 Feng Shao
Feng Shao Huamei Pan
Huamei Pan Ping Li1,2
Ping Li1,2 Zuogang Peng
Zuogang Peng- 1Key Laboratory of Freshwater Fish Reproduction and Development (Ministry of Education), Southwest University School of Life Sciences, Chongqing, China
- 2College of Fisheries, Southwest University, Chongqing, China
Introduction
Aquaculture plays a vital role in food security and economic stability worldwide (Houston et al., 2020). Selective breeding to genetically improve production traits has great potential in increasing the efficiency of aquaculture and reducing its environmental footprint (such as habitat destruction and infectious disease outbreaks). To achieve this, we require fish species with excellent economic traits and high-quality genomic data that can be applied at all stages of domestication for ongoing genetic improvement (Houston, et al., 2020).
Siluriformes (catfish) is an order of major aquaculture species worldwide, especially in China, the United States, and Vietnam (De Silva and Phuong, 2011; Zhong et al., 2016; Kumar et al., 2020). Red-tail catfish (Hemibagrus wyckioides), belonging to the family Bagridae, initially possess a white caudal fin that becomes bright red when it reaches approximately 15 cm. The red-tail catfish is the largest Bagridae fish in body size and weight, reaching 130 cm and 80 kg, respectively (Ng, 1999). Red-tail catfish were originally distributed throughout the Mekong River. In China, they are now only distributed in Yunnan Province. Here, it is a famous indigenous fish due to a variety of excellent economic traits such as high protein content, strong disease resistance, easy domestication, and better production performance; it also has ornamental value (Zhou et al., 2021). Therefore, it has recently become an important aquaculture species in China (Zhou et al., 2019; Zhou et al., 2021). H. wyckioides exhibits marked sex dimorphism in growth, with the males growing much faster than females. Sexual maturation takes over 3 years and a lack of selective breeding has resulted in a decline in the growth rate of red-tail catfish (Zhou et al., 2021). Thus, it is essential to construct a reference genome of H. wyckioides and establish a breeding program to improve the economic characteristics, maintaining and developing the industry, to ultimately create an economically viable local fishing industry.
Data
In total, 41 Gb of clean reads (Illumina reads after quality control and trimming, the sequencing data used in the study detailed in Supplementary Table S1) were used to analyze the genome size and heterozygosity in H. wyckioides using k-mer analysis. Based on 26, 507, 871, 386 17-mers and a peak 17-mer depth of 34, the estimated heterozygosity rate was ∼0.3%, and the estimated genome size of H. wyckioides was ∼779 Mb (Supplementary Figure S1). It is the largest published genome size of Bagridae fish, as compared to that of the other two species; that of Pseudobagrus fulvidraco is ∼718 Mb (Gong et al., 2018) and that of Leiocassis longirostris is ∼689 Mb (He et al., 2021).
We produced 74.9 Gb of ONT (Oxford Nanopore Technologies) long reads and 6.7 Gb of PacBio HiFi reads. We used these data to construct our initial assembly. We obtained a 789.8 Mb genomic DNA sequence via assembly with a contig N50 length of 22.1 Mb (Supplementary Table S2). The long read assembly results consisted of 176 contigs, and the longest contig was 37.9 Mb (Supplementary Table S2). BUSCO (Simao et al., 2015) was used to assess the completeness of the assembled genome. Approximately 95.9% of the complete genes were detected in the genome of H. wyckioides (Supplementary Table S3). In addition, the average proportion of RNA-seq short reads were mapped to the assembled genome from different tissues is over 90% (Supplementary Table S4). Finally, we used the Hi-C technique to anchor the assembly contigs at the 29 chromosome level for H. wyckioides. We found that 136 contigs were successfully anchored in 29 chromosomes (Figure 1A). This result is consistent with the chromosome number records obtained by cytogenetic analysis (Supiwong et al., 2014), representing 97.7% of all scaffold nucleotide bases. The total assembly size of the chromosomes was ∼771.6 Mb (Supplementary Table S5).
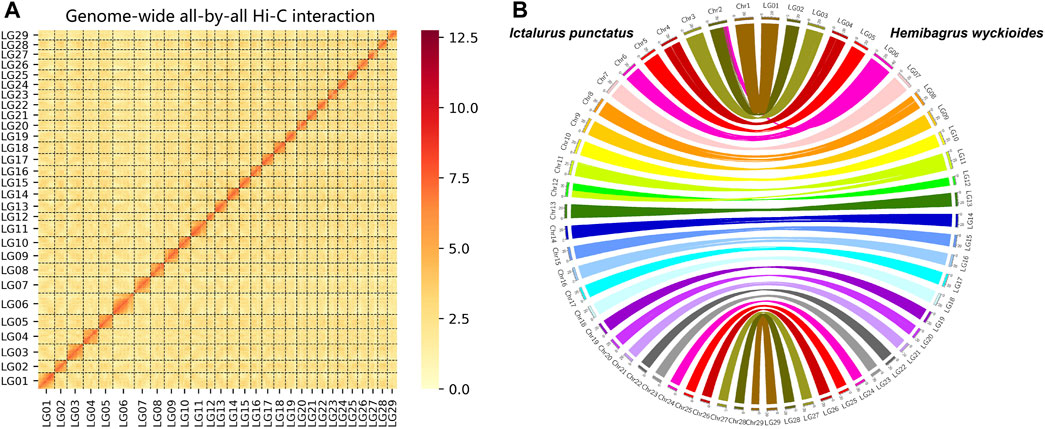
FIGURE 1. Pseudo-chromosome construct and comparative genomic analysis. (A) Hemibagrus wyckioides genome contig contact matrix using Hi-C data. (B) Genomic synteny of H. wyckioides and Ictalurus punctatus.
We aligned the entire genomic DNA sequences from I. punctatus and H. wyckioides to create the same chromosome numbering system for both species. More importantly, this greatly improves the usability of the data in future comparative genomics analyses, and the results showed that these species possess good collinearity (Figure 1B), which further demonstrates the reliability of the genomic data produced in this study. In total, 316, 847, 809 bp repeat sequences (40.12%) were identified. Overall, the combined homology-based and de novo prediction results indicated that TEs accounted for 34.59% of the assembled genome (Supplementary Table S6). Additionally, long terminal repeats, long interspersed nuclear elements, short interspersed nuclear elements, and DNA transposons occupied 4.84, 1.17, 6.13, and 19.09% of the assembled genome, respectively. Similar to most fish genomes, DNA transposons constituted a large proportion (Shao et al., 2019). H. wyckioides has the highest content of transposons among the published fish of the family Bagridae (Gong et al., 2018; He et al., 2021), which is consistent with the view that the larger the fish genome, the higher the content of transposons (Shao et al., 2019).
For genome annotation, 22,794 protein-coding genes were predicted in the H. wyckioides genome. Compared with other previously published catfish annotated information, the statistical results of the distribution showed that the CDS length, the number of exons, the length of exons, and the length of introns in the genes of H. wyckioides were consistent with the distribution trends of related species such as G. maculatum, P. fulvidraco, and B. yarrelli (Supplementary Figure S2). The BUSCO gene prediction of the existing genome sequence utilized the Actinopterygii_odb10 single-copy homologous gene. Approximately 95% of complete gene components were found in this gene set. This result indicates that most of the conserved genes were well predicted and the prediction results were relatively reliable (Supplementary Table S7). Finally, 21,142 genes were annotated in ≥1 of the databases (KOG, KEGG, NR, SwissProt, GO), and up to 92.75% of the genes were functionally annotated (Supplementary Table S8).
To determine the evolutionary relationships among H. wyckioides and other vertebrates, a phylogenetic tree was constructed using the 507 single-copy orthologous genes from 17 other vertebrate genomes (Figure 2A). L. oculatus was used as the outgroup. H. wyckioides and P. fulvidraco (which belong to the Bagridae family) formed a branch. Nine species of Siluriformes formed a monophyletic group. Siluriformes and Gymnotiformes formed sister groups. Otophysa fish were grouped together. Asian species (H. wyckioides, P. fulvidraco, G. maculatum, and B. yarrelli) clustered into one group and North American species (I. punctatus and A. melas) clustered into sister groups. This result is consistent with the “Big Asia” branch views suggested by Sullivan (Sullivan et al., 2006). We then created a time tree, and the estimated divergence time between H. wyckioides and P. fulvidraco was ∼41.73 Mya (Figure 2B; Supplementary Figure S3). In addition, the divergence time between Siluriformes and Gymnotiformes was ∼117.74 Mya. The divergence time between Anotophysi and Otophysa was ∼235.12 Mya.
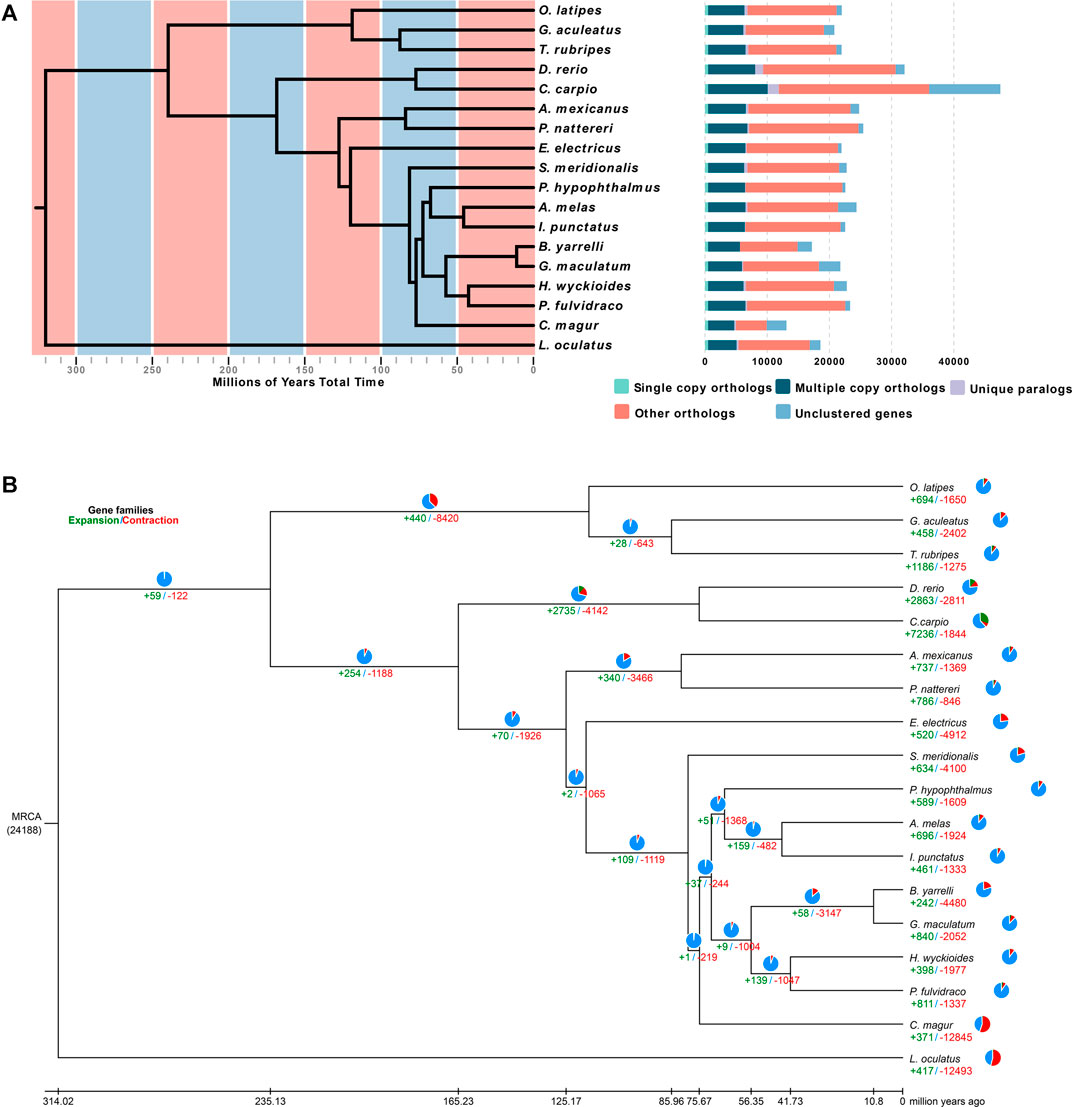
FIGURE 2. Phylogenetic and evolutionary analysis of Hemibagrus wyckioides. (A) Divergence time estimates and gene clusters in H. wyckioides and other species. (B) Expansion and contraction of H. wyckioides gene families. MRCA: most recent common ancestor; pie charts and numbers below represent the proportion and specific values of the gene families of expansion (green) and contraction (red), respectively.
We identified 398 expansion gene families and 1,977 contraction gene families in H. wyckioides (Figure 2B). Expansion gene families were enriched in 22 GO (Supplementary Table S9) categories and 33 KEGG pathways (Supplementary Table S10), most of which were related to nutrient metabolism (carbohydrates, proteins, and fats). This result provides insight for future studies on H. wyckioides growth and nutrient metabolism. Contraction gene families were enriched in 19 GO (Supplementary Table S11) categories and nine KEGG pathways (Supplementary Table S12), most of which were related to ion transport, cell interaction, and proteolysis.
Materials and Methods
Sample Collection, Library Construction, and Sequencing
Samples for genome sequencing of female H. wyckioides were collected from the Lancang River System, Xishuangbanna Prefecture, Yunnan Province, China (21°29′30.74″ N, 101°34′14.28″ E). The fin, blood, brain, gills, heart, head kidney, liver, muscle, and spleen were collected and immediately frozen in liquid nitrogen. Blood samples were collected and DNA was prepared using the QIAGEN® Genomic kit (Cat NO./ID: 13343, QIAGEN). The qualified libraries (200–400 bp) were sequenced using MGISEQ 2000. Raw reads were first filtered using a fastp (Chen et al., 2018) preprocessor (set to default parameters). For the ONT library preparations, the long DNA fragments were selected using the BluePippin system (Sage Science, United States). Sequencing was performed using a Nanopore PromethION sequencer (Oxford Nanopore Technologies, United Kingdom). The files were initially converted from FAST5 into FASTQ format using Guppy (Wick et al., 2019). The raw reads in fastq format with a mean_qscore_template <7 were then filtered. SMRTbell target size libraries (10–20 kb) were constructed for sequencing according to PacBio’s standard protocol (Pacific Biosciences, CA, United States). Sequencing was performed using a PacBio Sequel II. Raw data were analyzed using Smrtlink software (https://www.pacb.com/support/software-downloads/).
The RNA analyses involved nine tissues (fin, brain, gills, heart, head kidney, liver, muscle, and spleen) that were extracted using an RNeasy Plus Mini Kit (Qiagen). The Illumina paired-end sequencing (validated RNA samples, Illumina HiSeq4000, 150 bp) involved preparing a complementary DNA (cDNA) library using a TruSeq Sample Preparation Kit (Illumina). The qualified RNA from the nine tissues was mixed in equal amounts and reverse-transcribed using SQK-PCS109 (Oxford Nanopore Technologies) for the ONT library preparations and sequencing (Nanopore PromethION). The raw data were filtered in the same manner as DNA.
Genomic Features From K-Mer Analysis and Long Read Assembly
Quality-filtered reads were subjected to 17-mer frequency distribution analysis using the Jellyfish program (Marcais and Kingsford, 2011). We analyzed the 17-mer depth distribution from clean sequencing reads in FindGSE software (Sun et al., 2018). The simulation data results of Arabidopsis thaliana were further combined with different heterozygosity levels and the frequency peak distribution of the 17 k-mer using pIRS (Hu et al., 2012) to estimate the heterozygosity and the repeat content within the H. wyckioides genome. The long read assembly was constructed using NextDenovo (reads_cutoff:1k, seed_cutoff:32k, https://github.com/Nextomics/NextDenovo). To improve the accuracy of the assembly, the contigs were refined with default parameters in Racon for the long reads and Nextpolish using Illumina for the short reads.
Chromosomal-Level Genome Assembly by Hi-C and Assessment
To anchor the hybrid scaffolds onto the chromosome, genomic DNA was extracted from the blood of H. wyckioides for the Hi-C library, and sequencing (Illumina MGI-2000) was performed. The scaffolds were further clustered, ordered, and oriented onto chromosomes with LACHESIS (Korbel and Lee, 2013), with parameters CLUSTER_MIN_RE_SITES = 100, CLUSTER_MAX_LINK_DENSITY = 2.5, CLUSTER NONINFORMATIVE RATIO = 1.4, ORDER MIN N RES IN TRUNK = 60, and ORDER MIN N RES IN SHREDS = 60. Finally, the placement and orientation errors exhibited by obvious discrete chromatin interaction patterns were manually adjusted.
BUSCO and RNA-seq data mapping were used to evaluate our genome assembly. BUSCO was used to assess the completeness of the genome assembly by searching for the single-copy genes conserved across Actinopterygii in the H. wyckioides genome. We used hisat2 (Kim et al., 2015) to map RNA-seq short reads to the H. wyckioides genome. Furthermore, to compare the chromosome-level genome of H. wyckioides in this study with a reported chromosome-level genome of I. punctatus (Liu et al., 2016), we also performed a synteny analysis of these two genome assemblies using MUMmer (Marcais et al., 2018), only considering the reliable aligned regions more than 1 Mb in length. Circos plot distributions of homologous sequence pairs were plotted using Circos (Krzywinski et al., 2009).
Annotation of Repetitive Elements
We first annotated the tandem repeats using the software GMATA (Wang and Wang 2016) and TRF (Benson, 1999). For transposable element (TE) annotation, an ab inito repeat library for H. wyckioides was initially predicted using MITE-hunter (Han and Wessler, 2010) and RepeatModeler (Flynn et al., 2020) with default parameters, and LTR_FINDER (Xu and Wang, 2007), LTRharverst (Ellinghaus et al., 2008), and LTR_retriver (Ellinghaus et al., 2008) were also included in the H. wyckioides genome. The obtained library was then aligned to repbase (Bao et al., 2015) using TEclass (Abrusan et al., 2009) to classify the type of each repeat family. RepeatMasker (https://www.repeatmasker.org/) was applied to search for known and novel TEs by mapping sequences against the de novo repeat library and repbase TE library.
Gene Prediction and Functional Annotation
GeMoMa (Keilwagen et al., 2016) was used to align the homologous peptides from related species (Danio rerio, Oryzias latipes, Takifugu rubripes, Homo sapiens, P. fulvidraco, Glyptosternon maculatum, Bagarius yarrelli, and Ictalurus punctatus) to the assembly to obtain the gene structure information using homolog prediction. The RNA-seq-based gene prediction involved filtered RNA-seq short reads being aligned to the reference genome using STAR (Dobin et al., 2013) with default parameters. The transcripts were assembled using StringTie (Pertea et al., 2015). Full-length reads were identified and oriented from sequencing reads using the Pychopper tool (https://github.com/nanoporetech/pychopper) with default parameters. Full-length reads were aligned to the H. wyckioides reference genome using minimap2 (Li, 2018) with “-ax splice -uf”. The aligned full-length reads were clustered using pinfish software (https://github.com/nanoporetech/pinfish) following Nanopore’s Official recommendation. Redundancy was removed using cDNA_Cupcake software (https://github.com/Magdoll/cDNA_Cupcake) and polished using a reference genome sequence. The assembled transcripts based on full-length and short reads were merged with the open reading frames (ORFs) and were predicted using PASA (Haas et al., 2008). The de novo prediction involved RNA-seq reads being assembled for de novo using StringTie (Pertea et al., 2015) and analyzed with PASA (Haas et al., 2008) to produce a training set. AUGUSTUS (Stanke et al., 2008) with default parameters was used for the ab initio gene prediction with the training set. Finally, EVidenceModeler (Haas et al., 2008) was used to produce an integrated gene set in which genes with TEs were removed using the TransposonPSI package (http://transposonpsi.sourceforge.net/) and the miscoded genes were further filtered. BUSCO was used to assess the accuracy of gene prediction by searching for the single-copy genes conserved across Actinopterygii among the predicted genes in the assembly.
Gene function information, motifs, and domains of their proteins were assigned and compared with public databases, including SwissProt (UniProt Consortium, 2021), NR (https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins/), KEGG (Kanehisa et al., 2014), KOG (Tatusov et al., 2003), and GO (The Gene Ontology Consortium, 2017). The putative domains and GO terms of the genes were identified using the InterProScan (Jones et al., 2014) program with default parameters. For the other four databases, BLASTP (Camacho et al., 2009) was used to compare the EvidenceModeler-integrated protein sequences against the four well-known public protein databases with an E-value cutoff of 1e-5.
Evolutionary and Comparative Genomic Analyses
We identified homologous relationships among H. wyckioides and other species (I. punctatus, G. maculatum, Pangasianodon hypophthalmus, P. fulvidraco, Clarias magur, B. yarrelli, Ameiurus melas, Silurus meridionalis, Electrophorus electricus, Astyanax mexicanus, Pygocentrus nattereri, Cyprinus carpio, D. rerio, O. latipes, T. rubripes, Lepisosteus oculatus, and Gasterosteus aculeatus) by downloading their protein sequences and aligned them using OrthoMCL (Li et al., 2003).
Based on the orthologous gene sets identified with OrthoMCL (Li et al., 2003), molecular phylogenetic analysis was performed using the shared single-copy genes. Each ortholog group was multiple aligned using MAFFT (Yamada et al., 2016). Poorly aligned sequences were then eliminated using Gblocks (http://molevol.cmima.csic.es/castresana/Gblocks.html), and the GTRGAMMA substitution model in RAxML (Stamatakis, 2014) were used for phylogenetic tree construction with 1,000 bootstrap replicates. Three fossil calibration times were obtained from the TimeTree database (http://www.timetree.org/), the control time with the divergence times are provided in Supplementary Table S13. According to the results of OrthoMCL (Li et al., 2003), expansions and contractions of orthologous gene families were detected using CAFE (De Bie et al., 2006), and enrichment tests were performed using information from the homologs in the GO (The Gene Ontology Consortium, 2017) and KEGG (Kanehisa et al., 2014) databases.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by the Ethics committee of Southwest University.
Author Contributions
FS performed the major part of data analysis and drafted the manuscript. HP contributed to sample collection and drafted the manuscript. PL, LN, and YX contributed to sample collections. ZP contributed to the research design and final edits to the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported by grants from the Natural Science Foundation of China (31872204) to ZP and the Fundamental Research Funds for the Central Universities (SWU120049) to FS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Thanks for the support of the Fish10K Genome Project (Fish10K).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.747684/full#supplementary-material
References
Abrusan, G., Grundmann, N., DeMester, L., and Makalowski, W. (2009). TEclass--a Tool for Automated Classification of Unknown Eukaryotic Transposable Elements. Bioinformatics 25, 1329–1330. doi:10.1093/bioinformatics/btp084
Bao, W., Kojima, K. K., and Kohany, O. (2015). Repbase Update, a Database of Repetitive Elements in Eukaryotic Genomes. Mobile DNA 6, 11. doi:10.1186/s13100-015-0041-9
Benson, G. (1999). Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 27, 573–580. doi:10.1093/nar/27.2.573
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: Architecture and Applications. BMC Bioinformatics 10, 421. doi:10.1186/1471-2105-10-421
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: An Ultra-fast All-In-One FASTQ Preprocessor. Bioinformatics 34, i884–i890. doi:10.1093/bioinformatics/bty560
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: A Computational Tool for the Study of Gene Family Evolution. Bioinformatics 22, 1269–1271. doi:10.1093/bioinformatics/btl097
De Silva, S. S., and Phuong, N. T. (2011). Striped Catfish Farming in the Mekong Delta, Vietnam: A Tumultuous Path to a Global success. Rev. Aquacult. 3, 45–73. doi:10.1111/j.1753-5131.2011.01046.x
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 29, 15–21. doi:10.1093/bioinformatics/bts635
Ellinghaus, D., Kurtz, S., and Willhoeft, U. (2008). LTRharvest, an Efficient and Flexible Software for De Novo Detection of LTR Retrotransposons. BMC Bioinformatics 9, 18. doi:10.1186/1471-2105-9-18
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for Automated Genomic Discovery of Transposable Element Families. Proc. Natl. Acad. Sci. USA 117, 9451–9457. doi:10.1073/pnas.1921046117
Gene Ontology Consortium (2017). Expansion of the Gene Ontology Knowledgebase and Resources. Nucleic Acids Res. 45, D331–D338. doi:10.1093/nar/gkw1108
Gong, G., Dan, C., Xiao, S., Guo, W., Huang, P., Xiong, Y., et al. (2018). Chromosomal-Level Assembly of Yellow Catfish Genome Using Third-Generation DNA Sequencing and Hi-C Analysis. Gigascience 7, giy120. doi:10.1093/gigascience/giy120
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated Eukaryotic Gene Structure Annotation Using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7. doi:10.1186/gb-2008-9-1-r7
Han, Y., and Wessler, S. R. (2010). MITE-Hunter: A Program for Discovering Miniature Inverted-Repeat Transposable Elements from Genomic Sequences. Nucleic Acids Res. 38, e199. doi:10.1093/nar/gkq862
He, W. P., Zhou, J., Li, Z., Jing, T. S., Li, C. H., Yang, Y. J., et al. (2021). Chromosome-Level Genome Assembly of the Chinese Longsnout Catfish Leiocassis Longirostris. Zool. Res. 42, 417–422. doi:10.24272/j.issn.2095-8137.2020.327
Houston, R. D., Bean, T. P., Macqueen, D. J., Gundappa, M. K., Jin, Y. H., Jenkins, T. L., et al. (2020). Harnessing Genomics to Fast-Track Genetic Improvement in Aquaculture. Nat. Rev. Genet. 21, 389–409. doi:10.1038/s41576-020-0227-y
Hu, X., Yuan, J., Shi, Y., Lu, J., Liu, B., Li, Z., et al. (2012). pIRS: Profile-Based Illumina Pair-End Reads Simulator. Bioinformatics 28, 1533–1535. doi:10.1093/bioinformatics/bts187
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 30, 1236–1240. doi:10.1093/bioinformatics/btu031
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, Information, Knowledge and Principle: Back to Metabolism in KEGG. Nucl. Acids Res. 42, D199–D205. doi:10.1093/nar/gkt1076
Keilwagen, J., Wenk, M., Erickson, J. L., Schattat, M. H., Grau, J., and Hartung, F. (2016). Using Intron Position Conservation for Homology-Based Gene Prediction. Nucleic Acids Res. 44, e89. doi:10.1093/nar/gkw092
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 12, 357–360. doi:10.1038/nmeth.3317
Korbel, J. O., and Lee, C. (2013). Genome Assembly and Haplotyping with Hi-C. Nat. Biotechnol. 31, 1099–1101. doi:10.1038/nbt.2764
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 19, 1639–1645. doi:10.1101/gr.092759.109
Kumar, G., Engle, C., Hegde, S., and Senten, J. (2020). Economics of U.S. Catfish Farming Practices: Profitability, Economies of Size, and Liquidity. J. World Aquacult Soc. 51, 829–846. doi:10.1111/jwas.12717
Li, H. (2018). Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 34, 3094–3100. doi:10.1093/bioinformatics/bty191
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 13, 2178–2189. doi:10.1101/gr.1224503
Liu, Z., Liu, S., Yao, J., Bao, L., Zhang, J., Li, Y., et al. (2016). The Channel Catfish Genome Sequence Provides Insights into the Evolution of Scale Formation in Teleosts. Nat. Commun. 7, 11757. doi:10.1038/ncomms11757
Marçais, G., Delcher, A. L., Phillippy, A. M., Coston, R., Salzberg, S. L., and Zimin, A. (2018). MUMmer4: A Fast and Versatile Genome Alignment System. Plos Comput. Biol. 14, e1005944. doi:10.1371/journal.pcbi.1005944
Marçais, G., and Kingsford, C. (2011). A Fast, Lock-Free Approach for Efficient Parallel Counting of Occurrences of K-Mers. Bioinformatics 27, 764–770. doi:10.1093/bioinformatics/btr011
Ng, H. (1999). The Bagrid Catfish Genus Hemibagrus (Teleostei: Siluriformes) in Central Indochina with a New Species from the Mekong River. Raffles B. Zool. 47, 555–576.
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 33, 290–295. doi:10.1038/nbt.3122
Shao, F., Han, M., and Peng, Z. (2019). Evolution and Diversity of Transposable Elements in Fish Genomes. Sci. Rep. 9, 15399. doi:10.1038/s41598-019-51888-1
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 31, 3210–3212. doi:10.1093/bioinformatics/btv351
Stamatakis, A. (2014). RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 30, 1312–1313. doi:10.1093/bioinformatics/btu033
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using Native and Syntenically Mapped cDNA Alignments to Improve De Novo Gene Finding. Bioinformatics 24, 637–644. doi:10.1093/bioinformatics/btn013
Sullivan, J. P., Lundberg, J. G., and Hardman, M. (2006). A Phylogenetic Analysis of the Major Groups of Catfishes (Teleostei: Siluriformes) Using Rag1 and Rag2 Nuclear Gene Sequences. Mol. Phylogenet. Evol. 41, 636–662. doi:10.1016/j.ympev.2006.05.044
Sun, H., Ding, J., Piednoël, M., and Schneeberger, K. (2018). findGSE: Estimating Genome Size Variation within Human and Arabidopsis Using K-Mer Frequencies. Bioinformatics 34, 550–557. doi:10.1093/bioinformatics/btx637
Supiwong, W., Liehr, T., Cioffi, M. B., Chaveerach, A., Kosyakova, N., Pinthong, K., et al. (2014). Chromosomal Evolution in Naked Catfishes (Bagridae, Siluriformes): A Comparative Chromosome Mapping Study. Zoologischer Anzeiger - A J. Comp. Zoolog. 253, 316–320. doi:10.1016/j.jcz.2014.02.004
UniProt Consortium (2021). UniProt: the Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi:10.1093/nar/gkaa1100
Wang, X., and Wang, L. (2016). GMATA: An Integrated Software Package for Genome-Scale SSR Mining, Marker Development and Viewing. Front. Plant Sci. 7, 1350. doi:10.3389/fpls.2016.01350
Wick, R. R., Judd, L. M., and Holt, K. E. (2019). Performance of Neural Network Basecalling Tools for Oxford Nanopore Sequencing. Genome Biol. 20, 129. doi:10.1186/s13059-019-1727-y
Xu, Z., and Wang, H. (2007). LTR_FINDER: An Efficient Tool for the Prediction of Full-Length LTR Retrotransposons. Nucleic Acids Res. 35, W265–W268. doi:10.1093/nar/gkm286
Yamada, K. D., Tomii, K., and Katoh, K. (2016). Application of the MAFFT Sequence Alignment Program to Large Data-Reexamination of the Usefulness of Chained Guide Trees. Bioinformatics 32, 3246–3251. doi:10.1093/bioinformatics/btw412
Zhong, L., Song, C., Chen, X., Deng, W., Xiao, Y., Wang, M., et al. (2016). Channel Catfish in China: Historical Aspects, Current Status, and Problems. Aquaculture 465, 367–373. doi:10.1016/j.aquaculture.2016.09.032
Zhou, Y.-L., Wang, Z.-W., Guo, X.-F., Wu, J.-J., Lu, W.-J., Zhou, L., et al. (2021). Construction of a High-Density Genetic Linkage Map and fine Mapping of QTLs for Growth and Sex-Related Traits in Red-Tail Catfish (Hemibagrus Wyckioides). Aquaculture 531, 735892. doi:10.1016/j.aquaculture.2020.735892
Keywords: Hemibagrus wyckioides, genome, HiFi reads, comparative genomics, asssembly
Citation: Shao F, Pan H, Li P, Ni L, Xu Y and Peng Z (2021) Chromosome-Level Genome Assembly of the Asian Red-Tail Catfish (Hemibagrus wyckioides). Front. Genet. 12:747684. doi: 10.3389/fgene.2021.747684
Received: 26 July 2021; Accepted: 27 September 2021;
Published: 12 October 2021.
Edited by:
James Reecy, Iowa State University, United StatesReviewed by:
Aleksey V Zimin, Johns Hopkins University, United StatesGeoff Waldbieser, Agricultural Research Service (USDA), United States
Copyright © 2021 Shao, Pan, Li, Ni, Xu and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Shao, c2hhb2ZlbmdAc3d1LmVkdS5jbg==; Zuogang Peng, cHpnQHN3dS5lZHUuY24=