center for Medical, Agricultural and Veterinary Entomology, USDA Agricultural Research Service, Gainesville, FL, United States
- 5Department of Plastic and Reconstructive Surgery, Shanghai Ninth People’s Hospital, Shanghai Institute of Precision Medicine, Shanghai JiaoTong University School of Medicine, Shanghai, China
- 6Laboratory of Entomology, Wageningen University, Wageningen, Netherlands
- 7Institute for Biodiversity and Ecosystem Dynamics, University of Amsterdam, Amsterdam, Netherlands
- 8Department of Biology, University of New Mexico, Albuquerque, NM, United States
- 9Institute for Evolution & Biodiversity, University of Münster, Münster, Germany
- 10Alabama Agricultural Experiment Station, Center for Advanced Science, Innovation and Commerce, Auburn, AL, United States
- 11HudsonAlpha Institute for Biotechnology, Huntsville, AL, United States
The parasitoid wasp Muscidifurax raptorellus (Hymenoptera: Pteromalidae) is a gregarious species that has received extensive attention for its potential in biological pest control against house fly, stable fly, and other filth flies. It has a high reproductive capacity and can be reared easily. However, genome assembly is not available for M. raptorellus or any other species in this genus. Previously, we assembled a complete circular mitochondrial genome with a length of 24,717 bp. Here, we assembled and annotated a high-quality nuclear genome of M. raptorellus, using a combination of long-read (104× genome coverage) and short-read (326× genome coverage) sequencing technologies. The assembled genome size is 314 Mbp in 226 contigs, with a 97.9% BUSCO completeness score and a contig N50 of 4.67 Mb, suggesting excellent continuity of this assembly. Our assembly builds the foundation for comparative and evolutionary genomic analysis in the genus of Muscidifurax and possible future biocontrol applications.
Introduction
Muscidifurax (Hymenoptera: Pteromalidae) is a chalcid wasp genus with nine characterized species, all of which are pupal parasitoids. Muscidifurax raptor was the first species described in the genus, in 1910 by Girault and Sanders (Girault and Sanders, 1910). In 1970, four sibling species were described: M. zaraptor Kogan and Legner, collected from the southwestern United States; M. raptoroides Kogan and Legner collected from Central America and Mexico; M. raptorellus Kogan and Legner collected from Uruguay and Chile; and a thelytokous species M. uniraptor Kogan and Legner collected from the central mountain range of the island of Puerto Rico (Kogan and Legner, 1970). Based on the mitochondrial gene sequence alignment in this genus, the most closely related sexual species to the asexual M. uniraptor is M. raptorellus (Taylor et al., 1997). Four additional Muscidifurax species were identified in China (Xiao et al., 2018).
Muscidifurax raptorellus (Chilean strain) is a gregarious parasitoid that typically produces 2–10 offspring per parasitized host pupa (Geden and Moon, 2009). The number of eclosed offspring depends on the host size (Seidl and King, 1993). The population found in Uruguay is partially gregarious (Legner, 1969). Females can lay 16–20 eggs per day during their peak ovipositional period and about 150 eggs during their lifetime (Petersen and Currey, 1996). In sharp contrast, M. zaraptor only deposits one egg per host, and the first larva will eliminate subsequent larvae or eggs deposited by superparasitism (McKay and Broce, 2004). M. uniraptor only produces a single female offspring from each host, and the parthenogenesis is caused by the infection of A strain Wolbachia bacteria (Zchori-Fein et al., 2000; Newton et al., 2016). The diverse reproductive strategies make this genus an excellent model system for the study of sexual vs. asexual evolution.
M. raptorellus is an effective biological control agent of dipteran filth flies, including house fly (Musca domestica L.), stable fly (Stomoxys calcitrans L.), horn fly (Haematobia irritans L.), black dump fly [Hydrotaea aenescens (Weidemann)], and flesh fly (Sarcophaga bullata Parker) (Petersen and Currey, 1996; Geden and Hogsette, 2006; Geden and Moon, 2009). Application of insecticide, which is the primary control strategy, is of limited effectiveness due to the evolution of resistant genes in these pests. Parasitoid wasps have great potential as an alternative management strategy that is more environmentally friendly and sustainable (Heraty, 2009). Wasps in the genus Muscidifurax are also of interest for comparative genomic studies, due to their close relationship to the model parasitoid genus Nasonia, which currently has genome assemblies for three species (Werren et al., 2010; Wang et al., 2020), with Muscidifurax estimated to be 15 million years divergent (Martinson et al., 2017a). Here, we report the first draft genome assembly of M. raptorellus using PacBio long-read sequencing. This well-assembled and annotated genome will provide an essential genetic toolkit for functional and evolutionary genomic studies in M. raptorellus and its sibling species. The high-quality reference genome could also inform and facilitate future genome manipulation in parasitoid wasps for more effective biological control strategies (Leung et al., 2020).
Materials and Methods
Sample Source and Insect Rearing
The source of M. raptorellus used in this study was derived from a colony maintained by Dr. Chris Geden at the Center for Medical, Agricultural and Veterinary Entomology, USDA Agricultural Research Service (Gainesville, FL). Genomic sequencing samples were collected from two independent colonies, both derived from the same USDA colony: one maintained at the Auburn University College of Veterinary Medicine in Auburn, Alabama, since 2019 (Aub sample) and the other one maintained at Koppert Biological Systems in the Netherlands (Kop sample) since 20 years ago. M. raptorellus was originally collected in 1965 from Chile but was referred to as M. raptor (Legner et al., 1967); subsequently described as M. raptorellus in 1970 (Kogan and Legner, 1970); and afterward distributed in North America for biological control efforts. The current colony was originally established from field-collected specimens on a New York poultry farm (Kaufman et al., 2001) and maintained in the Geden laboratory on housefly pupae. Samples from the colony were obtained from the Werren laboratory in 2016 and maintained on Sarcophaga bullata pupae and then sent to the Wang laboratory in Auburn, Alabama, in 2019 and maintained on commercial Sarcophaga bullata pupae (flesh fly pupae) at a constant temperature of 25°C and 24 h constant light. The Kop sample was maintained on Lucilia spp. pupae for 20 years and was sent to the Verhulst laboratory in 2014 and maintained on Calliphora spp. pupae at 25°C and 18 h/6 h light/dark conditions. Both the Aub and Kop samples were from the same fully inbred strain of M. raptorellus.
Genomic DNA Extraction, Library Preparation, and Sequencing
High-molecular-weight (HMW) genomic DNA (gDNA) was extracted from adults of the M. raptorellus Aub sample using the Genomic-tip 20/G kit (Qiagen, Catalog No. 10223) with DNA concentration checked on a Qubit 3.0 Fluorometer (Thermo Fisher Scientific, United States). The size distribution and gDNA quality were assessed on an Agilent TapeStation 4200 machine (Agilent Technologies, CA) using the genomics kit (Agilent, Catalog No. 5067-5366). A total of 10 μg high-quality M. raptorellus genomic DNA was sheared into 20 kb fragments, and the end damage was repaired. After sequencing adapter ligation, the DNA fragment was annealed with Sequencing Primer v2 and Sequel II DNA Polymerase and bound to the SMRTbell templates, and the library was constructed following SMRTbell Template Prep Kit v2 following the CCS HiFi library protocol (Pacific Biosciences, CA). The size distribution of the constructed library was assessed using LabChip GX Touch HT (PerkinElmer, MA, United States), and the final library quantity was examined with a Qubit 3.0 Fluorometer (Thermo Fisher Scientific, United States). The PacBio library was sequenced on a PacBio Sequel II System at the HudsonAlpha Genome Sequencing Center (Supplementary Table S1).
HMW genomic DNA was diluted to ∼ 0.8 ng/μl with elution buffer for 10x Genomics library preparation using Chromium Genome Reagent Kit v2 (10× Genomics, Inc., CA). The diluted denatured gDNA, sample master mix, and gel beads were loaded to the genomic chip following the protocol and then ran on a 10× Chromium Controller to generate Gel Bead-In-EMulsions (GEMs). The obtained GEMs were used for the subsequent incubation and cleanup. The Chromium i7 Sample Index served as the library barcode to provide linked information. After quality control with a Qubit 3.0 Fluorometer (Thermo Fisher Scientific, MA, United States) and Agilent TapeStation 4200 (Agilent Technologies, CA), the 10× genomic sequencing was performed on an Illumina NovaSeq 6000 machine.
HMW gDNA was extracted from a pool of thirty females of the M. raptorellus Kop sample that were collected at the black pupal stage (∼16 days after egg-laying), using the Genomic-tip 100/G kit (Qiagen, Catalog No. 10243) combined with the Genomic DNA Buffer Set (Qiagen, Catalog No. 19060). The sample was ground to fine powder in liquid nitrogen by a plastic pestle, and the total DNA was extracted following the protocol provided by the manufacturer. After extraction, genomic DNA was sheared into 8–30 kb range by using g-TUBE (Covaris) following the manufacturer’s protocol. The quality and quantity of sheared genomic DNA were checked by gel electrophoresis with 1.5% TAE agarose gel stained with Midori Green (NIPPON Genetics) and by spectrophotometry (Nanodrop™ 2000, Thermo Fisher). The genomic DNA was measured and quality controlled at Novogene Co., Ltd. (Beijing, China). SMRTbell library templates were prepared for long-read sequencing on the PacBio Sequel system using three flow cells, to generate up to 70 kb long reads with an average read length of 12–15 kb. A total of 1.57 million high-quality subreads were obtained, with an estimated read depth of 55.8× (Supplementary Table S1).
Genome Assembly, Polishing, and Assessment
The raw sequencing reads (Aub sample) from both PacBio library and 10× Genomics library were checked for sequencing quality using FastQC (Andrews et al., 2010) before genome assembly. De novo genome assembly for the M. raptorellus Aub sample was performed by a Supernova 2.1.1 (Weisenfeld et al., 2017) assembler using 400 million reads subsampled from the total amount of reads generated from the 10× Genomics library. Filtered HiFi PacBio reads were assembled by hifiasm v0.13 (Cheng et al., 2021) and HiCanu v2.1.1 (Nurk et al., 2020), dedicated assemblers using long-read sequencing. The Kop CLS PacBio data were assembled using Canu v2.1 (Koren et al., 2017). The Kop CANU assembly was polished with Pilon (version 1.22; parameter settings: fix = all) (Walker et al., 2014) to correct small errors based on high-quality 150 bp paired-end Illumina short reads (Table 1). A final round of polishing with Arrow (VariantCaller version 2.1.0) was performed to correct large structural errors, based on the raw PacBio reads that were aligned with Minimap2 (Li, 2018). Aub and Kop cultures have identical mitochondrial genomes (100% sequence identity) with only one 11 bp indel. The Aub 10× Genomics reads were aligned to the repeat-masked Kop assembly using the Longranger v2.1.6 (Zheng et al., 2016) software suite with the ALIGN pipeline. 58,350 SNPs were called by UnifiedGenotyper in the Genome Analysis Toolkit (GATK) (McKenna et al., 2010; DePristo et al., 2011). SNP positions in repetitive regions and variants outside the coverage depth threshold (120–500 bp) were filtered out using BEDTools v2.30.0 (Quinlan, 2014). A total of 11,523 homozygote SNPs between Aub and Kop were identified, and the percentage of fixed differences in the nuclear genome was estimated to be 0.0038%. To achieve the best assembly, these draft assemblies with different assemblers from both Aub and Kop samples were merged into a draft assembly using an assembly combination tool quickmerge v0.3.0 (Chakraborty et al., 2016). Potential bacterial contaminations were checked using a pipeline described in our previous research (Wang et al., 2020), and no bacteria contig contamination was discovered. The draft assembly was polished to yield a final high-quality assembly with the 10× Genomics Illumina short reads for indel correction using Pilon v1.23.0 (Walker et al., 2014). The final genome assembly was evaluated based on the N50 size of contigs and RNA-seq read mapping percentages, and genome completeness was assessed by BUSCO version 4.0.6 (Seppey et al., 2019). The BUSCO scores were calculated using arthropoda_odb10 with a total of 1,013 orthologs.
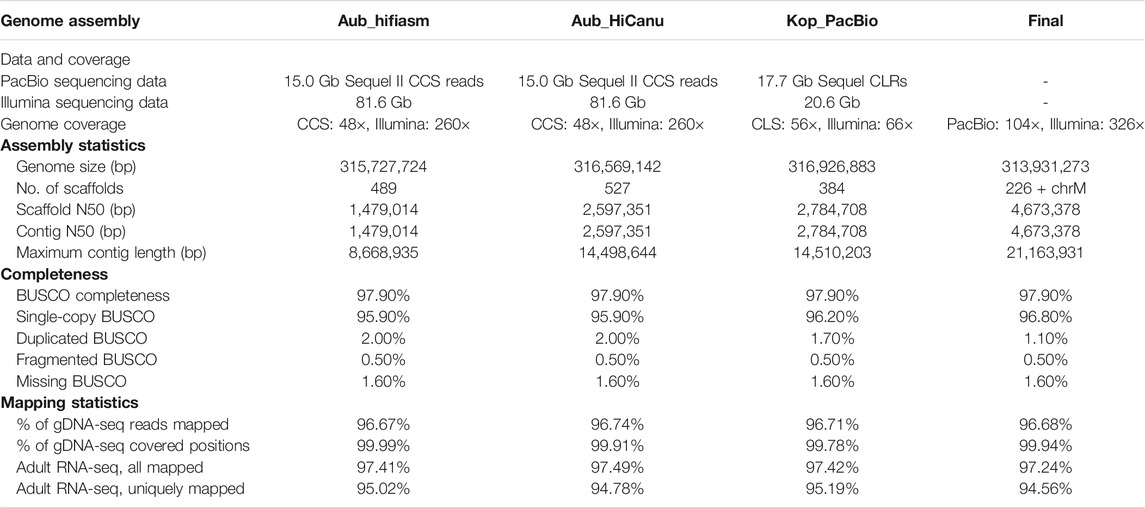
TABLE 1. Summary statistics of the Muscidifurax raptorellus genome assemblies.
RNA-Seq Data Processing and Transcriptome Assembly
Total RNA was isolated from adult whole-body samples of adult male and female M. raptorellus in three biological replicates for each sex from samples collected in the Werren laboratory. The RNA extraction, quantification, library preparation, and sequencing protocol were performed as previously described (Martinson et al., 2017b). A total of 308,475,537 reads were obtained from six samples. FastQC (Andrews et al., 2010) was used for quality control of raw RNA-seq data. The paired-end RNA-seq reads were processed with Trimmomatic v0.38 (Bolger et al., 2014). After trimming the potential adapter sequences, we performed de novo assembly of the M. raptorellus transcriptome using Trinity v2.4.0 (Haas et al., 2013), and pre-aligned transcripts were annotated by Cufflinks v2.2.1 (Trapnell et al., 2012).
Repeat Annotation
A de novo M. raptorellus repeat database was constructed using RepeatModeler v2.0.1 (Flynn et al., 2020) with the default parameters, which employs three complementary computational programs, RECON v1.0.8 (Bao and Eddy, 2002), RepeatScout v1.0.5 (Price et al., 2005), and Tandem Repeats Finder (TRF) (Benson, 1999), to annotate repetitive elements in our genome assembly. RepeatScout is a de novo repeat finder to identify highly conserved repetitive elements, while RECON can find less conserved elements. TRF is a program to locate and display tandem repeats. The high-quality library of transposable element (TE) families was then used to mask homologous repeats and low-complexity DNA sequences using RepeatMasker v4.0.6 (Chen, 2004) with RMBlast v2.10.0 as the default search engine.
Gene Prediction and Functional Annotation
To annotate the structures and functions of the M. raptorellus genome, we integrated ab initio and RNA-seq based methods to predict the genes in repeat-masked assembly. For RNA-seq prediction, the trimmed RNA-seq reads were aligned to the repeat-masked genome assembly using Tophat v2.1.1 (Kim et al., 2013) and then assembled into transcripts using cufflinks v2.2.1 (Trapnell et al., 2012) with default parameters. In addition, de novo assembly of M. raptorellus transcriptomes was achieved by Trinity v2.4.0 (Haas et al., 2013). The annotation of the genome assembly was performed using the MAKER v2.31.9 (Cantarel et al., 2008) annotation pipeline. Gene models were predicted using ab initio gene prediction algorithms with protein and transcriptome evidence by EST2GENOME and PROTEIN2GENOME procedures in MAKER (Data S1). The generated GFF3 file and assembled transcriptome from RNA-seq prediction were provided as expressed sequence tag (EST) evidence. The Arthropoda_odb10 dataset served as protein homology evidence. After evaluation and filtering with evidence scores, the predicted genes were used to train both SNAP (Korf, 2004) and AUGUSTUS (Stanke and Waack, 2003; Stanke et al., 2006) gene predictors. Two additional iterations were performed to generate the final predicted gene models for the M. raptorellus genome. A homology-based gene prediction tool, Gene Model Mapper (GeMoMa) (Keilwagen et al., 2019), was also utilized to annotate the coding genes in M. raptorellus using well-annotated Nasonia vitripennis OGS2 (official gene set 2) (Rago et al., 2016) as the protein reference.
Comparative Genome Analysis
To compare the genome structure between M. raptorellus and N. vitripennis genomes, the homologous regions in these two genomes were identified using MCScanX (Wang et al., 2012) with default parameters, which is a Python package for synteny detection and evolutionary analysis. The inferred gene pairs and linked relationships were visualized and placed in the context of whole-genome collinearity using a genomic circle generated by Circos (Krzywinski et al., 2009). The chromosome-level genome assembly of N. vitripennis (Nvit_psr_1.1) (Dalla Benetta et al., 2020) was downloaded at NCBI Assembly with accession number GCA_009193385.2.
Phylogenetic Analysis
To investigate the phylogenetic relationship between M. raptorellus and other Hymenoptera insect species, nine representative species (jewel wasp Nasonia vitripennis, honey bee Apis mellifera, turnip sawfly Athalia rosae, fig wasp Ceratosolen solmsi marchali, Indian jumping ant Harpegnathos saltator, Braconid wasp Microplitis demolitor, wood wasp Orussus abietinus, red paper wasp Polistes canadensis, and minute polyphagous wasp Trichogramma pretiosum) were selected from 40 Hymenoptera species in OrthoDB v10.1 (https://www.orthodb.org/) (Kriventseva et al., 2018). A total of 4,390 1:1 single-copy orthologs among these nine genomes were identified. The protein sequences for M. raptorellus were aligned to N. vitripennis using BLASTp alignments with a minimum of 60% sequence identity, and 3,662 1:1 orthologs were identified. The detailed information of 3,662 1:1 single-copy genes in the M. raptorellus genome and the nine representative Hymenoptera genomes is provided in Data S1. Subsequently, the protein sequences of the single-copy orthologs in the nine species were extracted from the OrthoDB fasta file, and M. raptorellus protein sequences of these genes were extracted from our genome assembly. The protein sequences across the selected Hymenoptera species and M. raptorellus were independently aligned with MAFFT v7.407 (Katoh and Standley, 2014). The protein alignments were concatenated for phylogenomic analysis. ProtTest 3 (Darriba et al., 2011) was used to estimate the best protein model of protein evolution. The maximum-likelihood (ML) phylogenetic tree was finally built with the concatenated protein sequence by using RAxML v8.2 (Stamatakis, 2014) with the best JTT protein model. 1,000 rapid bootstrap replicates were applied for evaluation of their branch supports. The tree was displayed by FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Results and Discussion
Genome Assembly and Assessment
Two independent PacBio libraries were constructed for the assembly of M. raptorellus genome (see Materials and Methods). The PacBio Sequel II HiFi reads (14,992,520,996 bp) generated from the Aub sample were assembled using hifiasm and HiCanu, and the Kop PacBio data (17,675,696,457 bp) were assembled using Canu (see Materials and Methods). The genome size of all three assemblies ranges from 315.7 to 316.9 Mbp (Table 1), which is very close to the estimated size from 10× Genomics data using Supernova based on K-mer profiles (315 Mbp), indicating high confidence in the genome size. The merged genome has significant improvement over individual assemblies, in terms of reduction in the number of contigs (from 527 to 226), the increase in contig N50 (from 1.5 to 4.7 Mb), and the maximum contig length (from 8.7 to 21.2 Mb), as well as a reduced proportion of duplicated BUSCO (from 2 to 1.1%; Table 1), without sacrificing the DNA and RNA sequencing mapping rate (Table 1). The final assembled genome is 313,931,273 bp in length with 226 scaffolds (the GC content is 40.06%) and a circularized mitochondrial genome (GenBank accession number MT985329) (Lin et al., 2021). The contig N50 is 4,673,378 bp, and the BUSCO completeness score is 97.9% (96.8% single-copy, 1.1% duplicated, 0.5% fragmented, and 1.6% missing). The adult RNA-seq reads were aligned to the M. raptorellus assembly using Tophat (Trapnell et al., 2009), and 97% of the reads were mapped to the genome. The 10× Genomics short-read data were also mapped to the genome assembly, and the alignment rate was 96.68%. The proportion of the genome with zero depth was 0.06%. The assembly and mapping statistics suggest that the quality of our assembly is high in both genome completeness and continuity (Table 1).
Syntenic Analysis With Nasonia vitripennis Genome
N. vitripennis and the congeners of M. raptorellus, M. uniraptor, and M. zaraptor have a haploid karyotype of n = 5 (Gokhman and Westendorff, 2000; Goodpasture, 1974; Silva-Junior et al., 2000). A total of 25 scaffolds from our M. raptorellus assembly with a total length of 187.4 Mb (59.7% of the whole assembly) were unambiguously aligned to the five assembled chromosomes in the N. vitripennis genome (Figure 1). The N. vitripennis chromosome assembly was based on recombination data between two closely related species (N. vitripennis and N. giraulti) (Niehuis et al., 2010; Desjardins et al., 2013), with all non-repetitive and non-centromeric regions correctly assembled and oriented (total chromosome size 159.4 Mb, 55% of the genome). The remaining 40% repetitive regions (Table 2) were not assembled into N. vitripennis chromosomes. The majority of N. vitripennis chromosomal regions have a collinearity relationship with M. raptorellus scaffolds (Table 1), suggesting high evolutionary conservation. The synteny analysis results also identified regional inversion, translocation, and duplication events, which will shed light on the genome evolution in these two genera.
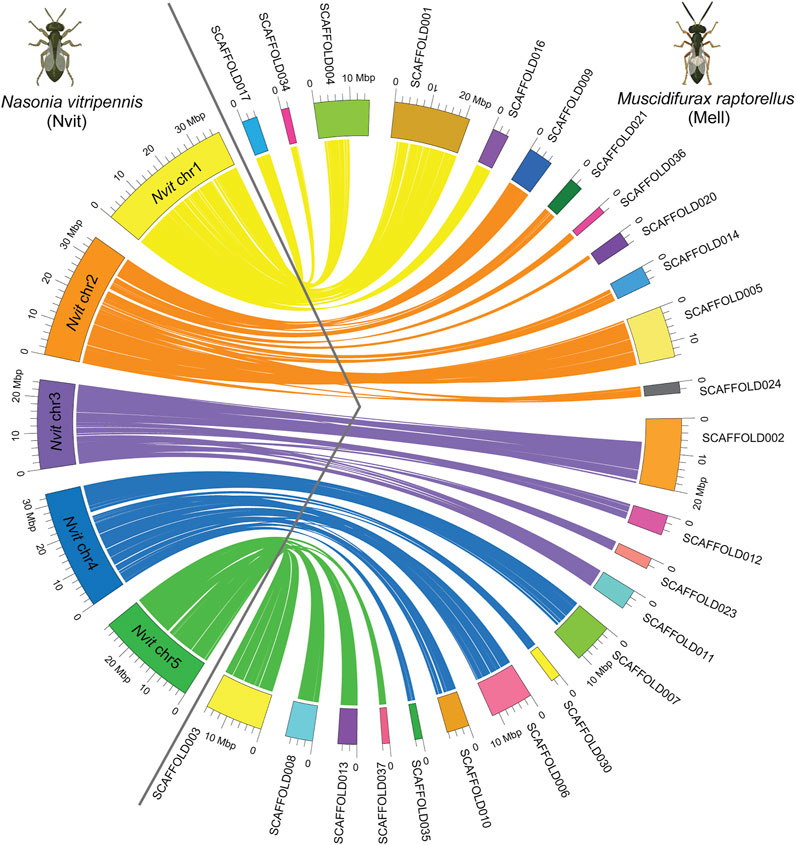
FIGURE 1. Genome comparisons between Muscidifurax raptorellus and Nasonia vitripennis. A total of 25 largest scaffolds in the M. raptorellus assembly showed a one-to-one relationship with the five chromosomes in the N. vitripennis genome. Chrs 1–5 on the left of the circle represent N. vitripennis chromosomes, and scaffolds on the right represent M. raptorellus assembled scaffolds. Parts of the figure were created with BioRender.com.
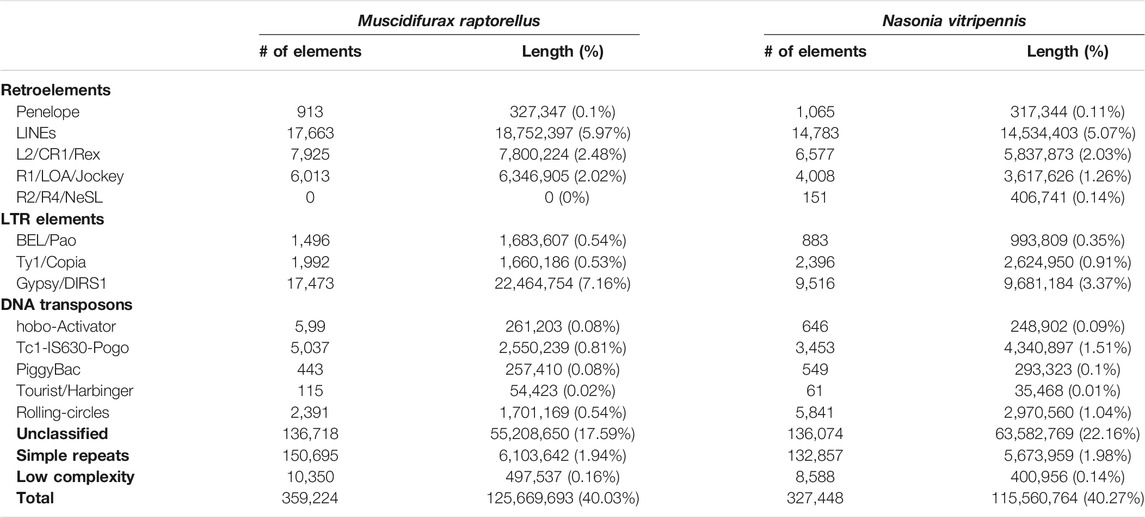
TABLE 2. Summary repeat element classes in Muscidifurax raptorellus and Nasonia vitripennis genomes.
Repeat Annotation
Repetitive regions accounted for 40% of the M. raptorellus genome with a total length of 126 Mbp based on the M. raptorellus specific repeat database (Table 2). The proportion of repeat regions is similar to that in Nasonia vitripennis, a jewel wasp species in the Nasonia genus (40.27%). LINEs (6.0%) and Gypsy (7.2%) elements are the most abundant classes in M. raptorellus, both with significantly higher abundance compared to those in N. vitripennis (Table 2).
Phylogeny With Hymenopteran Genomes
To construct the phylogenetic tree of M. raptorellus and other hymenopteran species, we used 3,662 single-copy 1:1 orthologs in nine species (turnip sawfly, parasitic wood wasp, Braconid wasp, minute polyphagous wasp, jewel wasp, fig wasp, paper wasp, ant, and honey bee). M. raptorellus clustered with the chalcid wasp species within the superfamily Chalcidoidea (Figure 2). M. raptorellus is the closest outgroup species to the jewel wasp Nasonia genus that has a high-quality reference genome, which will facilitate the evolutionary studies in the Nasonia subgroup and parasitoid wasp comparative genomics.
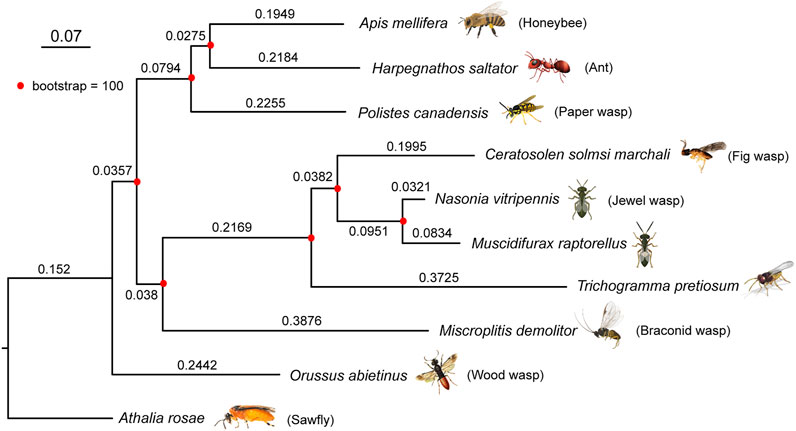
FIGURE 2. Phylogenetic relationship between M. raptorellus and nine representative hymenopteran species. A maximum-likelihood phylogenetic tree of M. raptorellus with nine other hymenopteran species was constructed based on 3,662 shared 1:1 single-copy proteins, using RAxML v8.2. The sawfly Athalia rosae was used as the outgroup. The bootstrap values were supported at 100/100. The length of each branch is shown on the branches. Parts of the figure were created with BioRender.com.
Gene Annotations
After repeat regions were soft-masked, the first-round MAKER annotation based on Trinity output generated 18,392 gene models (Supplementary Data S2). Subsequent MAKER iterations resulted in 10,362 protein-coding genes supported by both RNA-seq and gene prediction algorithms (Supplementary Data S2). Among them, 7,520 single-copy orthologs were identified between M. raptorellus and N. vitripennis (Supplementary Data S3). To evaluate the completeness and quality of predicted genes, we compared the gene length distributions of the 7,520 orthologs and found an average CDS length of 1,008 bp in M. raptorellus (standard deviation = 1,585) and 1,035 bp in N. vitripennis (standard deviation = 1,631). The 3,662 single-copy 1:1 orthologs between M. raptorellus and nine other hymenopteran species also have similar CDS length distributions (Supplementary Figure S1), indicating good gene model quality for these orthologs in M. raptorellus. To perform the gene annotation using an independent approach, 9,520 protein-coding genes (with 20,493 transcript isoforms) were annotated using the homology-based gene predictor GeMoMa (Keilwagen et al., 2016; Keilwagen et al., 2018) (Supplementary Data S2). 417 tRNA (transfer RNA) genes and 83 rRNA (ribosomal RNA) gene clusters were also annotated in the genome (Supplementary Data S4).
Data Availability Statement
The sequencing data generated for this study can be found in the NCBI Sequence Read Archive database with accession number SRR15058746. The draft genome assembly of M. raptorellus has been deposited at NCBI under Assembly accession number JAHUUD000000000. Supplementary Material is available at github.com/XuWangLab/MellV1_genome_assembly.
Author Contributions
XW, JW, ECV, and JG contributed to conception and design of the study. XX, YK, YW, EJ, EOM, and XW performed the experiments and data analysis. XW, JW, CJG, CZ, ECV, and JG provided samples, resources, and analysis tools. XX and XW wrote the first draft of the article. JW, ECV, YW, EJ, CJG, EOM, and JG wrote the sections of the article. All authors contributed to article revision and read and approved the submitted version.
Funding
This project was supported by an Auburn University Intramural Grant Program award to XW (AUIGP 180271) and the USDA National Institute of Food and Agriculture (Hatch project 1018100). XW was supported by the National Science Foundation EPSCoR RII Track-4 Research Fellowship (OIA1928770), an Alabama Agriculture Experiment Station (AAES) Agriculture Research Enhancement, Exploration, and Development (AgR-SEED) award, and a laboratory start-up fund from Auburn University College Veterinary Medicine. XX was supported by the Auburn University Presidential Graduate Research Fellowship and College of Veterinary Medicine Dean’s Fellowship. Contributions of JW (including RNA-seq) were supported by the National Institutes of Health (RO1GM098667), the National Science Foundation (1950078), and the Nathaniel and Helen Wisch Chair funds. JG acknowledges DFG grant SPP 1819: Rapid evolutionary adaptation (GA 661/4–1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Koppert Biological Systems, Netherlands, for kindly providing M. raptorellus to EV. We thank the HudsonAlpha Genome Sequencing Center for assistance with PacBio sequencing. We acknowledge the Auburn University Easley Cluster for support of this work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.748135/full#supplementary-material
References
Andrews, S., Krueger, F., Segonds-Pichon, A., Biggins, L., Krueger, C., and Wingett, S. (2010). FastQC. Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Bao, Z., and Eddy, S. R. (2002). Automated De Novo Identification of Repeat Sequence Families in Sequenced Genomes. Genome Res. 12, 1269–1276. doi:10.1101/gr.88502
Benson, G. (1999). Tandem Repeats Finder: a Program to Analyze DNA Sequences. Nucleic Acids Res. 27, 573–580. doi:10.1093/nar/27.2.573
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a Flexible Trimmer for Illumina Sequence Data. Bioinformatics. 30, 2114–2120. doi:10.1093/bioinformatics/btu170
Cantarel, B. L., Korf, I., Robb, S. M., Parra, G., Ross, E., Moore, B., et al. (2008). MAKER: an Easy-To-Use Annotation Pipeline Designed for Emerging Model Organism Genomes. Genome Res. 18, 188–196. doi:10.1101/gr.6743907
Chakraborty, M., Baldwin-Brown, J. G., Long, A. D., and Emerson, J. J. (2016). Contiguous and Accurate De Novo Assembly of Metazoan Genomes With Modest Long Read Coverage. Nucleic Acids Res. 44, e147. doi:10.1093/nar/gkw654
Chen, N. (2004). Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr Protoc Bioinformatics, 5, 1–4. doi:10.1002/0471250953.bi0410s05
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H., and Li, H. (2021). Haplotype-Resolved De Novo Assembly Using Phased Assembly Graphs With Hifiasm. Nat. Methods. 18, 170–175. doi:10.1038/s41592-020-01056-5
Dalla Benetta, E., Antoshechkin, I., Yang, T., Nguyen, H. Q. M., Ferree, P. M., and Akbari, O. S. (2020). Genome Elimination Mediated by Gene Expression From a Selfish Chromosome. Sci. Adv. 6, eaaz9808. doi:10.1126/sciadv.aaz9808
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). ProtTest 3: Fast Selection of Best-Fit Models of Protein Evolution. Bioinformatics. 27, 1164–1165. doi:10.1093/bioinformatics/btr088
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 43, 491–498. doi:10.1038/ng.806
Desjardins, C. A., Gadau, J., Lopez, J. A., Niehuis, O., Avery, A. R., Loehlin, D. W., et al. (2013). Fine-scale Mapping of the Nasonia Genome to Chromosomes Using a High-Density Genotyping Microarray. G3 (Bethesda). 3, 205–215. doi:10.1534/g3.112.004739
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). Repeat Modeler 2 for Automated Genomic Discovery of Transposable Element Families. Proc. Natl. Acad. Sci. USA. 117, 9451–9457. doi:10.1073/pnas.1921046117
Geden, C. J., and Hogsette, J. A. (2006). Suppression of House Flies (Diptera: Muscidae) in Florida Poultry Houses by Sustained Releases of Muscidifurax Raptorellus and Spalangia cameroni (Hymenoptera: Pteromalidae). Environ. Entomol. 35, 75–82. doi:10.1603/0046-225x-35.1.75
Geden, C. J., and Moon, R. D. (2009). Host Ranges of Gregarious Muscoid Fly Parasitoids:Muscidifurax raptorellus (Hymenoptera: Pteromalidae), Tachinaephagus zealandicus (Hymenoptera: Encyrtidae), and Trichopria nigra (Hymenoptera: Diapriidae). Environ. Entomol. 38, 700–707. doi:10.1603/022.038.0321
Girault, A. A., and Sanders, G. E. (1910). The Chalcidoid Parasites of the Common House or Typhoid Fly (Musca domestica Linn.) and its Allies. Psyche: A J. Entomol. 17, 9–28. doi:10.1155/1910/17925
Gokhman, V. E., and Westendorff, M. (2000). The Chromosomes of Three Species of the Nasonia Complex (Hymenoptera, Pteromalidae). contrib.entomol. 50, 193–198. doi:10.21248/contrib.entomol.50.1.193-198
Goodpasture, C. E. (1974). Cytological Data and Classification of the Hymenoptera. Davis.: University of California.
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De Novo transcript Sequence Reconstruction From RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 8, 1494–1512. doi:10.1038/nprot.2013.084
Heraty, J. (2009). Parasitoid Biodiversity and Insect Pest Management. Insect Biodiversity., 445–462. doi:10.1002/9781118945568.ch19
Katoh, K., and Standley, D. M. (2014). MAFFT: Iterative Refinement and Additional Methods. Mult. Seq. alignment Methods.. Methods in Molecular Biology 1079, 131–146. doi:10.1007/978-1-62703-646-7_8
Kaufman, P. E., Long, S. J., Rutz, D. A., and Waldron, J. K. (2001). Parasitism Rates of Muscidifurax Raptorellus and Nasonia vitripennis (Hymenoptera: Pteromalidae) After Individual and Paired Releases in New York Poultry Facilities. J. Econ. Entomol. 94, 593–598. doi:10.1603/0022-0493-94.2.593
Keilwagen, J., Hartung, F., and Grau, J. (2019). GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-Seq Data. Methods Mol. Biol. 1962, 161–177. doi:10.1007/978-1-4939-9173-0_9
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O., and Grau, J. (2018). Combining RNA-Seq Data and Homology-Based Gene Prediction for Plants, Animals and Fungi. BMC Bioinformatics. 19, 189. doi:10.1186/s12859-018-2203-5
Keilwagen, J., Wenk, M., Erickson, J. L., Schattat, M. H., Grau, J., and Hartung, F. (2016). Using Intron Position Conservation for Homology-Based Gene Prediction. Nucleic Acids Res. 44, e89. doi:10.1093/nar/gkw092
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol. 14, R36–R13. doi:10.1186/gb-2013-14-4-r36
Kogan, M., and Legner, E. F. (1970). A Biosystematic Revision of the Genus Muscidifurax (Hymenoptera: Pteromalidae) With Descriptions of Four New Species. Can. Entomol. 102, 1268–1290. doi:10.4039/ent1021268-10
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: Scalable and Accurate Long-Read Assembly via Adaptivek-Mer Weighting and Repeat Separation. Genome Res. 27, 722–736. doi:10.1101/gr.215087.116
Korf, I. (2004). Gene Finding in Novel Genomes. BMC Bioinformatics. 5, 59. doi:10.1186/1471-2105-5-59
Kriventseva, E. V., Kuznetsov, D., Tegenfeldt, F., Manni, M., Dias, R., Simão, F. A., et al. (2018). OrthoDB V10: Sampling the Diversity of Animal, Plant, Fungal, Protist, Bacterial and Viral Genomes for Evolutionary and Functional Annotations of Orthologs. Nucleic Acids Res. 47, D807–D811. doi:10.1093/nar/gky1053
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an Information Aesthetic for Comparative Genomics. Genome Res. 19, 1639–1645. doi:10.1101/gr.092759.109
Legner, E. F., Bay, E. C., and White, E. B. (1967). Activity of Parasites From Diptera: Musca domestica, Stomoxys Calcitrans, Fannia Canicularis, and F. Femoralis, at Sites in the Western Hemisphere1. Ann. Entomol. Soc. America. 60, 462–468. doi:10.1093/aesa/60.2.462
Legner, E. F. (1969). Reproductive Isolation and Size Variation in the Muscidifurax Raptor Complex1,2. Ann. Entomol. Soc. America. 62, 382–385. doi:10.1093/aesa/62.2.382
Leung, K., Ras, E., Ferguson, K. B., Ariëns, S., Babendreier, D., Bijma, P., et al. (2020). Next‐Generation Biological Control: the Need for Integrating Genetics and Genomics. Biol. Rev. 95, 1838–1854. doi:10.1111/brv.12641
Li, H. (2018). Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics. 34, 3094–3100. doi:10.1093/bioinformatics/bty191
Lin, Z. J., Wang, X., Wang, J., Tan, Y., Tang, X., Werren, J. H., et al. (2021). Comparative Analysis Reveals the Expansion of Mitochondrial DNA Control Region Containing Unusually High G-C Tandem Repeat Arrays in Nasonia vitripennis. Int. J. Biol. Macromolecules. 166, 1246–1257. doi:10.1016/j.ijbiomac.2020.11.007
Martinson, E. O., Mrinalini, K. Y. D., Kelkar, Y. D., Chang, C.-H., and Werren, J. H. (2017a). The Evolution of Venom by Co-Option of Single-Copy Genes. Curr. Biol. 27, 2007–2013. doi:10.1016/j.cub.2017.05.032
Martinson, E. O., Mrinalini, Y. D., Kelkar, Y. D., Chang, C.-H., and Werren, J. H. (2017b). The Evolution of Venom by Co-Option of Single-Copy Genes. Curr. Biol. 27, 2007–2013. doi:10.1016/j.cub.2017.05.032
McKay, T., and Broce, A. B. (2004). Discrimination of Self-Parasitized Hosts by the Pupal Parasitoid Muscidifurax Zaraptor (Hymenoptera: Pteromalidae). Ann. Entomol. Soc. Am. 97, 592–599. doi:10.1603/0013-8746(2004)097[0592:doshbt]2.0.co;2
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 20, 1297–1303. doi:10.1101/gr.107524.110
Newton, I. L. G., Clark, M. E., Kent, B. N., Bordenstein, S. R., Qu, J., Richards, S., et al. (2016). Comparative Genomics of Two Closely Related Wolbachia With Different Reproductive Effects on Hosts. Genome Biol. Evol. 8, 1526–1542. doi:10.1093/gbe/evw096
Niehuis, O., Gibson, J. D., Rosenberg, M. S., Pannebakker, B. A., Koevoets, T., Judson, A. K., et al. (2010). Recombination and its Impact on the Genome of the Haplodiploid Parasitoid Wasp Nasonia. PLoS One. 5, e8597. doi:10.1371/journal.pone.0008597
Nurk, S., Walenz, B. P., Rhie, A., Vollger, M. R., Logsdon, G. A., Grothe, R., et al. (2020). HiCanu: Accurate Assembly of Segmental Duplications, Satellites, and Allelic Variants From High-Fidelity Long Reads. Genome Res. 30, 1291–1305. doi:10.1101/gr.263566.120
Petersen, J., and Currey, D. (1996). Reproduction and Development of Muscidifurax Raptorellus (Hymenoptera: Pteromalidae), a Parasite Offilth Flies. J. Agric. Entomol. 13, 99–107.
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De Novo Identification of Repeat Families in Large Genomes. Bioinformatics. 21, i351–i358. doi:10.1093/bioinformatics/bti1018
Quinlan, A. R. (2014). BEDTools: the Swiss‐Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinformatics. 47, 11.12. 11–11.12. 34. doi:10.1002/0471250953.bi1112s47
Rago, A., Gilbert, D. G., Choi, J.-H., Sackton, T. B., Wang, X., Kelkar, Y. D., et al. (2016). OGS2: Genome Re-Annotation of the Jewel Wasp Nasonia vitripennis. BMC Genomics. 17, 678. doi:10.1186/s12864-016-2886-9
Seidl, S. E., and King, B. (1993). Sex‐Ratio Manipulation by the Parasitoid Wasp Muscidifurax Raptor in Response to Host Size. Evolution. 47, 1876–1882. doi:10.1111/j.1558-5646.1993.tb01276.x
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. (Clifton, NJ). 1962, 227–245. doi:10.1007/978-1-4939-9173-0_14
Silva-Junior, J., Pompolo, S., and Campos, L. (2000). “Cytogenetics of Some Species of Parasitic Wasps of the Families Pteromalidae and Eulophidae,” in Abstracts XXI International Congress of Entomology Brazil, August. 2000.
Stamatakis, A. (2014). RAxML Version 8: a Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics. 30, 1312–1313. doi:10.1093/bioinformatics/btu033
Stanke, M., and Waack, S. (2003). Gene Prediction With a Hidden Markov Model and a New Intron Submodel. Bioinformatics. 19 (Suppl. 2), ii215–25. doi:10.1093/bioinformatics/btg1080
Stanke, M., Schöffmann, O., Morgenstern, B., and Waack, S. (2006). Gene Prediction in Eukaryotes With a Generalized Hidden Markov Model that Uses Hints From External Sources. BMC Bioinformatics. 7, 62. doi:10.1186/1471-2105-7-62
Taylor, D. B., Peterson, R. D., Szalanski, A. L., and Petersen, J. J. (1997). Mitochondrial Dna Variation Among Muscidifurax Spp. (Hymenoptera: Pteromalidae), Pupal Parasitoids of Filth Flies (Diptera). Ann. Entomol. Soc. America. 90, 814–824. doi:10.1093/aesa/90.6.814
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: Discovering Splice Junctions With RNA-Seq. Bioinformatics. 25, 1105–1111. doi:10.1093/bioinformatics/btp120
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential Gene and Transcript Expression Analysis of RNA-Seq Experiments With TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi:10.1038/nprot.2012.016
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PloS one. 9, e112963. doi:10.1371/journal.pone.0112963
Wang, X., Kelkar, Y. D., Xiong, X., Martinson, E. O., Lynch, J., Zhang, C., et al. (2020). Genome Report: Whole Genome Sequence and Annotation of the Parasitoid Jewel Wasp Nasonia Giraulti Laboratory Strain RV2X[u]. Genetics. 10, 2565–2572. doi:10.1534/g3.120.401200
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 40, e49. doi:10.1093/nar/gkr1293
Weisenfeld, N. I., Kumar, V., Shah, P., Church, D. M., and Jaffe, D. B. (2017). Direct Determination of Diploid Genome Sequences. Genome Res. 27, 757–767. doi:10.1101/gr.214874.116
Werren, J. H., Richards, S., Desjardins, C. A., Niehuis, O., Gadau, J., Colbourne, J. K., et al. (2010). Functional and Evolutionary Insights From the Genomes of Three Parasitoid Nasonia Species. Science. 327, 343–348. doi:10.1126/science.1178028
Xiao, H., Zhou, S.-y., and Tong, Y.-f. (2018). A Taxonomic Study of Muscidifurax Girault & Sanders From China (Hymenoptera, Chalcidoidea, Pteromalidae). Zookeys. 776, 91–103. doi:10.3897/zookeys.776.25030
Zchori-Fein, E., Gottlieb, Y., and Coll, M. (2000). Wolbachia Density and Host Fitness Components in Muscidifurax Uniraptor (Hymenoptera: Pteromalidae). J. Invertebr. Pathol. 75, 267–272. doi:10.1006/jipa.2000.4927
Keywords: Muscidifurax, parasitoid wasp, biological control, housefly, linked-read technology, PacBio sequencing
Citation: Xiong X, Kelkar YD, Geden CJ, Zhang C, Wang Y, Jongepier E, Martinson EO, Verhulst EC, Gadau J, Werren JH and Wang X (2021) Long-Read Assembly and Annotation of the Parasitoid Wasp Muscidifurax raptorellus, a Biological Control Agent for Filth Flies. Front. Genet. 12:748135. doi: 10.3389/fgene.2021.748135
Received: 27 July 2021; Accepted: 04 October 2021;
Published: 12 November 2021.
Edited by:
Atashi Sharma, Virginia Tech, United StatesReviewed by:
Xueyan Li, Kunming Institute of Zoology, ChinaRichard John Edwards, University of New South Wales, Australia
Copyright © 2021 Xiong, Kelkar, Geden, Zhang, Wang, Jongepier, Martinson, Verhulst, Gadau, Werren and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xu Wang, eHp3MDA3MEBhdWJ1cm4uZWR1
†ORCID: Xu Wang, orcid.org/0000-0002-7594-5004