 Yilun Tao
Yilun Tao Dong Han1
Dong Han1 Yiju Wei
Yiju Wei- 1Medical Genetic Center, Changzhi Maternal and Child Health Care Hospital, Changzhi, China
- 2Department of Pediatrics, Penn State Health Hershey Medical Center, Penn State College of Medicine, Hershey, PA, United States
- 3Department of Pediatrics, Changzhi Maternal and Child Health Care Hospital, Changzhi, China
- 4Obstetrics Department, Changzhi Maternal and Child Health Care Hospital, Changzhi, China
Background: Infantile hypotonia with psychomotor retardation and characteristic facies 2 (IHPRF2) is a rare autosomal recessive neurodevelopmental disorder caused by mutations in the UNC80 gene. It is characterized by severe global developmental delay, poor or absent speech and absent or limited walking abilities. The current study explored a case of a Chinese patient with IHPRF2 caused by a novel splicing variant of UNC80.
Case Report: The proband is a 8-year-old Chinese male manifested with global developmental delay, severe truncal hypotonia, absent speech and intellectual disability. SNP array analysis revealed a uniparental isodisomy of the entire chromosome 2 [UPD(2)] in the proband. Whole exome sequencing (WES) subsequently identified a novel mutation c.5609-4G>A in the UNC80 gene, which was inherited from his mother and was confirmed by Sanger sequencing, indicating that UPD(2) was of maternal origin.
Conclusion: A novel UNC80 homozygous splicing variant c.5609-4G>A associated with maternal UPD(2) was identified. These findings indicate that UPD poses a high risk of autosomal recessive diseases, and provides information on the variant spectrum for UNC80. Our findings elucidate on understanding of the genotype-phenotype associations that occur in IHPRF2 patients.
Introduction
Infantile hypotonia with psychomotor retardation and characteristic facies 2 (IHPRF2, MIM 616801) is a rare autosomal recessive neurodevelopmental disorder with onset at birth or in early infancy (Shamseldin et al., 2016). It is a phenotypically heterogeneous disease characterized by severe global developmental delay, hypotonia, facial dysmorphism, intellectual disability, poor or absent speech and lack of or limited walking abilities (Perez et al., 2016; Shamseldin et al., 2016; He et al., 2018). The first case of IHPRF2 was reported in Bedouin families in 2016 (Perez et al., 2016). It has previously been reported that IHPRF2 is caused by homozygous or compound heterozygous mutation in UNC80 gene (MIM 612636) (Lu et al., 2010).
The UNC80 gene is located on chromosome 2 and comprises of 64 exons. UNC80 gene encodes a subunit of the non-selective sodium leak channel (NALCN) complex, consisting of 3,258 amino acids (Lu et al., 2010). This channel complex is a voltage-insensitive and nonselective sodium-leak channel, which is predominantly expressed in the brain and plays an important role in the establishment and maintenance of resting membrane potentials in neuron (Lu et al., 2007; Zhang et al., 2021). Proteins expressed from the UNC80 gene interacts with NALCN and acts as a scaffold protein for UNC79 (Lu et al., 2010; Wie et al., 2020). Recessive loss-of-function biallelic pathogenic variants of UNC80 cause UNC80 deficiency, which can result in dysfunctional NALCN complex leading to severe pathological phenotypes (Valkanas et al., 2016; Wie et al., 2020).
Uniparental disomy (UPD), a rare case that was first reported in 1980, occurs when a chromosome pair is derived from the same parent in a disomic cell with a balanced karyotype (Engel 1980). The prevalence of UPD ranges from 1 in 2000 to 1 in 5,000 people (Robinson, 2000; Liehr, 2010; Nakka et al., 2019). UPDs can cause clinical abnormalities, resulting in an aberrant dosage of imprinting genes or homozygosity of variants for recessive phenotypes (Yamazawa et al., 2010). Maternal and paternal UPDs have been reported in almost every human chromosome (Nakka et al., 2019). However, maternal UPD of chromosome 2 [UPD(2)mat] with a homozygous pathogenic variant in UNC80 has not been previously reported.
We report a case of IHPRF2 with a novel homozygous splicing variant c.5609-4G>A of UNC80 gene arising from UPD(2)mat, which expands on the disease spectrum associated with UNC80 mutations. In addition, previous genotypes and phenotypes of patients with IHPRF2 were reviewed, to help in understanding the genotype-phenotype relationship of UNC80.
Materials and Methods
Case Report
The proband is an 8-year-old male born as the sole child of healthy and non-consanguineous Chinese parents. The mother did not report any history of exposure to teratogenic pathogens or drugs during gestation. Birth weight of the child was 3,150 g (25th percentile), birth length was 51 cm (75th percentile), and occipitofrontal circumference (OFC) was 36 cm (>95th percentile) on week 39 of gestation. He has been suffering from severe hypotonia and feeding difficulty since birth. The proband was able to grasp at 6 months and could sit at 9 months. At 13 months of age, the patient was admitted to Beijing Children’s Hospital affiliated to Capital Medical University for astasia, development delay and failure to thrive with feeding difficulty. He manifested dystonia in the extremities, with complete inability to stand up and walk. He could not speak, even simple words such as “baba” and “mama”. Electroencephalograph (EEG) analysis did not reveal any abnormalities, whereas brain magnetic resonance imaging (MRI) showed bilateral dilation of lateral ventricles, periventricular leukomalacia and delayed myelination. No definite diagnosis existed to that time, and he did not undergo any adjuvant therapy. At the age of five, he presented to our hospital for severe psychomotor development. On examination, he had severe intellectual disability, hypotonia, strabismus and esotropia. He was unable to speak or communicate. He was able to walk with some aid but exhibited poor balance and he could not jump on one foot. In addition, the subject suffered from constipation. Facial dysmorphisms included a triangular face, microcephaly, low-set posterior rotated ears, and a thin and tented upper lip. He had long and slender fingers. His height, weight and head circumference were 107 cm (14th percentile), 15.5 kg (4th percentile) and 47.1 cm (<1st percentile), respectively.
In this family, we identified only one patient (proband) and proband’s father and mother is normal. We therefore used SNP array analysis and whole genome sequencing to search for any evidence of the disease.
SNP Array Analysis
A DNA sample (250 ng) was extracted from the peripheral blood of the patient and was hybridized using an Affymetrix CytoScan® 750K array kit (Affymetrix, Inc., Santa Clara, CA, United States) according to the manufacturer’s protocol. The SNP array data was analyzed for the presence of copy number variations (CNVs) using Affymetrix Chromosome Analysis Suite (ChAS) Software version 3.3. Pathogenicity of CNVs was evaluated based on published literature and public databases, including DGV (http://dgv.tcag.ca/dgv/app/home), Clingen (https://www.clinicalgenome.org/), DECIPHER (https://decipher.sanger.ac.uk/) and OMIM (https://www.omim.org/). This analysis was performed in accordance with the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) 2020 guidelines (Riggs et al., 2020).
Whole-Exome Sequencing
Peripheral blood sample of the patient was collected. Genomic DNA was extracted using a QIAamp DNA Mini Kit (Qiagen, China), according to the manufacturer’s instructions. DNA library preparation was performed following Illumina protocols, which included end repair, adapter ligation and PCR enrichment. The amplified DNA was then captured using Whole Exome Capture Kit (MyGenostics Inc, Beijing, China). Biotinylated capture probes were designed to tile all exons without repeated regions. The enriched libraries were sequenced for paired-end reads of 150 bp using the Illumina HiSeq X Ten platform.
The bioinformatics analyses were carried out utilizing established methods with some modifications (Zheng et al., 2018; Dai et al., 2019; Han et al., 2020; Zhang et al., 2020). After sequencing, clean reads were aligned to the UCSC hg19 human reference genome using the Burrows-Wheeler Alignment tool. Duplicated reads were removed using the Picard tool (http://broadinstitute.github.io/picard). The variants of SNP and small insertions or deletions (InDel) were detected by GATK HaplotypeCaller, then using GATK VariantFiltration to filter variant. The identified variants were then annotated using ANNOVAR. The variants with frequencies less than 0.05 in the normal population database were screened out, including the 1,000 Genomes, Exome Aggregation Consortium (ExAC), GnomAD and Inhouse database (MyGenostics Inc, Beijing, China). In addition, the identified variants were predicted using Mutation Taster (MT), Sorting Intolerant From Tolerant (SIFT), PolyPhen-2 (PP2) and Genomic Evolutionary Rate Profiling (GERP++), dbscSNV, SPIDEX, SPliceAI. Classification of variants (pathogenic, likely pathogenic, VUS and likely benign and benign) has been done according to the variant interpretation guidelines of American College of Medical Genetics and Genomics (Richards et al., 2015). Finally, we compared the variants found in patient and his parents. The function of the variant and their correlation with the disease phenotype was done by previously published articles and OMIM database.
Sanger Sequencing
Candidate variable sites were confirmed by Sanger sequencing for the patient and his parents. The primers are as follows: F 5′- CAACGAAGAGAACAAACACCTACG -3′, R 5′- TATTGGAGGGCATTGAGTTGC-3. The reference sequence NM_032,504 of UNC80 was used. Target sequences were sequenced on an ABI 3730 genetic analyzer (Applied Biosystems, Foster City Carlsbad, CA, United States) and identified using Chromas 2.6.5 (Technelysium Pty Ltd, Australia).
Results
Genome-wide SNP array analysis did not reveal any clinically significant copy number variations. However, it revealed an absence of heterozygosity (AOH) across the entire chromosome 2 (Figure 1A). There was no disease-related imprinting gene located on chromosome 2, therefore, whole exome sequencing (WES) of the patient was subsequently performed. A novel homozygous variant c.5609-4G> A in UNC80 was detected in the proband, which was only present in his mother as a heterozygous trait and was absent in the father (Figure 1B). This contradiction to Mendelian inheritance indicates that the AOH originated from maternal UPD. The variant c.5609-4G> A was located on intron 35, -4 bp to exon 36 splice acceptor site of the UNC80 gene. This variant has not been previously reported and was absent on the 1,000 Genome Project, the ExAC, the gnomAD database or our inhouse database. Furthermore, it has not been reported in HGMD. In silico analysis by MutationTaster and regSNP-intron showed that the variant may have an impact on pre-mRNA splicing (the score was 0.96 and 0.37), whereas SPliceAI indicated a prediction that is in total disagreement. Samples were not available for reanalysis, therefore, RT-PCR was not performed to explore the effect of splicing. Based on ACMG guidelines, the c.5609-4G>A variant was classified as variant of uncertain clinical significance (PM2). The diagnosis of IHPRF2 was confirmed by molecular and clinical findings.
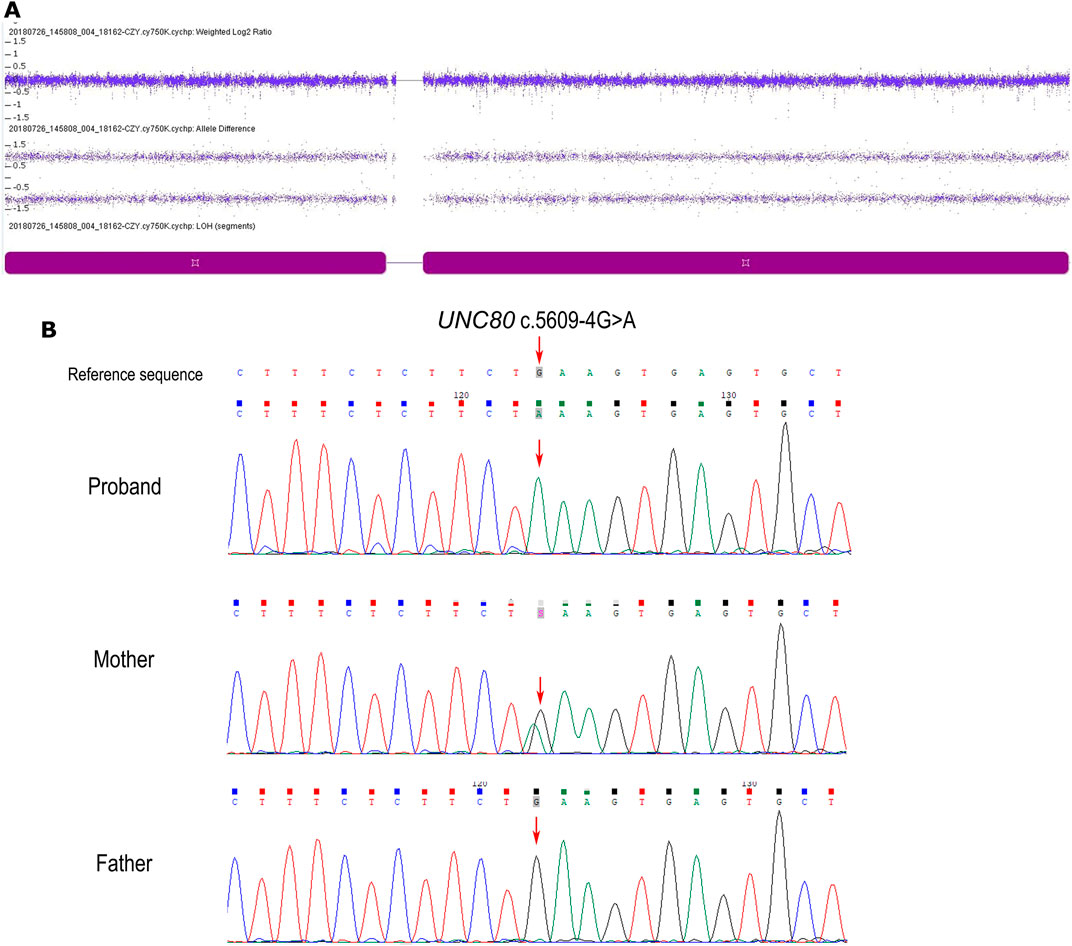
FIGURE 1. (A) Single nucleotide polymorphism microarray (SNP array, Affymetrix Cytoscan 750k) profiles of chromosome 2 in the patient. The dark purple block indicates complete UPD in chromosome 2, with two allele difference tracks where the plot has a line at + 1 and −1. (B) Sequence electropherogram of the c.5609-4G> A variation showing (arrow) the homozygous mutation in the patient, the heterozygous genotype in his mother, and wild-type in his father.
Discussion
The current study reports a case of an IHPRF2 patient with global developmental delay, truncal hypotonia, intellectual disability, and absent speech. A novel UNC80 homozygous splicing variant c.5609-4G> A originating from UPD(2)mat was identified, which has not been previously reported, and was absent in the general population. Bioinformatics analysis showed that the variant might be involved in splicing modulation. Similar variants, which lead to protein truncation have been previously reported (Anna and Monika, 2018; Li et al., 2019; Śmigiel et al., 2020). And in vitro cytology experiments have demonstrated that loss-of-function mutations in UNC80 are associated with IHPRF2 (Valkanas et al., 2016; Wie et al., 2020). Clinical presentation of the patient showed that he had IHPRF2. These findings indicate that the variant c.5609-4G> A is associated with pathogenesis of IHPRF2. However, further functional studies are needed to validate the pathogenicity of this variant.
The potential harmful effects of UPD include imprinted gene diseases, or activation of recessive pathogenic genes. Maternal and paternal UPD2, with normal phenotypes, have been reported previously, indicating the absence of genomic imprinting effects in UPD2 (Keller et al., 2009; Ou et al., 2013; Zhou et al., 2017; Zhang et al., 2019). Single conventional SNP-array analysis did not provide a conclusive diagnosis for the patient, therefore, an integrated approach is needed to determine the underlying genetic cause. This limitation has hindered genetic counseling for the family. Homozygosity of the UNC80 c.5609-4G>A variant resulted from UPD, thus the recurrence risk for IHPRF2 in this family is significantly reduced compared to a 25% risk expected when both parents are carriers of a UNC80 pathogenic variant. These findings indicate that UPD is a possible cause of autosomal recessive diseases and should be noticed, especially when patients have large chromosome segments of AOH or a rare homozygous pathogenic variant.
Comprehensive clinical and genetic analysis of 39 reported cases of IHPRF2 (including the present case) from 23 families are summarized in Supplementary Table S1. A total of 29 distinct UNC80 variants were identified from these patients (Supplementary Table S2). These variants included thirteen nonsense, eight missense, four frameshifts, three splice-site and one deletion. Approximately 65.5% (19/29) of the variants were nonsense, frameshift or splicing variants, indicating that loss of function is the mechanism behind the cause of IHPRF2 by UNC80 variants. Variants were unevenly distributed throughout the UNC80 gene (Figure 2), but were mainly concentrated in the exons (26/29, 89.66%). Twenty-three variants were observed once or twice, indicating high genetic heterogeneity among IHPRF2 patients. However, approximately 50% of the alleles in IHPRF2 patients were contributed by only 6 variants including p. Arg51*, p. Arg174*, p. Val189Met, c.601-1G>A, p. Thr561Arg fs*33 and p. Arg1265*. Almost all of the 6 variants were identified in the homozygous state in pedigrees with at least 2 patients. Only three genetically diagnosed IHPRF2 patients with compound heterozygous variants were reported from the Chinese population, and they possessed six variants including c.1447C>A, c.3719G>A, c.4926_4937del, c.4963C>T, c.8818C>T, and 8385C>G (Yang et al., 2017; He et al., 2018). These findings did not show a specific hotspot variant, indicating that most IHPRF2 patients in China do not undergo genetic testing.
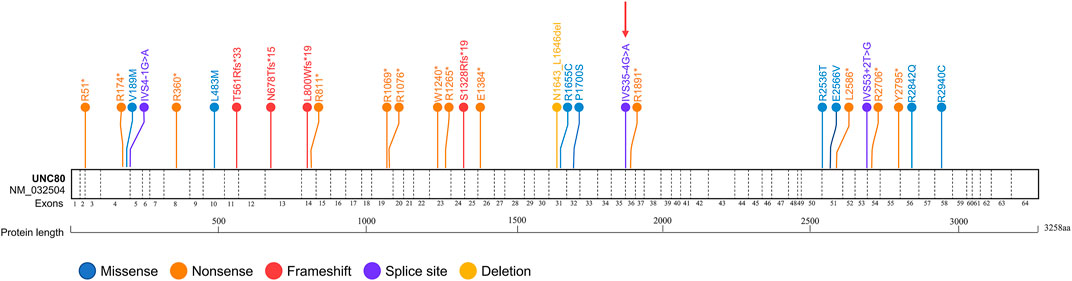
FIGURE 2. Reported variants in the UNC80 gene. Structure of UNC80 gene is shown, along with the 29 previously reported variants. The variant found in the current study is indicated by a red arrow.
Genotypes among the 23 genetically diagnosed families were highly heterogeneous, with each family presenting with a unique genotype. Only one variant, c.520C>T (p.Arg174*), was observed in 2 different families. This finding can be attributed to complex phenotypic heterogeneity of IHPRF2. Among the 39 genetically diagnosed patients, 29 (74.36%) presented with homozygous genotypes. Most patients were associated with consanguineous marriages (27/29, 93.10%), which explains why IHPRF2 is highly prevalent in Mediterranean and Arabic populations.
The 39 patients presented with intellectual disability, global developmental delay, absent or very poor speech and dyskinesias. Most patients had facial dysmorphisms including triangular face (31/35), strabismus (24/31), microcephaly (24/35), thin upper lip (16/22), prominent nasal bridge (10/14), short and smooth philtrum (9/13), frontal bossing (9/13), low set posterior rotated ears (9/13) and high forehead (8/9). Other common phenotypes included hypotonia (35/37), tapering fingers (21/24), birth length less than 3rd percentile (20/25), feeding difficulties (17/23), constipation (15/20), and esotropia (11/13). In addition, other neurodevelopmental abnormalities including seizures, abnormal EEG, abnormal brain MRI, and inability to walk independently were observed in 43.24% (16/37), 64.52% (20/31), 43.33% (13/30), and 83.33% (15/18) of IHPRF2 patients, respectively.
In this study, null UNC80 alleles were defined as those containing frameshift, splice-site, and/or nonsense mutations. Patients were split into two groups with different genotypes including: (i) N/N (null/null) (n = 30) and (ii) M/M (missense/missense) (n = 8). One individual with genotype of M/N (missense/null) was excluded. Incidences of most phenotypes of the individuals with genotype “N/N” werelarger compared to that of the “M/M” group (Table 1), indicating a relatively poor survival in patients with null variants. However, there was no significant difference observed between the two groups which can be attributed to the small sample size, challenges in phenotypic assessment, and other modifying factors that influence IHPRF2 phenotypes. In this study, the patient exhibited a homozygous splice site variant and was grouped in the “N/N” genotype class. He had been unwell since birth, and presented with severe congenital nervous system anomalies as well as global developmental delay. Seizures were not observed, and there was no way to verify that seizures may have actually ever occurred and were not captured. The patient presented with typical clinical manifestations of IHPRF2 and relatively poor prognosis, which can be associated with the null variant.
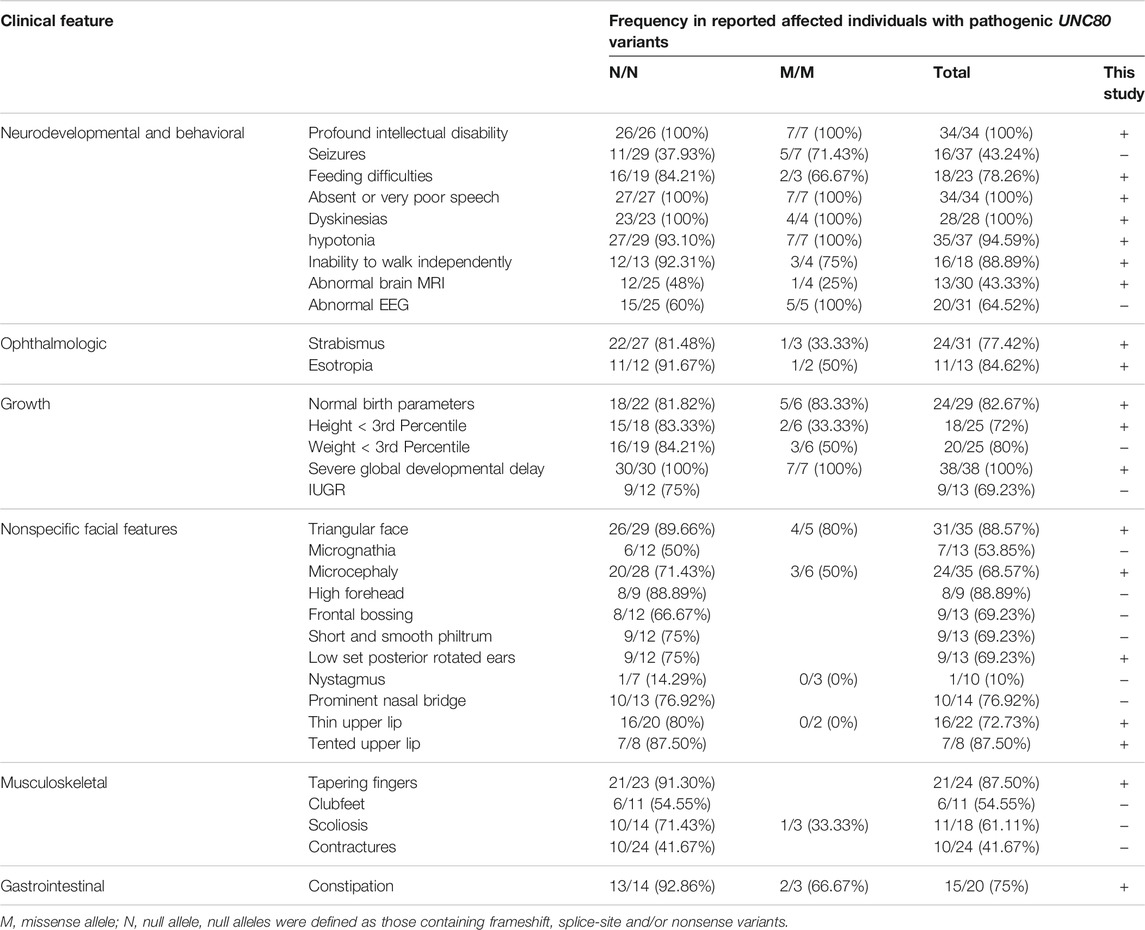
TABLE 1. Clinical features of IHPRF2 patients.
In summary, a novel splicing variant, c.5609-4G>A in UNC80 was identified in the current study. This finding elucidatesn on genetic variants that cause IHPRF2. The review section describes all known UNC80 variants, thus provides a basis for exploring genotype-phenotype correlations of IHPRF2. The findings of this current study indicate the importance of genetic testing in identifying the underlying molecular cause of the IHPRF2 disease and in providing adequate genetic counseling on possible recurrence risks.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI SRA BioProject, accession no: PRJNA757139.
Ethics Statement
The studies involving human participants were reviewed and approved by Changzhi Maternal and Child Health Care Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YT conceived and designed the experiment, conducted genetic data acquisition and interpretation, reviewed the published cases and wrote the manuscript. DH performed the experiment and patient follow-up. YW conducted the bioinformatics analysis and revised the manuscript. WS and LW performed in patient management. XL supervised the study. All authors have read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to the patient and his family in our research. We also express our gratitude to all the pediatricians that were involved in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.747422/full#supplementary-material
References
Anna, A., and Monika, G. (2018). Splicing Mutations in Human Genetic Disorders: Examples, Detection, and Confirmation. J. Appl. Genet. 59, 253–268. doi:10.1007/s13353-018-0444-7
Dai, Y., Liang, S., Dong, X., Zhao, Y., Ren, H., Guan, Y., et al. (2019). Whole Exome Sequencing Identified a Novel DAG1 Mutation in a Patient with Rare, Mild and Late Age of Onset Muscular Dystrophy-Dystroglycanopathy. J. Cel. Mol. Med. 23, 811–818. doi:10.1111/jcmm.13979
Han, P., Wei, G., Cai, K., Xiang, X., Deng, W. P., Li, Y. B., et al. (2020). Identification and Functional Characterization of Mutations in LPL Gene Causing Severe Hypertriglyceridaemia and Acute Pancreatitis. J. Cel. Mol. Med. 24, 1286–1299. doi:10.1111/jcmm.14768
He, Y., Ji, X., Yan, H., Ye, X., Liu, Y., Wei, W., et al. (2018). Biallelic UNC80 Mutations Caused Infantile Hypotonia with Psychomotor Retardation and Characteristic Facies 2 in Two Chinese Patients with Variable Phenotypes. Gene. 660, 13–17. doi:10.1016/j.gene.2018.03.063
Keller, M. C., McRae, A. F., McGaughran, J. M., Visscher, P. M., Martin, N. G., and Montgomery, G. W. (2009). Non-pathological Paternal Isodisomy of Chromosome 2 Detected from a Genome-wide SNP Scan. Am. J. Med. Genet. 149A, 1823–1826. doi:10.1002/ajmg.a.32973
Li, L., Cao, Y., Zhao, F., Mao, B., Ren, X., Wang, Y., et al. (2019). Validation and Classification of Atypical Splicing Variants Associated with Osteogenesis Imperfecta. Front. Genet. 10, 979. doi:10.3389/fgene.2019.00979
Liehr, T. (2010). Cytogenetic Contribution to Uniparental Disomy (UPD). Mol. Cytogenet. 3, 8. doi:10.1186/1755-8166-3-8
Lu, B., Su, Y., Das, S., Liu, J., Xia, J., and Ren, D. (2007). The Neuronal Channel NALCN Contributes Resting Sodium Permeability and Is Required for normal Respiratory Rhythm. Cell. 129, 371–383. doi:10.1016/j.cell.2007.02.041
Lu, B., Zhang, Q., Wang, H., Wang, Y., Nakayama, M., and Ren, D. (2010). Extracellular Calcium Controls Background Current and Neuronal Excitability via an UNC79-UNC80-NALCN Cation Channel Complex. Neuron. 68, 488–499. doi:10.1016/j.neuron.2010.09.014
Nakka, P., Pattillo Smith, S., O’Donnell-Luria, A. H., McManus, K. F., Mountain, J. L., Ramachandran, S., et al. (2019). Characterization of Prevalence and Health Consequences of Uniparental Disomy in Four Million Individuals from the General Population. Am. J. Hum. Genet. 105, 921–932. doi:10.1016/j.ajhg.2019.09.016
Ou, X., Liu, C., Chen, S., Yu, J., Zhang, Y., Liu, S., et al. (2013). Complete Paternal Uniparental Isodisomy for Chromosome 2 Revealed in a Parentage Testing Case. Transfusion. 53, 1266–1269. doi:10.1111/j.1537-2995.2012.03863.x
Perez, Y., Kadir, R., Volodarsky, M., Noyman, I., Flusser, H., Shorer, Z., et al. (2016). UNC80mutation Causes a Syndrome of Hypotonia, Severe Intellectual Disability, Dyskinesia and Dysmorphism, Similar to that Caused by Mutations in its Interacting Cation channelNALCN. J. Med. Genet. 53, 397–402. doi:10.1136/jmedgenet-2015-103352
Richards, S., Aziz, N., Aziz, N., Bale, S., Bick, D., Das, S., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423. doi:10.1038/gim.2015.30
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical Standards for the Interpretation and Reporting of Constitutional Copy-Number Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22, 245–257. doi:10.1038/s41436-019-0686-8
Robinson, W. P. (2000). Mechanisms Leading to Uniparental Disomy and Their Clinical Consequences. Bioessays. 22, 452–459. doi:10.1002/(sici)1521-1878(200005)22:5<452:aid-bies7>3.0.co;2-k
Shamseldin, H. E., Faqeih, E., Alasmari, A., Zaki, M. S., Gleeson, J. G., and Alkuraya, F. S. (2016). Mutations in UNC80, Encoding Part of the UNC79-UNC80-NALCN Channel Complex, Cause Autosomal-Recessive Severe Infantile Encephalopathy. Am. J. Hum. Genet. 98, 210–215. doi:10.1016/j.ajhg.2015.11.013
Śmigiel, R., Biela, M., Szmyd, K., Błoch, M., Szmida, E., Skiba, P., et al. (2020). Rapid Whole-Exome Sequencing as a Diagnostic Tool in a Neonatal/Pediatric Intensive Care Unit. J. Clin. Med. 9, 2220. doi:10.3390/jcm9072220
Valkanas, E., Schaffer, K., Dunham, C., Maduro, V., du Souich, C., Rupps, R., et al. (2016). Phenotypic Evolution of UNC80 Loss of Function. Am. J. Med. Genet. 170, 3106–3114. doi:10.1002/ajmg.a.37929
Wie, J., Bharthur, A., Bharthur, A., Wolfgang, M., Narayanan, V., Ramsey, K., et al. (2020). Intellectual Disability-Associated UNC80 Mutations Reveal Inter-subunit Interaction and Dendritic Function of the NALCN Channel Complex. Nat. Commun. 11, 3351. doi:10.1038/s41467-020-17105-8
Yamazawa, K., Ogata, T., and Ferguson-Smith, A. C. (2010). Uniparental Disomy and Human Disease: an Overview. Am. J. Med. Genet. 154C, 329–334. doi:10.1002/ajmg.c.30270
Yang, X., Pan, G., Li, W. H., Zhang, L. M., Wu, B. B., Wang, H. J., et al. (2017). Analysis of Gene Mutation of Early Onset Epileptic Spasm with Unknown Reason. Zhonghua Er Ke Za Zhi. 55, 813–817. doi:10.3760/cma.j.issn.0578-1310.2017.11.004
Zhang, D., Zhao, W., Liu, J., Ou, M., Liang, P., Li, J., et al. (2021). Sodium Leak Channel Contributes to Neuronal Sensitization in Neuropathic Pain. Prog. Neurobiol. 202, 102041. doi:10.1016/j.pneurobio.2021.102041
Zhang, R., Chen, S., Han, P., Chen, F., Kuang, S., Meng, Z., et al. (2020). Whole Exome Sequencing Identified a Homozygous Novel Variant in CEP290 Gene Causes Meckel Syndrome. J. Cel. Mol. Med. 24, 1906–1916. doi:10.1111/jcmm.14887
Zhang, X., Ding, Z., He, R., Qi, J., Zhang, Z., and Cui, B. (2019). Complete Paternal Uniparental Disomy of Chromosome 2 in an Asian Female Identified by Short Tandem Repeats and Whole Genome Sequencing. Cytogenet. Genome Res. 157, 197–202. doi:10.1159/000499893
Zheng, Y., Xu, J., Liang, S., Lin, D., and Banerjee, S. (2018). Whole Exome Sequencing Identified a Novel Heterozygous Mutation in HMBS Gene in a Chinese Patient with Acute Intermittent Porphyria with Rare Type of Mild Anemia. Front. Genet. 9, 129. doi:10.3389/fgene.2018.00129
Keywords: maternal uniparental disomy, chromosome 2, UNC80 gene, SNP array, whole exome sequencing
Citation: Tao Y, Han D, Wei Y, Wang L, Song W and Li X (2021) Case Report: Complete Maternal Uniparental Disomy of Chromosome 2 With a Novel UNC80 Splicing Variant c.5609-4G> A in a Chinese Patient With Infantile Hypotonia With Psychomotor Retardation and Characteristic Facies 2. Front. Genet. 12:747422. doi: 10.3389/fgene.2021.747422
Received: 26 July 2021; Accepted: 02 September 2021;
Published: 14 September 2021.
Edited by:
Santasree Banerjee, Beijing Genomics Institute (BGI), ChinaReviewed by:
Keiko Matsubara, National Center for Child Health and Development (NCCHD), JapanMohammad Faghihi, Express Gene Molecular Diagnostics Laboratory, United States
Copyright © 2021 Tao, Han, Wei, Wang, Song and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoze Li, bGl4aWFvemU1MjBAMTI2LmNvbQ==