 Lu-Xi Chen
Lu-Xi Chen Hai-Feng Xu
Hai-Feng Xu Pei-Shan Wang
Pei-Shan Wang Zhi-Ying Wu
Zhi-Ying Wu Hong-Fu Li
Hong-Fu Li- Department of Neurology and Research Center of Neurology in Second Affiliated Hospital and Key Laboratory of Medical Neurobiology of Zhejiang Province, Zhejiang University School of Medicine, Hangzhou, China
Background: Mutations in superoxide dismutase 1 gene (SOD1) are the most frequent high penetrant genetic cause for amyotrophic lateral sclerosis (ALS) in the Chinese population. A detailed natural history of SOD1-mutated ALS patients will provide key information for ongoing genetic clinical trials.
Methods: We screened for SOD1 mutations using whole exome sequencing (WES) in Chinese ALS cases from 2017 to 2021. Functional studies were then performed to confirm the pathogenicity of novel variants. In addition, we enrolled previously reported SOD1 mutations in our centers from 2007 to 2017. The SOD1 mutation spectrum, age at onset (AAO), diagnostic delay, and survival duration were analyzed.
Results: We found two novel SOD1 variants (p.G17H and p.E134*) that exerted both gain-of-function and loss-of-function effects in vitro. Combined with our previous SOD1-mutated patients, 32 probands with 21 SOD1 mutations were included with the four most frequently occurring mutations of p.V48A, p.H47R, p.C112Y, and p.G148D. SOD1 mutations account for 58.9% of familial ALS (FALS) cases. The mean (SD) AAO was 46 ± 11.4 years with a significant difference between patients carrying mutations in exon 1 [n = 5, 34.6 (12.4) years] and exon 2 [n = 8, 51.4 (8.2) years] (p = 0.038). The mean of the diagnostic delay of FALS patients is significantly earlier than the sporadic ALS (SALS) patients [9.5 (4.8) vs. 20.3 (9.3) years, p = 0.0026]. In addition, male patients survived longer than female patients (40 vs. 16 months, p = 0.05).
Conclusion: Our results expanded the spectrum of SOD1 mutations, highlighted the mutation distribution, and summarized the natural history of SOD1-mutated patients in southeastern China. Male patients were found to have better survival, and FALS patients received an earlier diagnosis. Our findings assist in providing a detailed clinical picture, which is important for ongoing genetic clinical trials.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by the involvement of both upper and lower motor neurons in the spinal cord, brainstem, and motor cortex with or without cognitive dysfunction. ALS insidiously begins with focal weakness and muscle atrophy but relentlessly spreads to the diaphragm. Eventually the paralysis typically causes death in 3–5 years as a result of respiratory failure. Approximately 90% of ALS cases are sporadic (SALS), and the remaining cases are inherited (familial ALS or FALS) with a Mendelian pattern of inheritance, which implies that a single gene mutation can drive the pathogenesis of ALS.
The advancement of genetic technologies, such as whole exome sequencing (WES), has facilitated the identification of genes associated with ALS. Currently, more than 50 genes have been implicated in ALS. With joint efforts, genetic analysis has added valuable pieces to the ALS puzzle. Superoxide dismutase 1 gene (SOD1) was the first ALS-associated gene dating back to 1993 (Rosen et al., 1993) and led to engineering of the first transgenic model of SOD1-G93A mice (Gurney et al., 1994). It ushered in a new era of ALS research and set the stage for future genetic breakthroughs.
SOD1 is a ubiquitously expressed protein, existing as a homodimer of 32 kDa. Each monomer is highly structured, and intramolecular disulfide bridges increase their stability. However, mutations in SOD1 can destabilize the protein and contribute to the collapse of the homodimeric structure and its subsequent aggregation (Brasil et al., 2019). It is well established that SOD1 mutations lead to toxic gain of function of SOD1 proteins, but the effects of loss of function remain controversial (Gurney et al., 1994).
To date, more than 200 mutations in SOD1 have been reported (http://www.hgmd.cf.ac.uk/). ALS patients carrying SOD1 mutations present with a highly heterogenous phenotype that is clinically indistinguishable in SALS and FALS. SOD1 gene mutations are the leading cause of FALS in the Chinese population. Therefore, it is important to clinically summarize both the mutation spectrum of the SOD1 gene and the natural history of SOD1-mutated ALS patients in southeastern China.
Materials and Methods
Participants
Patients were recruited from the Second Affiliated Hospital of Zhejiang University School of Medicine from May 2017 to April 2021. A total of 114 patients with SALS and 15 with FALS were screened by WES. In addition, we performed the C9orf72 gene test, and all the hexanucleotide expansions of patients except one were within 30 repeats. For further phenotype–genotype analysis, we included previously reported SOD1-mutated probands in our centers from December 2007 to April 2017. All patients were diagnosed by at least two senior neurologists as having ALS according to the Revised El Escorial criteria (Brooks et al., 2000). All participants were of Han Chinese descent and came from southeastern China. The ethics committee of each participating center approved the study, and written consents were obtained from all participants during their first hospital visit. The clinical data of ALS patients, including gender, family history, age at onset (AAO), diagnostic delay, site of onset, side of onset, disease duration, and clinical manifestations were collected upon the first visit to the hospital.
Whole Exome Sequencing, Bioinformatic Analysis, and Sanger Sequencing
Genomic DNA was extracted from blood using a DNA Extraction Kit (Qiagen, Hilden, Germany). The samples were captured by the Agilent Sure Select Human All Exon V6 products and sequenced on the Illumina HiSeq X Ten platform (XY Biotechnology Co. Ltd., Hangzhou, China). Further bioinformatic analysis has been performed according to our previously reported protocol (Dong et al., 2021). Briefly, all variants were annotated by ANNOVAR. SIFT and PolyPhen-2 software were used to predict the functional changes in proteins caused by the variants. Single Nucleotide Polymorphism (dbSNP) Database, the 1,000 Genomes Project, and the ExAC database were used to check the frequency in the general population. Variants were finally classified according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines. Sanger sequencing was performed to validate the potential variants. All primers covering the five exons in SOD1 were seen in our previous report (Niu et al., 2011).
Plasmid Construction
The coding sequence of human wild-type (WT) SOD1 gene (RefSeq NM_000454.5) was cloned into the pFlag-CMV-4 vector using a ClonExpress II One Step Cloning Kit (Vazyme). Plasmids with mutant SOD1 (c.400G > T and c.49_50del insCA) were created by PCR mutagenesis (Toyobo, Osaka, Japan) and were verified by Sanger sequencing. Four plasmids expressing Vector, WT, p.G17H, and p.E134* were constructed.
Cell Culture and Transfection
HEK293T cells were maintained in Dulbecco’s modified essential medium DMEM-supplemented 10% fetal bovine serum stored in a humidified incubator under 5% CO2 at 37°C. Transient transfection was performed using Lipofectamine 3,000 (Invitrogen, Life Technologies, Grand Island, NY, United States ).
Immunofluorescence Confocal Microscopy
HEK293T cells were seeded on poly-D-lysine (PDL)-treated coverslips (NEST, China) and transiently transfected with various expression plasmids (pFLAG-SOD1-WT, pFLAG-SOD1-G17H, pFLAG-SOD1-E134*). Twenty-4 hours later, the cells were rinsed with 1 × phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 8 min, and then permeabilized with 1 × PBS supplemented with 0.01% Triton X100. The cells were blocked with 5% donkey serum and 1% bovine serum albumin (BSA; sigma, St. Louis, MO) in PBS for 1 h at room temperature. The primary antibody was rabbit antibody anti-flag (1:800, Cell Signaling Technology, 14793S). The secondary antibody was Alexa Fluor 488 donkey anti-rabbit. The cells were incubated with NucBlue Live Reagent (Hoechst 33342; Thermo Fisher Scientific, Oregon, United States) and visualized using Olympus FluoView FV3000 confocal microscopy with a ×63 objective.
Western Blot
Forty-eight hours after transfection, HEK293T cells over-expressing pFlag-SOD1-WT, pFlag-SOD1 mutants, or the empty vector were lysed and harvested. The protein samples were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride (PVDF) membrane, and blotted with the 5% non-fat milk. The antibodies against flag (1:1,000, CST) and β-tubulin (1:5,000) were used. All immunoblotting images were acquired using a BioRad system.
Quantitative Real-Time PCR
Total RNA was extracted using Trizol reagent (Takara, Kusatsu, Japan), which was then reverse transcribed to cDNA by the PrimeScript RT reagent Kit (Takara, Kusatsu, Japan) in accordance with the manufacturer’s instructions. Reverse transcription (RT)-PCR was further performed using a SYBR Premix Ex Taq Kit (Takara, Kusatsu, Japan). The PCR conditions are listed as follows: incubation for 3 min at 50°C followed by incubation for 3 min at 95°C, and finally 40 cycles at a duration of 10 s each while at 95°C and then 30 s at 30°C. The following primers were used: target SOD1 and human GAPDH. Detection and data analysis were conducted using an ABI StepOnePlus sequence detection system (Thermo Fisher Scientific, Oregon, United States ), and endogenous glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. Expression levels were quantified by threshold cycle values.
Statistical Analysis
Descriptive statistics were provided for the site of onset, heredity of disease (FALS or SALS), gender, AAO, diagnostic delay, and survival time. Besides, continuous data were compared using Student’s t-test or the Mann–Whitney test, while dichotomous variables, such as gender and site of onset, were analyzed using either the standard chi-square test or Fisher’s exact test. Survival duration was determined by Kaplan-Meier analysis, and differences were determined by log-rank testing. A two-tailed p < 0.05 was considered statistically significant. All analyses were performed using GraphPad Prism 8.0 (GraphPad Software, CA, United States).
Results
Whole Exome Sequencing Screen of ALS Patients and Pathogenicity Classification of Novel SOD1 Mutations
From 2017 to 2021, a total of eight different variants in SOD1 were found, including six known pathogenic variants (p.G38R, p.V48A, p.N87S, p.C112Y, p.I114T, and p.L145S) and two novel variants (p.G17H and p.E134*). Two heterozygous variants, c.49_50del insCA (p.G17H) and c.400G > T (p.E134*), were detected in two families with a positive family history (Figures 1A,B), which was confirmed with Sanger sequencing (Figures 1C,D). Both novel variants were absent in ESP6500 and ExAC database and were highly conserved from chimpanzee to zebrafish (Figures 1E,F). The nonsense variant, p.E134*, generates the truncated protein caused by premature termination. However, the nonsense variant was not regarded as very strong evidence of pathogenicity because loss of function is not the primary mechanism in SOD1-related ALS. In addition, p.E134* was predicted to be detrimental by Mutation Taster and CADD. The variants p.G17H and p.E134* were classified as likely pathogenic and pathogenic, respectively, according to ACMG criteria obtained using Varsome.
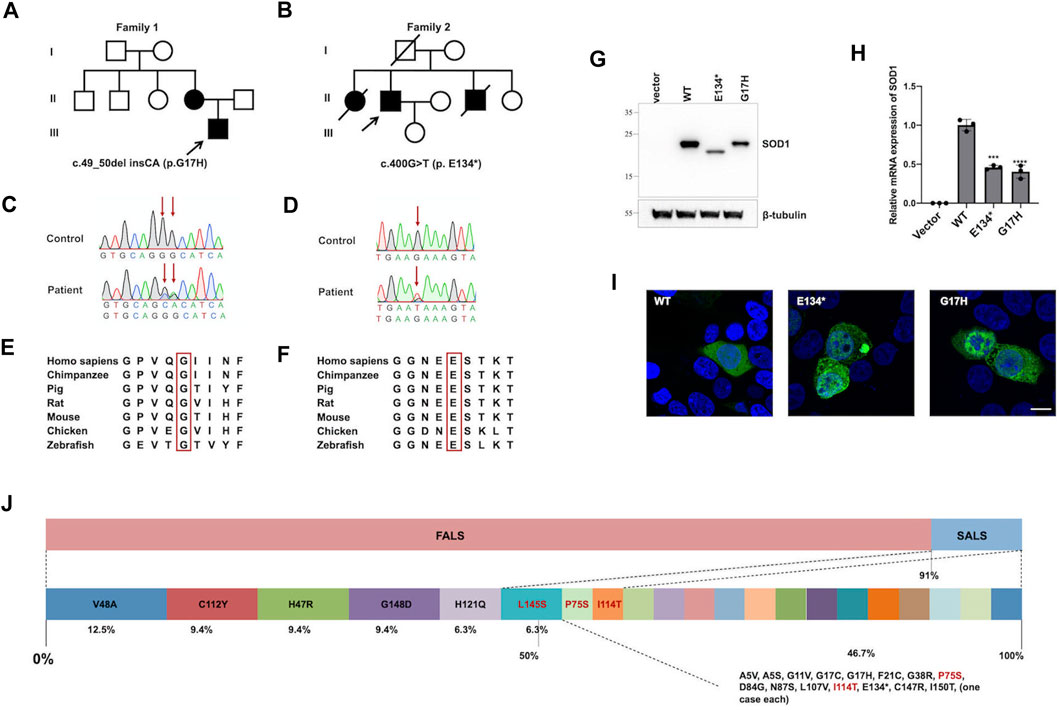
FIGURE 1. SOD1 novel variants identified in patients, functional study, and SOD1 mutation distribution. (A,B) Pedigrees of two ALS families carried missense variants of the SOD1 gene; arrows indicate the proband of the family. (C,D) Sequence of c.49_50del insCA (p.G17H) and c.400G > T (p.E134*) variants in SOD1 are shown; arrows indicate variant sites. (E,F) The p.G17H and p.E134* variants were highly conserved from chimpanzee to zebrafish. (G) HEK293T cells were transiently transfected with plasmids expressing Vector, p.WT, p.G17H, and p.E134*. The SOD1 protein was detected by anti-Flag antibody. (H) Quantitative real-time PCR of SOD1 mRNA levels in HEK293T cells transfected with empty vector, WT, or mutant flag-tagged SOD1 vectors. GAPDH was used as an internal control. The data shown are representative of three independent experiments. Error bars indicate SDs, ***p < 0.001, ****p < 0.0001 (one-way ANOVA). (I) Immunofluorescence confocal of HEK293T cells expressing WT or mutant SOD1. (J) Mutation spectrum in SOD1 gene of southeastern Chinese ALS patients in our ALS cohort.
To further elucidate the biological effects of these two variants, SOD1 protein and mRNA levels of mutants were markedly reduced compared to WT (Figures 1G,H), indicating that the reduced protein expression was caused by reduced mRNA synthesis. The in vitro results showed that the mRNA levels of mutant SOD1 were degraded more rapidly than WT, suggesting a loss-of-function effect. The aggregation propensity assay showed that the cells overexpressing WT plasmid showed diffuse cytoplasmic SOD1 protein, while the misfolded aggregates were seen in cells transfected with p.G17H and p.E134* mutants (Figure 1I). The soluble physiological SOD1 protein had a strong tendency to become toxic aggregates due to mutation, implying a gain-of-function effect.
Mutation Frequency of SOD1 in Our ALS Patients From Southeastern China
In this study, a total of 114 patients with SALS and 15 patients with FALS were screened by WES. Among them, 12 subjects were genetically identified as having SOD1 variants, 10 (10/15, 66.7%) of whom were FALS patients and 2 (2/114, 1.8%) of whom were apparently SALS patients. We previously reported 20 probands with SOD1 mutations in 36 unrelated FALS patients (Niu et al., 2011; Liu et al., 2014; Lin et al., 2019; Liu et al., 2019). When integrating these results into the current study, SOD1 mutations account for 58.9% (32/51) of our FALS cases.
A total of 21 SOD1 mutations, including 1 nonsense and 1 deletion/insertion mutations, were found spanning all five exons, but only one mutation was in exon 3 (Table 1). The most frequent SOD1 mutation was p.V48A (4/32, 12.5%) in four FALS probands, followed by p.H47R, p.C112Y, and p.G148D [each were found in three probands (3/32, 9.4%)], as well as p.H121Q and p.L145S [each were found in two probands (2/32, 6.3%)], and the remaining mutations were each found in one proband (Figure 1J). Among all the mutations, p.L145S, p.P75S, and p.I114T are found in SALS patients (Figure 1J).
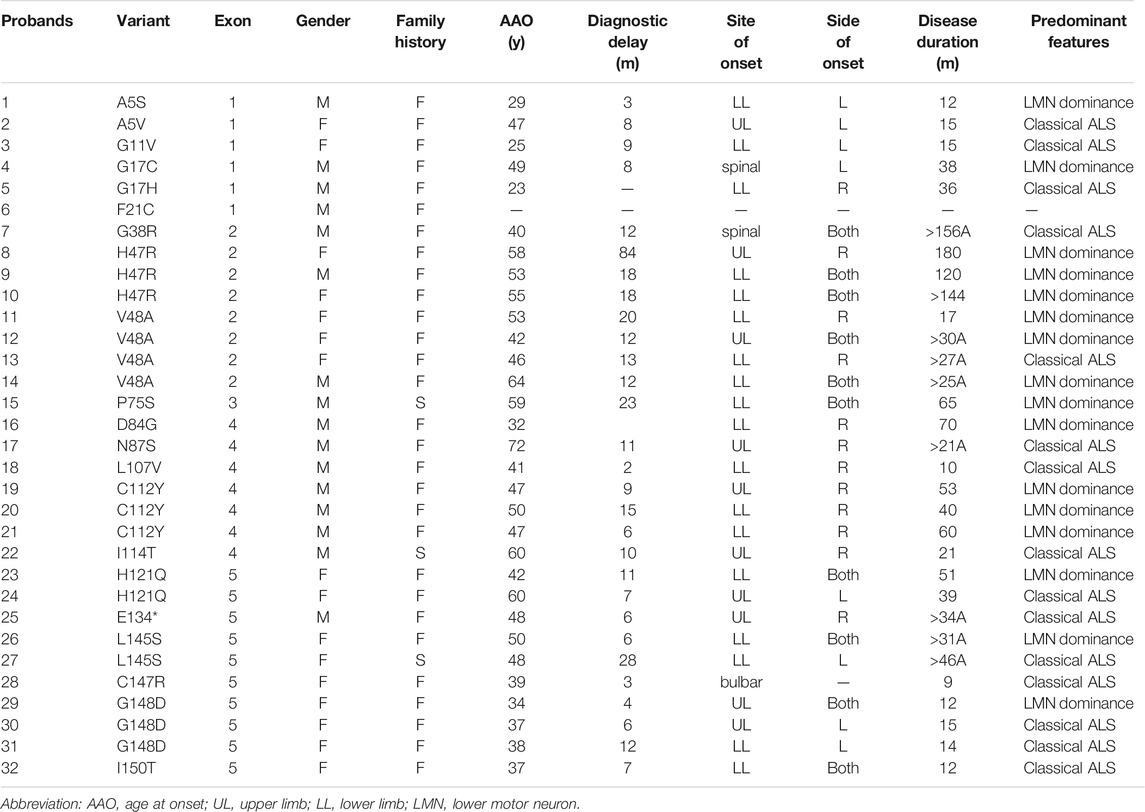
TABLE 1. The clinical features of the probands carrying mutations in SOD1.
Clinical Features and Natural History of ALS Patients Carrying the SOD1 Mutation
Combining our previously reported SOD1-mutated patients in our centers, a total of 32 probands were genetically identified with SOD1 mutations (Table 1). Twenty-nine (29/32, 90.6%) of the probands were FALS patients, and three (3/32, 9.4%) were apparently SALS patients. The gender ratio (M:F) was 1:1. With the exception of 1 case with missing data, only 1 patient among 31 available probands exhibited bulbar onset. The majority of them (18/31, 58%) presented with lower limb onset, but some (10/31, 33.3%) presented with upper limb onset. Sixteen probands presented with a predominantly lower motor neuron phenotype, while another 16 probands were the classical ALS phenotype.
With the exception of the patient with missing data, 31 patients had available AAO data for analysis. The mean (SD) AAO was 46 ± 11.4 years, and the median AAO was 47 years (Figure 2A). The mean AAO of patients carrying mutations in exon 1 [n = 5, 34.6 (12.4)] was statistically significantly earlier than those with mutations in exon 2 [n = 8, 51.4 (8.2)] (p = 0.038) (Figure 2B). There was no difference in the mean (SD) AAO between male and female patients [47.6 (12.2) vs. 45.25 (11.2), p = 0.58] (Figure 2C). The onset of FALS in patients is earlier than that of SALS, albeit without statistical significance [45.4 (11.4) vs. 55.7 (10.2), p = 0.15] (Figure 2D). The AAOs of ALS patients were highly heterogenous with the 25th and 75th percentiles at 38 and 53 years, respectively. Six mutations (p.A5S, p.G11V, p.G17H, p.D84G, p.G148D, and p.I150T) showed relatively younger AAOs, which was lower than the first quartile (25%, <38 years), and another six mutations (p.H47R, p.V48A, p.P75S, p.N87S, p.I114T, and p.H121Q) presented with relatively older AAOs, which were higher than the third quartile (75%, >53 years). The youngest and oldest AAOs are p.G17H and p.N87S, which were found in patients in their twenties and seventies, respectively.
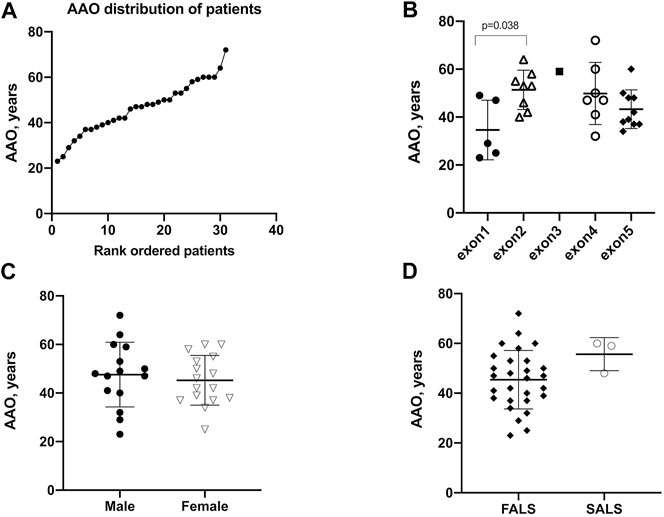
FIGURE 2. Age at onset (AAO) of patients with SOD1 mutations. (A) Plot of rank ordered SOD1-mutant patients showing the median AAO of 47 years (B) Plot comparing the AAO among patients carrying mutations in different exons of the SOD1 gene. Patients (n = 24) harboring mutations in exon 1 were older than those (n = 18) harboring mutations in exon 2 (34.6 vs. 51.4 years, p = 0.038). (C) Plots comparing male and female patients and (D) plots comparing FALS patients with SALS patients; neither comparison identified a significant difference in AAO (p = 0.58 and 0.15, respectively).
Twenty-nine probands patients were accessible for data regarding diagnostic delay. With the exception of one case with an extremely long diagnostic delay of 84 months, the mean (SD) diagnostic delay of the remaining was 10.7 ± 6.2 months (Figure 3A). The diagnostic delay showed no differences in different exons (Figure 3B). The time to diagnosis of male patients exhibited no difference compared to female patients [10.4 (5.9) vs. 11.0 (6.7) months, p = 0.82] (Figure 3C). It is worth noting that the mean of the diagnostic delay of FALS patients is less than that of SALS patients and was found to be statistically significant [9.5 (4.8) vs. 20.3 (9.3) months, p = 0.0026] (Figure 3D). Diagnostic delay varied greatly across different mutations. Four mutations of four probands, namely, p.A5S, p.L107V, p.C147R, and p.G148D, showed a diagnostic delay less than the first quartile (25%, <6 months). Another five mutations of eight probands, namely, p.H47R, p.V48A, p.P75S, p.C112Y, and p.L145S, showed a diagnostic delay more than the third quartile (75%, >12 months).
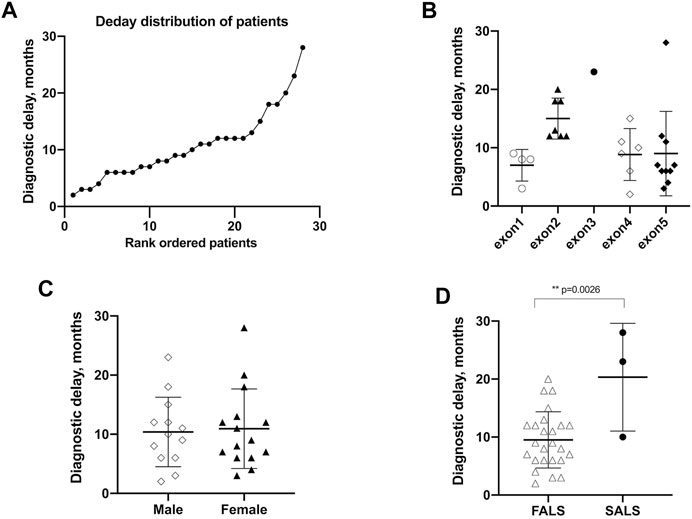
FIGURE 3. Diagnostic delay of patients with SOD1 mutations. (A) Plot of rank ordered SOD1-mutant patients showing the median diagnostic delay of 9.5 months (B) Plot comparing the diagnostic delay showed no differences in different exons of SOD1 gene. (C) Plots comparing the diagnostic delay exhibited no differences between male and female patients. (D) plots comparing FALS patients with SALS patients in the diagnostic delay time; neither comparison identified a significant difference in AAO (p = 0.58 and 0.15, respectively). FALS patients (n = 25) received an earlier diagnosis than SALS patients (n = 3) [9.5 (4.8) vs. 20.3 (9.3), p = 0.0026].
Disease duration data were available for 31 probands, and 9 patients still survive at the censoring date. The median survival time was 40.0 months, and the 5-years survival rate was 30.7% for all subjects (Figure 4A). The survival time showed no difference between all male and female patients (Figure 4B). Given the extremely long survival time in mutation of p.H47R, we excluded the p.H47R mutation in both male and female patients for statistical analysis. Strikingly, male patients showed significantly longer survival time than female patients (40 vs. 16 months, p = 0.05) (Figure 4C).
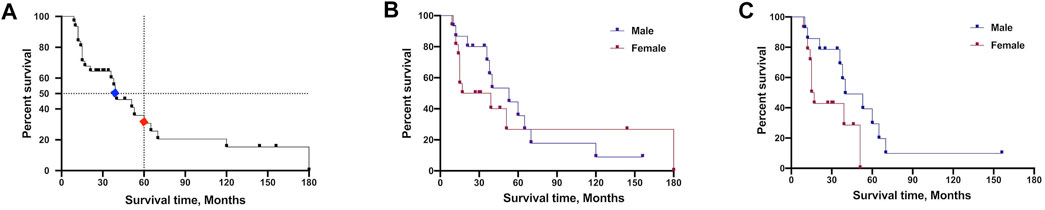
FIGURE 4. Survival analysis comparison of patients carrying SOD1 mutations. (A) Plot of survival probabilities for all patients with SOD1 mutations. The overall median survival time is seen in blue mark (40 months), and the 5-years survival rate is seen in red (30.7%). (B) Plot of survival probabilities between all male and female patients (median survival 53 vs. 28 months, p = 0.63). (C) Plot of survival probabilities between male and female patients after excluding the H47R mutation data, indicating that males had a longer survival time than females (median survival 40 vs. 16 months, p = 0.05).
Discussion
In this study, which was combined with our previously reported SOD1 mutations, 32 ALS probands were harboring 21 confirmed SOD1 mutations. This is the first study to study SOD1 mutations to date in the southeastern Chinese ALS population. The extensive analysis of SOD1 mutation distribution and natural history could serve ongoing clinical trials targeted for patients with SOD1 mutations.
Both novel variants (p.G17H and p.E134*) could form toxic aggregates in vitro, indicating that they caused loss of protein intrinsic stability. Interestingly, a loss-of-function effect was also shown in both variants, given the reduced protein expression and mRNA synthesis. Many SOD1 mutations, such as p.G93A, p.G37R, and p.H48Q, would increase protein expression, which is in accordance with the gain-of-function presumption (Tang et al., 2019). However, overloading misfolded SOD1 proteins would induce endoplasmic reticulum (ER) stress, subsequently activating the unfold protein response (UPR) to maintain homeostasis. Once the UPR is activated, overall protein translation would shut down to alleviate ER stress by stopping the translation of misfolded proteins. Our results highlight that albeit with reduced expression of toxic protein level, neurons still fail to control the homeostasis because of toxic SOD1 aggregated protein.
In southeastern Chinese SOD1 mutant patients with ALS, the four most frequent mutations in SOD1 gene were p.V48A (4/32, 12.5%), H47R, C112Y, and G148D (3/32, 9.4%). The p.V48A has been reported by another team from southern China (Chen et al., 2020), implying that there might be a founder effect existing for p.V48A in southeastern China. According to a recent nationwide survey on SOD1 in China by Fan’s team, p.H47R was the most frequent mutation in China (Tang et al., 2019) and Japan (Yamashita and Ando, 2015). In our study, patients with p.H47R and p.C112Y variants presented with the typical SOD1-related phenotype (lower limbs onset and lower motor neuron involvement) and mild disease course (longer diagnostic delay and extended survival time) (Table 1). The most predominant SOD1 gene mutations in total were p.D91A, followed by p.I114T and p.A5V (Yamashita and Ando, 2015). The mutation p.D91A was absent in our study and absent in southeastern Chinese demographic, and only one case with p.A5V mutation was found. However, approximately 30% of all Finnish ALS patients have p.D91A mutation, and the p.A5V accounts for almost half in SOD1 mutations in the United States (Bali et al., 2017; Korpi et al., 2020). Notably, SOD1A5V is a relatively homogeneous form exclusively with aggressive progression and short disease duration (Bali et al., 2017). Therefore, the frequency of specific mutations can vary among different regions.
ALS predominance in males was reported by several teams in Mainland and Taiwan in China (Soong et al., 2014; Wei et al., 2017; Tang et al., 2019), as well as the largest cohort study from North America (Bali et al., 2017). However, there was no gender difference in our patients harboring SOD1 mutations probably due to the limited sample size. The median survival time of SOD1-mutated patients in our study is 3.3 years, while it is 2.7 years in American patients (Bali et al., 2017). The aggressive mutation p.A5V is an unignorable bias for survival time calculation which accounts for 38% of participants in the cohort of North America (Bali et al., 2017). And the benign form p.H47R was the most frequently identified mutation in SOD1-mutated Chinese patients. Therefore, it is reasonable to find different survival times in Chinese and American patients. The 5-years survival rate was 30.7% for all subjects in our study compared to 55% in Fan’s team (Tang et al., 2019). In addition, our results revealed that male SOD1-mutated patients had longer survival time, which is not consistent with the finding by Fan’s team (Tang et al., 2019). At first, we found that the survival time showed no difference between genders when including all variants (Figure 4B). Because the variant p.H47R is associated with long survival time and high frequency in Chinese SOD1-mutated patients, we think it would be a potential bias to affect the statistical analysis. Therefore, we further analyzed the survival time by excluding the p.H47R variant in one male and two female patients, and the results revealed increased survival in male patients (Figure 4C). Given the variant p.H47R predominance in females in the study conducted by Fan’s team (Tang et al., 2019), this may be the possible reason for this conflicting result. Thus, it deserves our attention when combining p.H47R into the overall results, especially when considering the limited sample size. One possible explanation for gender differences might be the effect of gonadal hormones. A very recent report demonstrated that dihydrotestosterone (DHT) levels were significantly decreased in all ALS patients, indicating that DHT is probably integral to motor neuron survival (Sawal et al., 2020).
The mean (SD) AAO of the SOD1-mutant patients in this study was 46 ± 11.4 years, which is older than the finding from the largest study of a Chinese population (43.92 ± 9.24) but younger than the multi-center cohort from North America (49.7 ± 12.3) (Bali et al., 2017; Tang et al., 2019). Moreover, the mean AAO of the SOD1-mutant patients is younger than that of the overall Chinese ALS population (55.5–58.76 years) (Liu et al., 2018). SOD1-mutant patients were a subgroup in the overall ALS population, and similar findings could be seen in Japan (62.1 years) (Furuta et al., 2013), Europe (62.1–66.3 years) (Al-Chalabi et al., 2014), and Canada (48.9 years) (Eisen et al., 2008). Interestingly, the overall AAO in India is 46.2 years old, which is lower than many regions in the world (Nalini et al., 2008). The mean (SD) diagnostic delay of SOD1-mutant patients was 10.7 ± 6.2 months, which is shorter than 14 months of overall Chinese ALS patients (Chen et al., 2021). The SOD1-mutant FALS patients received an earlier diagnosis than the SALS patients (Figure 3D), which is not consistent with the finding of Fan’s team. All ALS patients in our cohort carried one mutation in the SOD1 gene. However, it should be mentioned that 3.8–4.7% of ALS patients carried potentially pathogenic variants in more than one ALS genes (Cady et al., 2015; Liu et al., 2016). Most FALS patients carried missense mutations in SOD1 concomitantly with the variants in other genes, including SETX, OPTN, DAO, GRN, ANG, and DCTN1 (Cady et al., 2015; Liu et al., 2016). The average AAO was less than 10 years earlier in patients harboring multiple variants compared with those harboring one (Cady et al., 2015; Liu et al., 2016). However, the burden of variants in ALS-related genes seemingly did not influence the site of onset or survival (Cady et al., 2015; Liu et al., 2016).
Limitations
The SOD1-mutant ALS patients in this cohort all come from southeastern China; thus, it should be mentioned that these data are not representative of all Chinese populations. Given the limited sample size, the frequency of SOD1 for FALS probably was overestimated. Moreover, our center is still working on the enrollment of the ALS patients and screening for the genetic causes. In addition, for those patients with family history but negative genetic results, we would need a larger scale sequence such as whole genome sequencing (WGS) to define the genetic information of Chinese ALS cases.
Conclusion
Overall, our work provides an updated natural history of SOD1-mutated ALS patients in southeastern China, which can serve as a supplementary reference database for SOD1-targeted therapy in clinical trials.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors. The data presented in the study are deposited in the Sequence Read Archive repository, accession number SAMN21561907.
Ethics Statement
The studies involving human participants were reviewed and approved by the ethics committee of Second Affiliated Hospital of Zhejiang University School of Medicine. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Authors Contributions
L-XC, study concept and design, genetic analysis, interpretation of data, experiments, statistical analysis, and drafting of the article and figures. H-FX, validation of data and experiments. P-SW, patient enrollment. X-XY, patient enrollment. Z-YW, study concept and design, acquisition of data, study supervision, and critical revision of the article. H-FL, study concept and design, study supervision, and critical revision of the article. All authors read and approved the article.
Funding
This work was supported by the grant (81671245) to Z-YW from the National Natural Science Foundation of China and the Fundamental Research Funds for the Central Universities (2019XZZX001-01-04).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank all participants for their willingness to participate in this study. In particular, we are thankful for the efforts of Cameron Lenahan in helping to revise the language.
References
Al-Chalabi, A., Calvo, A., Chio, A., Colville, S., Ellis, C. M., Hardiman, O., et al. (2014). Analysis of Amyotrophic Lateral Sclerosis as a Multistep Process: a Population-Based Modelling Study. Lancet Neurol. 13, 1108–1113. doi:10.1016/s1474-4422(14)70219-4
Bali, T., Self, W., Liu, J., Siddique, T., Wang, L. H., Bird, T. D., et al. (2017). Defining SOD1 ALS Natural History to Guide Therapeutic Clinical Trial Design. J. Neurol. Neurosurg. Psychiatry 88, 99–105. doi:10.1136/jnnp-2016-313521
Brasil, A. d. A., de Carvalho, M. D. C., Gerhardt, E., Queiroz, D. D., Pereira, M. D., Outeiro, T. F., et al. (2019). Characterization of the Activity, Aggregation, and Toxicity of Heterodimers of WT and ALS-Associated Mutant Sod1. Proc. Natl. Acad. Sci. USA 116, 25991–26000. doi:10.1073/pnas.1902483116
Brooks, B. R., Miller, R. G., Swash, M., Munsat, T. L., and World Federation of Neurology Research, D. (2000). El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 293–299. doi:10.1080/146608200300079536
Cady, J., Allred, P., Bali, T., Pestronk, A., Goate, A., Miller, T. M., et al. (2015). Amyotrophic Lateral Sclerosis Onset Is Influenced by the burden of Rare Variants in Known Amyotrophic Lateral Sclerosis Genes. Ann. Neurol. 77, 100–113. doi:10.1002/ana.24306
Chen, L., Xu, L., Tang, L., Xia, K., Tian, D., Zhang, G., et al. (2021). Trends in the Clinical Features of Amyotrophic Lateral Sclerosis: A 14-year Chinese Cohort Study. Eur. J. Neurol. 28, 2893. doi:10.1111/ene.14943
Chen, W., Xie, Y., Zheng, M., Lin, J., Huang, P., Pei, Z., et al. (2020). Clinical and Genetic Features of Patients with Amyotrophic Lateral Sclerosis in Southern China. Eur. J. Neurol. 27, 1017–1022. doi:10.1111/ene.14213
Dong, H. L., Ma, Y., Yu, H., Wei, Q., Li, J. Q., Liu, G. L., et al. (2021). Bi-allelic Loss of Function Variants in COX20 Gene Cause Autosomal Recessive Sensory Neuronopathy. Brain 144, 2457. doi:10.1093/brain/awab135
Eisen, A., Mezei, M. M., Stewart, H. G., Fabros, M., Gibson, G., and Andersen, P. M. (2008). SOD1 Gene Mutations in ALS Patients from British Columbia, Canada: Clinical Features, Neurophysiology and Ethical Issues in Management. Amyotroph. Lateral Scler. 9, 108–119. doi:10.1080/17482960801900073
Furuta, N., Makioka, K., Fujita, Y., and Okamoto, K. (2013). Changes in the Clinical Features of Amyotrophic Lateral Sclerosis in Rural Japan. Intern. Med. 52, 1691–1696. doi:10.2169/internalmedicine.52.0195
Gurney, M., Pu, H., Chiu, A., Dal Canto, M., Polchow, C., Alexander, D., et al. (1994). Motor Neuron Degeneration in Mice that Express a Human Cu,Zn Superoxide Dismutase Mutation. Science 264, 1772–1775. doi:10.1126/science.8209258
Korpi, E. R., Lindholm, D., Panula, P., Tienari, P. J., and Haltia, M. (2020). Finnish Neuroscience from Past to Present. Eur. J. Neurosci. 52, 3273–3289. doi:10.1111/ejn.14693
Lin, H.-X., Tao, Q.-Q., Wei, Q., Chen, C.-X., Chen, Y.-C., Li, H.-F., et al. (2019). Identification and Functional Analysis of Novel Mutations in the SOD1 Gene in Chinese Patients with Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 20, 222–228. doi:10.1080/21678421.2019.1582668
Liu, Q., Liu, F., Cui, B., Lu, C. X., Guo, X. N., Wang, R. R., et al. (2016). Mutation Spectrum of Chinese Patients with Familial and Sporadic Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 87, 1272–1274. doi:10.1136/jnnp-2016-313337
Liu, X., He, J., Gao, F.-B., Gitler, A. D., and Fan, D. (2018). The Epidemiology and Genetics of Amyotrophic Lateral Sclerosis in China. Brain Res. 1693, 121–126. doi:10.1016/j.brainres.2018.02.035
Liu, Z.-J., Lin, H.-X., Wei, Q., Zhang, Q.-J., Chen, C.-X., Tao, Q.-Q., et al. (2019). Genetic Spectrum and Variability in Chinese Patients with Amyotrophic Lateral Sclerosis. Aging Dis. 10, 1199–1206. doi:10.14336/ad.2019.0215
Liu, Z. J., Li, H. F., Tan, G. H., Tao, Q. Q., Ni, W., Cheng, X. W., et al. (2014). Identify Mutation in Amyotrophic Lateral Sclerosis Cases Using HaloPlex Target Enrichment System. Neurobiol. Aging 35, 2881–e15. doi:10.1016/j.neurobiolaging.2014.07.003
Nalini, A., Thennarasu, K., Gourie-Devi, M., Shenoy, S., and Kulshreshtha, D. (2008). Clinical Characteristics and Survival Pattern of 1153 Patients with Amyotrophic Lateral Sclerosis: Experience over 30 Years from India. J. Neurol. Sci. 272, 60–70. doi:10.1016/j.jns.2008.04.034
Niu, Y.-F., Xiong, H.-L., Wu, J.-J., Chen, Y., Qiao, K., and Wu, Z.-Y. (2011). Screening of Mutations in SOD1 Gene and Analysis of Geno-Type-Phenotype Correlation in Chinese Patients with Amyotrophic Lateral Sclerosis. Hereditas (Beijing) 33, 720–724. doi:10.3724/sp.j.1005.2011.00720
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 362, 59–62. doi:10.1038/362059a0
Sawal, N., Kaur, J., Kaur, K., and Gombar, S. (2020). Dihydrotestosterone in Amyotrophic Lateral Sclerosis-The Missing Link?. Brain Behav. 10, e01645. doi:10.1002/brb3.1645
Soong, B. W., Lin, K. P., Guo, Y. C., Lin, C. C., Tsai, P. C., Liao, Y. C., et al. (2014). Extensive Molecular Genetic Survey of Taiwanese Patients with Amyotrophic Lateral Sclerosis. Neurobiol. Aging 35, 2423–2426. doi:10.1016/j.neurobiolaging.2014.05.008
Tang, L., Ma, Y., Liu, X.-l., Chen, L., and Fan, D.-s. (2019). Better Survival in Female SOD1-Mutant Patients with ALS: a Study of SOD1-Related Natural History. Transl. Neurodegener. 8, 2. doi:10.1186/s40035-018-0142-8
Wei, Q., Zhou, Q., Chen, Y., Ou, R., Cao, B., Xu, Y., et al. (2017). Analysis of SOD1 Mutations in a Chinese Population with Amyotrophic Lateral Sclerosis: a Case-Control Study and Literature Review. Sci. Rep. 7, 44606. doi:10.1038/srep44606
Keywords: amyotrophic lateral sclerosis, Cu/Zn superoxide dismutase 1, Chinese, Southeastern, mutation, natural history
Citation: Chen L-X, Xu H-F, Wang P-S, Yang X-X, Wu Z-Y and Li H-F (2021) SOD1 Mutation Spectrum and Natural History of ALS Patients in a 15-Year Cohort in Southeastern China. Front. Genet. 12:746060. doi: 10.3389/fgene.2021.746060
Received: 28 July 2021; Accepted: 14 September 2021;
Published: 14 October 2021.
Edited by:
Massimo Zeviani, University of Padua, ItalyReviewed by:
Adriano Chio, University of Turin, ItalySerena Lattante, Catholic University of the Sacred Heart, Italy
Copyright © 2021 Chen, Xu, Wang, Yang, Wu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi-Ying Wu, emhpeWluZ3d1QHpqdS5lZHUuY24=; Hong-Fu Li, aG9uZ2Z1bGlAemp1LmVkdS5jbg==