
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 09 September 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.744884
This article is part of the Research TopicInherited Protein Glycosylation Defects in Human DiseasesView all 15 articles
 Carlos Alberto González-Domínguez1,2
Carlos Alberto González-Domínguez1,2 Moisés O. Fiesco-Roa3,4
Moisés O. Fiesco-Roa3,4 Samuel Gómez-Carmona5Anke Paula Ingrid Kleinert-Altamirano5,7Miao He6Earnest James Paul Daniel6
Samuel Gómez-Carmona5Anke Paula Ingrid Kleinert-Altamirano5,7Miao He6Earnest James Paul Daniel6 Kimiyo M. Raymond8Melania Abreu-González9
Kimiyo M. Raymond8Melania Abreu-González9 Sandra Manrique-Hernández1,2Ana González-Jaimes1
Sandra Manrique-Hernández1,2Ana González-Jaimes1 Roberta Salinas-Marín1
Roberta Salinas-Marín1 Carolina Molina-Garay10
Carolina Molina-Garay10 Karol Carrillo-Sánchez10
Karol Carrillo-Sánchez10 Luis Leonardo Flores-Lagunes10
Luis Leonardo Flores-Lagunes10 Marco Jiménez-Olivares10
Marco Jiménez-Olivares10 Anallely Muñoz-Rivas10
Anallely Muñoz-Rivas10 Mario E. Cruz-Muñoz11
Mario E. Cruz-Muñoz11 Matilde Ruíz-García12
Matilde Ruíz-García12 Hudson H. Freeze13
Hudson H. Freeze13 Héctor M. Mora-Montes14
Héctor M. Mora-Montes14 Carmen Alaez-Verson10
Carmen Alaez-Verson10 Iván Martínez-Duncker1,15*
Iván Martínez-Duncker1,15*This study reports on a Mexican mestizo patient with a multi-systemic syndrome including neurological involvement and a type I serum transferrin profile. Clinical exome sequencing revealed complex alleles in ALG1, the encoding gene for the chitobiosyldiphosphodolichol beta-mannosyltransferase that participates in the formation of the dolichol-pyrophosphate-GlcNAc2Man5, a lipid-linked glycan intermediate during N-glycan synthesis. The identified complex alleles were NM_019109.5(ALG1): c.[208 + 16_208 + 19dup; 208 + 25G > T] and NM_019109.5(ALG1): c.[208 + 16_208 + 19dup; 1312C > T]. Although both alleles carried the benign variant c.208 + 16_208 + 19dup, one allele carried a known ALG1 pathogenic variant (c.1312C > T), while the other carried a new uncharacterized variant (c.208 + 25G > T) causing non-functional alternative splicing that, in conjunction with the benign variant, defines the pathogenic protein effect (p.N70S_S71ins9). The presence in the patient’s serum of the pathognomonic N-linked mannose-deprived tetrasaccharide marker for ALG1-CDG (Neu5Acα2,6Galβ1,4-GlcNAcβ1,4GlcNAc) further supported this diagnosis. This is the first report of an ALG1-CDG patient from Latin America.
Approximately 140 inborn errors of metabolism have been classified as congenital disorders of glycosylation (CDG), a rapidly expanding group of diseases caused by defects in the synthesis and attachment of glycans to glycoproteins and glycolipids (Ondruskova et al., 2021). ALG1-CDG is a subtype with severe multiorgan involvement (OMIM 608540) caused by pathogenic variants in ALG1. This gene encodes for a transmembrane chitobiosyldiphosphodolichol beta-mannosyltransferase that participates in the first steps of N-glycan biosynthesis that occur in the cytosolic side of the endoplasmic reticulum involving dolichol-pyrophosphate-GlcNAc2Man5 synthesis, an intermediate of the lipid-linked precursor oligosaccharide that is subsequently transferred to nascent glycoproteins for protein N-glycosylation. ALG1 extends GlcNAc2-PP-dolichol by adding the first mannose in ß1,4-linkage using GDP-mannose as a substrate donor to generate Man1GlcNAc2-PP-dolichol (Couto et al., 1984). ALG1 is part of a multi-mannosyltransferase complex that includes ALG1, ALG2, and ALG11 involved in a five-reaction process (Gao et al., 2004; O’Reilly et al., 2006; Aebi, 2013).
In contrast to PMM2-CDG, the most prevalent CDG affecting N-glycosylation with >1,000 patients reported worldwide, <80 cases of ALG1-CDG have been reported in the literature since 2004, none from Latin America (Grubenmann et al., 2004; Kranz et al., 2004; Schwarz et al., 2004; Dupré et al., 2010; Morava et al., 2012; Rohlfing et al., 2014; Barba et al., 2016; Harshman et al., 2016; Ng et al., 2016; Marques-da-Silva et al., 2017; Pérez-Cerdá et al., 2017; Medrano et al., 2019; Lei et al., 2020; Bogdañska et al., 2021; Quelhas et al., 2021). According to the Human Gene Mutation Database (HGMD), 50 disease-causing mutations have been reported in ALG1, including 41 missense/non sense, 6 splicing mutations, 1 small insertion, 1 small indel, and 1 gross deletion (as of July 2021).
Here, we describe a Mexican mestizo patient with ALG1-CDG who is a compound heterozygous of two pathogenic variants. One of these variants (NM_019109.5(ALG1):c.208 + 25G > T), previously uncharacterized, in combination with a known cis benign variant NM_019109.4(ALG1): c.208 + 16_208 + 19dup, causes, a new intronic donor splice site resulting in partial intron 1 retention (+27 bp), producing an aminoacid substitution and insertion (p.N70S_S71ins9).
The patient was a 15-year-old male of Mexican ancestry with intellectual disability, seizures, hypotonic quadriplegia, and disproportionated microcephaly (height in normal centile). He was born in Tabasco State (in the southern part of Mexico). No consanguinity or endogamy was reported. The patient’s parents and an older sister were healthy, but he had another sister who died at 15 years old with similar manifestations. The mother reported a history of a first-trimester spontaneous abortion. There was no other relevant family history (Figure 1). The patient was the product of a naturally conceived 38-week singleton pregnancy to a 33-year-old mother and 37-year-old father, uneventful pregnancy with two normal prenatal ultrasounds. Delivery was via emergency cesarean section for prolonged rupture of membranes. At birth weight the patient was 3,100 g [46th centile; −0.1 standard deviation scores (SDS)] and length was 50 cm (50th centile; 0.5 SDS); additional birth parameters and APGAR score were unknown. He did not require respiratory support or supplementary interventions. He left the hospital 2 days after delivery. He was hospitalized for 30 days at 9 months of age due to seizures. During this hospitalization he was first noted to have some developmental delay; the mother reported the loss of some abilities after the onset of seizures.
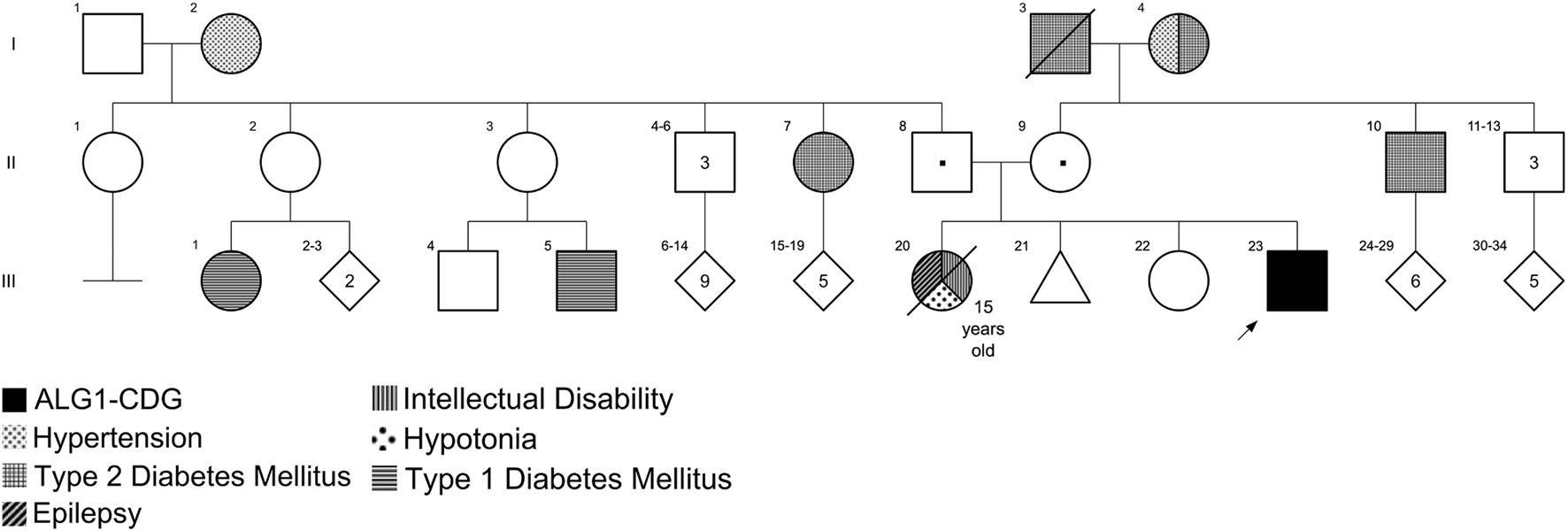
Figure 1. Pedigree of the family with ALG1-CDG. Individual III:23 is the patient reported in this work with ALG1-CDG. Individual III:20 had an intellectual disability, epilepsy, and hypotonia; however, genotyping was not possible because she died at 15 years old.
He was first seen by a geneticist at 5 years old; however, no diagnosis was integrated. He was re-evaluated by geneticists at 10.8 years old. The anthropometric features were 32.3 kg of weight (24th centile; −0.71 SDS), 151 cm of height (85th centile; + 1.05 SDS), and 49.2 cm of head circumference (< 3rd centile; −3.01 SDS) (HP:0000252); the head circumference measurement at 4 years old was 46.3 cm (< 3rd centile; −2.72 SDS), no previous head circumference evaluation was available. After a physical examination, the following findings were found: Short forehead (HP:0000350), eyebrow slightly sparse (HP:0000535), slightly prominent nasal bridge (HP:0000426), prominent cheeks (HP:0000293), wide mouth (HP:0000154), and widely spaced and misplaced teeth (HP:0000687 and HP:0006316, respectively). He had premature gray hair (HP:0002216) and one café-au-lait spot (HP:0000957) on his left forearm. The neurological evaluation found profound intellectual disability (HP:0002187), stereotypical and self-injurious behaviors (HP:0000733 and HP:0100716, respectively), sialorrhea (HP:0002307), central hypotonia (HP:0011398), hypotonic quadriplegia (HP:0002273), and generalized hyperreflexia (HP:0007034); no major motor milestones (gross motor function classification system level IV), and only guttural sounds [speech delay (alalia)] (HP:0001344). No other physical abnormality was detected.
During genetics follow-up, several investigations were performed. Metabolic screening for inborn errors of metabolism (including ammonia, lactate, plasma amino acids, and urine organic acids) was normal. Blood counts, INR, and prothrombin time were normal. No other laboratory tests were performed. An EEG performed at 10 years old showed diffuse slowing (HP:0010845), slow spike-and-wave activity (HP:0010850), and frontal intermittent rhythmic delta activity (FIRDA). The visual and brainstem-auditory evoked potentials revealed bilateral integrity of visual and auditory pathways, respectively. MRI was abnormal (Figure 2), showing generalized and asymmetric cortical-subcortical frontotemporal atrophy (HP:0006892) predominantly on the left, hypoplasia of the frontal lobes (HP:0007333), enlarged Sylvian cistern (HP:0100952), frontotemporal cortical dysplasia [pachygyria (HP:0001302) and polymicrogyria (HP:0002126)], asymmetric temporo-occipital ventriculomegaly (HP:0006956) predominantly on the left, hypoplastic corpus callosum (HP:0002079), and enlargement of cisterna magna (HP:0002280) without cerebellar atrophy. For the phenotype description, the Human Phenotype Ontology was used (Köhler et al., 2021).
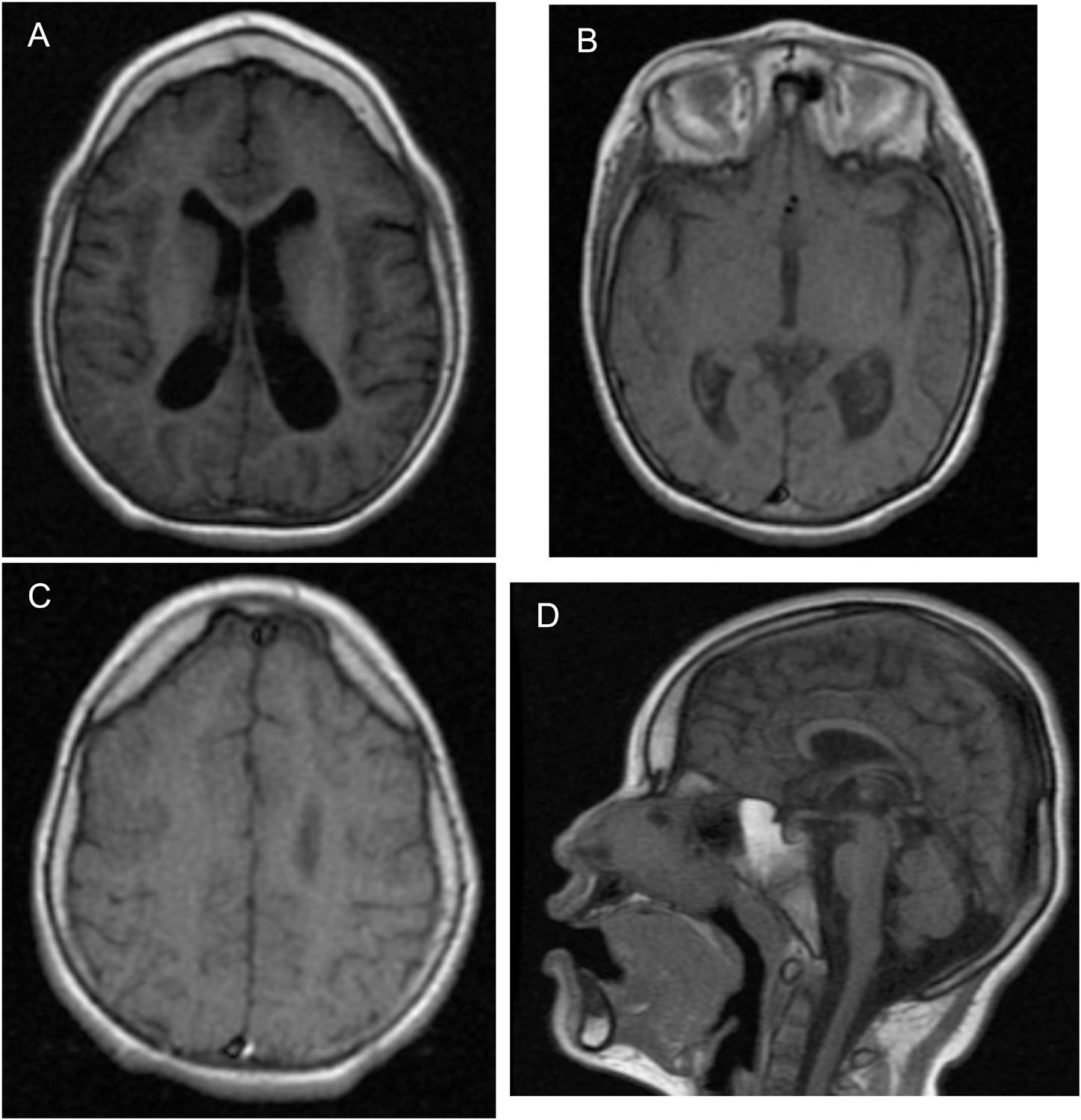
Figure 2. Abnormal brain MRI of the patient at 5 years old. (A) Cortical–subcortical frontotemporal atrophy, hypoplasia of the frontal lobes, and ventriculomegaly predominantly on the left on axial on axial T1-weighted FLAIR MR image. (B) Enlarged Sylvian cistern and ventriculomegaly predominantly on the left on axial T1-weighted MR image. (C) Frontotemporal cortical dysplasia pachygyria and polymicrogyria on axial T1-weighted MR image. (D) Hypoplastic corpus callosum and enlargement of cisterna magna without cerebellar atrophy on sagittal T1-weighted MR image.
Based on the clinical phenotype, electrospray ionization mass spectrometry (ESI-MS) analysis of serum transferrin (Tf) was performed and a type I profile was established (mono-oligo/di-oligo = 0.90) (Figure 3A). To determine the genetic basis of the disease, a skin biopsy was performed to obtain a fibroblast culture. The genomic DNA (gDNA) obtained from the patient’s fibroblasts was sent to clinical exome sequencing (CES), and three ALG1 variants were found in the form of two complex alleles: one complex allele composed by a benign variant NM_019109.4(ALG1): c.208 + 16_208 + 19dup in cis with the known pathogenic variant NM_019109.5(ALG1):c.1312C > T (p.R438W) defined as NM_019109.5(ALG1): c.[208 + 16_208 + 19dup; 1312C > T] and a second complex allele composed by the same benign variant NM_019109.4(ALG1): c.208 + 16_208 + 19dup in cis with a previously not described variant c.208 + 25G > T that we termed NM_019109.5(ALG1): c.[208 + 16_208 + 19dup; 208 + 25G > T].
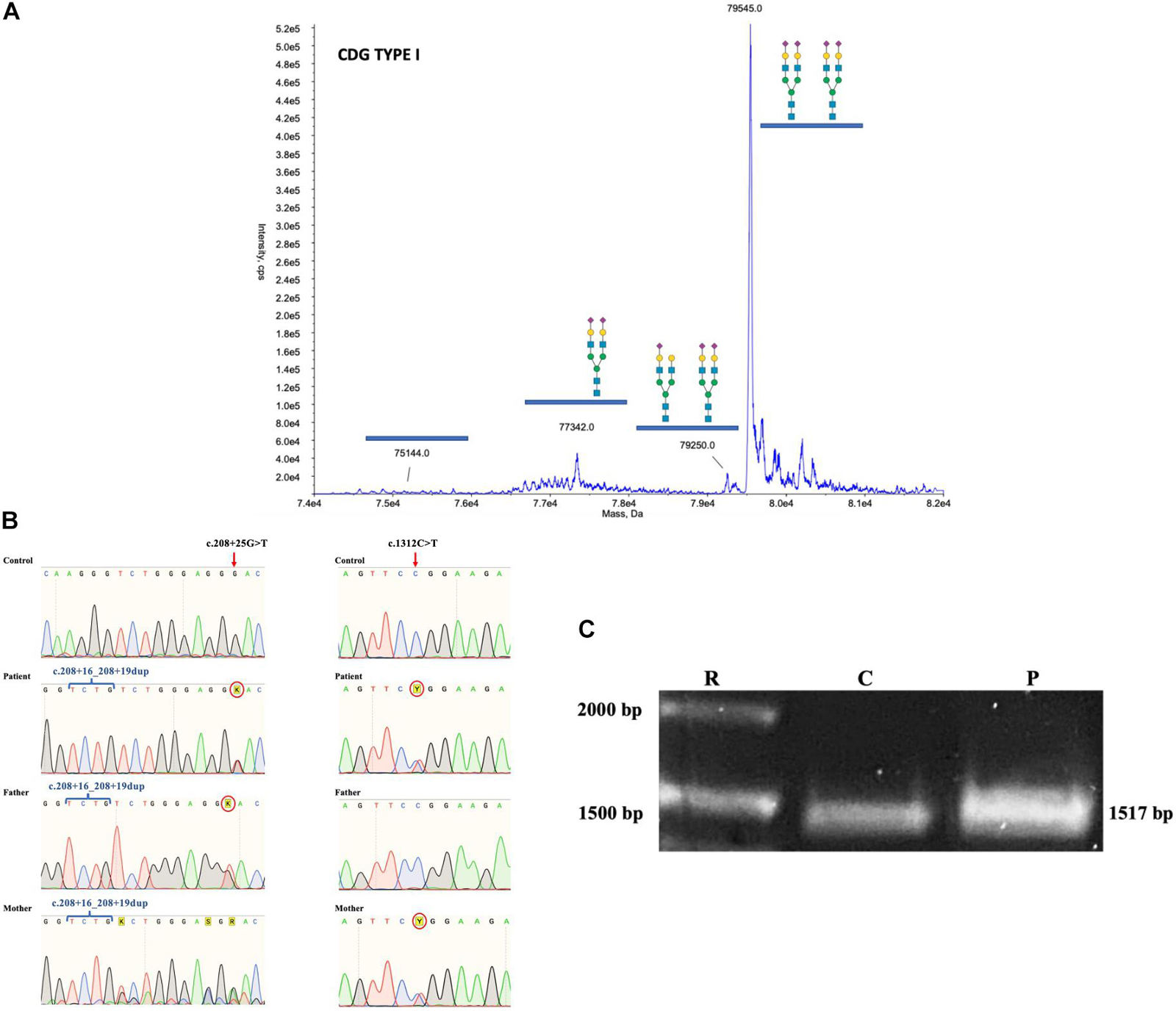
Figure 3. (A) ESI-MS analysis of serum Tf isoforms. Profile of the patient showing an increase of mono-oligo Tf, peaks are annotated with the glycosylation moiety structure with the following meaning: blue box = N-acetylglucosamine; green circle = mannose; yellow circle = galactose; purple diamond = N-acetylneuraminic acid (sialic acid). (B) Chromatograms of gDNA sequencing from healthy control, mother, father, and patient showing the inheritance of the ALG1 c.208 + 16_208 + 19dup, the c.208 + 25G > T, and the c.1312C > T variants. (C) PCR of the coding sequence of ALG1 showing the expected wt 1517 bp size in healthy control and the patient’s amplicon showing a slightly thicker band.
Sequencing of the parents’ gDNA showed that the c.[208 + 16_208 + 19dup; 1312C > T] complex allele was inherited from the mother and the c.[208 + 16_208 + 19dup; 208 + 25G > T] from the father (Figure 3B). The Sanger sequencing of the mother’s ALG1 shows an overlap that is the result of heterozygosity for the benign c.208 + 16_208 + 19dup variant, in contrast to the father that is homozygote.
A consideration in the case of the complex allele c.[208 + 16_208 + 19dup; 208 + 25G > T] is that the c.208 + 25G > T is located + 29 bp from the donor splicing site in exon 1 due to a four base duplication (TCTG) caused by the benign variant c.208 + 16_208 + 19dup (chr16:5122072); ClinVar Variation ID 95935, dbSNP:rs35400794 (Figure 4C).
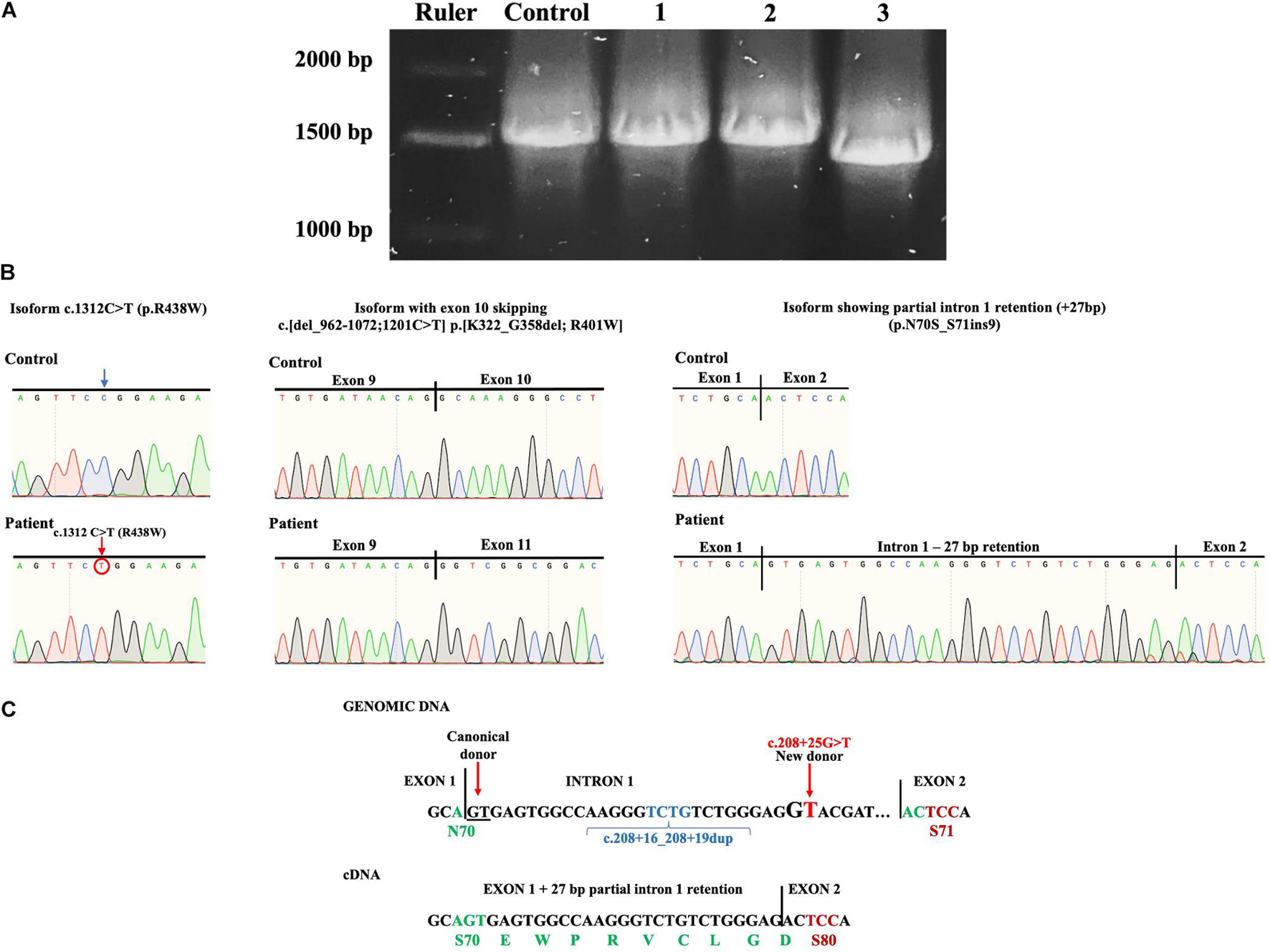
Figure 4. (A) Isoforms found after PCR subcloning of the patient’s ALG1 amplicon. R, ruler; C, healthy wt control; 1, a constitutively spliced isoform bearing the c.1312C > T (p.R438W) variant; 2, an alternatively spliced isoform with partial intron 1 retention (+27bp); 3, an alternatively spliced isoform bearing the c.1312C > T variant with exon 10 skipping c.[del_962-1072;1201C > T] p.[K322_G358del; R401W]. (B) Sanger sequencing chromatograms of patient’s isoforms showing the c.1312C > T (p.R438W) variant; exon 10 skipping c.[del_962-1072;1201C > T] p.[K322_G358del; R401W] and partial intron 1 retention (+ 27 bp) (p.N70S_S71ins9). (C) Splicing mechanism that induces the + 27 bp partial intron 1 retention caused by the complex allele c.[208 + 16_208 + 19dup; 208 + 25G > T]. The new donor splicing site (GG to GT) causes partial intron 1 retention (+ 27 bp) that when translated would result in aminoacid substitution N70S and insertion of nine aminoacids.
The c.208 + 25G > T variant was considered potentially pathogenic because it could induce a new donor splice site (GG to GT) and cause non-functional alternative splicing. Using the Human Splicing Finder prediction (HSFPro, Genomnis), it was found that this change significantly alters splicing with the following values [WT-Mut%variation] [HSF Donor site (matrix GT) 61.35 > 88.49 (44.24%) and MaxEnt Donor site 1.01 > −8.65 (756.44%)].
To further investigate the presence of alternative splicing, the patient’s complementary DNA (cDNA) was synthesized and ALG1 was polymerase chain reaction (PCR) amplified. The ALG1 has 13 exons and codes for a 464 aminoacid protein. Amplification of ALG1 with primers ALG1s and ALG1as encompasses the coding sequence plus short stretches from the 5′- and 3′-UTR for a predicted amplicon of 1517 bp. PCR product analysis in an agarose gel showed a slightly thicker amplicon in the patient compared to the healthy control (Figure 3C). Because this suggested potential splicing isoforms in the patient, subcloning and screening were performed. Three types of isoforms were identified: one with the c.1312C > T variant and constitutive splicing (c.1312C > T; p.R438W) or with the same variant (shifted to position 1201) and exon 10 skipping c.[del_962-1072;1201C > T] translated as p.[K322_G358del; R401W] and a third isoform without the c.1312C > T variant, but that presented partial intron 1 retention (+ 27 bp) without causing a frameshift and that theoretically results when translated in substitution of N70S and insertion of nine aminoacids EWPRVCLGD (p.N70S_S71ins9) (Figures 4A–C).
To further biochemically confirm the ALG1-CDG diagnosis, previous works have shown that ALG1-CDG patients accumulate a novel N-linked mannose-deprived tetrasaccharide considered as a pathognomonic marker for ALG1-CDG (Neu5Acα2,6Galβ1,4-GlcNAcβ1,4GlcNAc) on serum glycoproteins (Bengtson et al., 2016; Ng et al., 2016; Zhang et al., 2016; Chen et al., 2019). Total serum N-gycans were analyzed by electrospray ionization–quadrupole time-of-flight mass spectrometry (ESI-QTOF) to detect the presence of the mannose-deprived N-tetrasaccharide, finding it significantly elevated at 0.23% of total glycan (normal 0–0.03); Gal1GlcNAc2 was also increased at 0.12% (normal 0) (Figure 5).
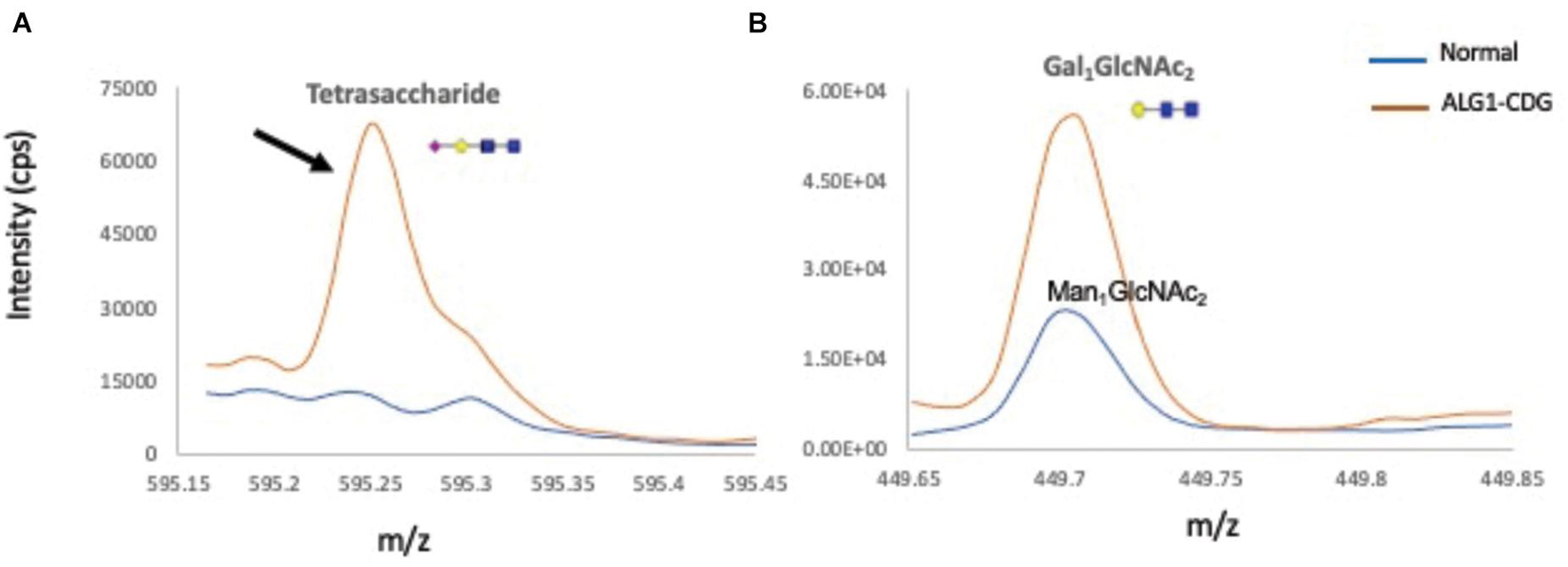
Figure 5. Total ion chromatograms of N-tetrasaccharide (NeuAc1Gal1GlcNAc2) and Gal1 or Man1GlcNAc2 in serum N-glycan profiles from a normal control and the ALG1-CDG patient. Overlay of total ion chromatograms of N-linked mannose deprived tetrasaccharide (A), Man1/Gal1GlcNAc2 (B) of a normal control plasma (in blue), and ALG1-CDG (red). Gal1GlcNAc2 is absent in total plasma proteins from normal controls, which instead have traces of Man1GlcNAc2 present. Markedly increased tetrasaccharide was detected in the ALG1-CDG patient as shown with a black arrow.
This report presents a patient with a severe clinical multisystemic phenotype with neurological involvement and an ESI-MS Tf with a type I profile consistent with CDG. CES revealed two complex alleles: The NM_019109.5(ALG1): c.[208 + 16_208 + 19dup; 1312C > T], inherited from the mother, and the NM_019109.5(ALG1): c.[208 + 16_208 + 19dup; 208 + 25G > T] inherited from the father. We considered that the latter could cause non-functional alternative splicing as predicted by HSFPro (Genomnis). PCR-based analysis of the patient’s ALG1 amplicon further supported this hypothesis (Figure 3C).
To determine the effects of the complex alleles, the patient’s ALG1 PCR amplicon was subcloned and screened, identifying three types of isoforms (Figure 4A), two derived from the c.[208 + 16_208 + 19dup; 1312C > T] complex allele and the third isoform with partial intron 1 retention (+ 27 bp) that did not present the c.1312C > T variant and that we conclude is derived from the c.[208 + 16_208 + 19dup; 208 + 25G > T] complex allele.
Regarding the isoforms derived from the c.[208 + 16_208 + 19dup; 1312C > T] complex allele, inherited by the mother, one presented constitutive splicing (p.R438W) and another an unexpected alternative splicing (exon 10 skipping) that translates into a 427 aa protein lacking 37 aminoacids p.[K322_G358del;R401W]. Because no variants were found in intron/exon junctions of exon 10, we consider that the c.1312C > T variant located in exon 13 could be responsible for altering distant splicing events, although further analysis is required to make a definitive conclusion. The c.1312C > T pathogenic variant included in this complex allele has been reported in six patients as compound heterozygotes with the following pathogenic variants, c.866A > G, c.450C > A, c.1145T > C, and c.1236A > G, and has been demonstrated to have reduced function (Dupré et al., 2010; Rohlfing et al., 2014; Ng et al., 2016; Zhang et al., 2016). No deleterious effects due to the benign intronic variant were observed.
For the third isoform without the c.1312C > T variant and with partial intron 1 retention (+ 27 bp) we conclude that it results from the paternally inherited complex allele c.[208 + 16_208 + 19dup; 208 + 25G > T] where the c.208 + 25G > T variant induces a new donor splice site that causes partial intron 1 retention (+ 27 bp). The partial intron 1 retention does not cause a frameshift but results in aminoacid substitution (N70S) and insertion of nine aminoacids (EWPRVCLGD) (p.N70S_S71ins9) (Figure 4C). It is important to note that the duplication of the TCTG bases occurring at the c.208 + 16_208 + 19dup variant (chr16:5122072) shifts the position c.208 + 25G > T to + 29 bp from the donor splicing site in exon 1 which determines the length of intron retention and preservation of the ORF (Figure 4C). We consider that the resulting p.N70S_S71ins9 has a reduced function based on the different physicochemical properties at position 70 [acidic (N) to hydroxylic (S)] as well as the insertion of nine aminoacids, including a P residue. Also, a previously reported p.S71F variant has been shown to have reduced function (Ng et al., 2016).
The presence of the ALG1-CDG pathognomonic N-linked mannose-deprived tetrasaccharide detected by ESI-QTOF when analyzing serum total N-glycans further supports the conclusion that the resulting protein variant p.N70S_S71ins9 does not have a normal function as it is not able to compensate for the decreased function of the p.R438W pathogenic variant. The fact that the N-tetrasaccharide was not detected by ESI-MS of serum Tf (Figure 3A) could be explained by differential sensitivity related to types of equipment and techniques, as well as a possible increased sensitivity to detect the N-tetrasaccharide when analyzing total N-glycans versus Tf glycans alone.
It is noteworthy that the c.208 + 16_208 + 19dup variant is considered a benign variant but that the additional presence of the c.208 + 25G > T variant results in a pathogenic complex allele c.[208 + 16_208 + 19dup; 208 + 25G > T]. According to gnomAD database (Lek et al., 2016) the variant c.208 + 16_208 + 19dup is very frequent in all ethnic groups (gnomAD ExomesVersion: 2.1.1 global frequency f = 0.558), as of July 2021. Concerning the c.208 + 25G > T variant, it is absent from the genomes and exomes in the genomAD database (as of July 2021). Interestingly, in the absence of the c208 + 16_208 + 19dup, the c.208 + 25G > T variant would cause a frameshift and a premature stop codon in exon 2 which would also be pathogenic and probably more severe.
Given these results, ALG1-CDG diagnosis was clinically, biochemically, and genetically established. In most disease-related genes, variants affecting splicing are not fully characterized because variant screening is restricted to gDNA. In our experience involving ATP6V0A2-CDG and more recently PMM2-CDG, amplification of cDNA transcripts is an invaluable tool to demonstrate non-functional alternative splicing, identifying the consequence on the protein and establishing the pathogenicity mechanism (Bahena-Bahena et al., 2014; González-Domínguez et al., 2020, 2021). Of the six reported disease-causing mutations in HGMD that potentially affect splicing of ALG1, only one has been experimentally confirmed (Table 1). The c.208 + 25G > T reported in this work would be the second variant confirmed to cause alternative non-functional splicing.
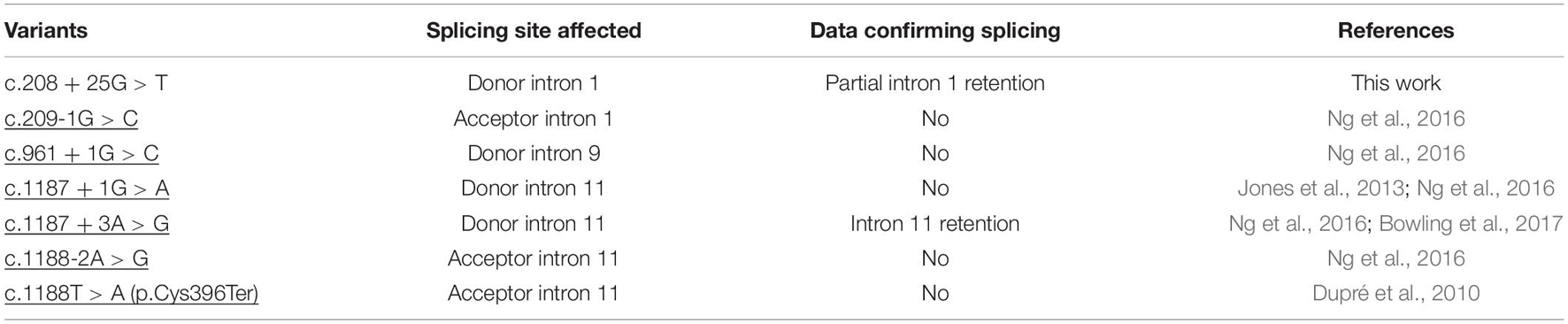
Table 1. Variants of ALG1 affecting splicing classified as disease-causing mutations in the Human Gene Mutation Database (HGMD) and the novel variant reported in this work.
This case also highlights the importance of increasing awareness and availability of technological platforms for biochemical and genetic diagnosis of rare diseases. This patient and his family had to wait 15 years to obtain a diagnosis, with a sibling who died at the same age without diagnosis. This time to diagnosis is not adequate for ALG1 patients that have an estimated premature death rate of 44% of which 65% occur at < 12 months of age (Ng et al., 2016). This is a lengthy diagnostic odyssey that is unfortunately too frequent in Latin American and most underdeveloped countries that in the era of exome sequencing can no longer be acceptable. It is necessary that academic and family organizations around the world pressure for a shift in global health public policy to guarantee that all patients affected by rare diseases have access to an interdisciplinary approach as well as free exome sequencing (Manickam et al., 2021).
The complex allele c.[208 + 16_208 + 19dup; 208 + 25G > T] (p.N70S_S71ins9) is pathogenic by causing non-functional alternative splicing of ALG1. Variants should be studied concerning their potential disruption of splicing, particularly if they affect canonical splicing sites. Increased awareness of rare diseases, including CDGs as well as the availability of technological platforms for genetic diagnosis, must be an international standard in public health policy.
Informed consent was obtained from both parents to perform a skin biopsy, fibroblast cultures, and all required research to obtain a molecular diagnosis and to publish other data on the patient and parents. All procedures followed were in accordance with national and institutional ethical standards on human experimentation and with the Helsinki Declaration of 1975 and revised in 2000.
On-column immunoaffinity ESI-MS analysis of serum Tf isoforms was performed using an API-5000 triple quadrupole mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA, United States).
The patient’s serum total N-glycans were analyzed by ESI-QTOF to detect the (Neu5Acα2,6Galβ1,4-GlcNAcβ1,4GlcNAc) N-tetrasaccharide as previously described (Chen et al., 2019).
From the patient’s skin biopsy, a primary culture of fibroblasts was obtained in AmnioMAXTM C-100 Basal Medium (Gibco® by Life TechnologiesTM; Life Technologies, Rockville, MD, United States) supplemented with 15% AmnioMAXTM C-100 Supplement (Gibco® by Life TechnologiesTM) and 1% penicillin/streptomycin antibiotic (Gibco® by Life TechnologiesTM). Fibroblast cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. Fibroblasts were further processed to obtain genetic material.
The gDNA was extracted from fibroblasts using Maxwell® 16 Blood DNA Purification Kit (Promega, Madison, WI, United States). The purity and concentration of the DNA samples were measured using NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States) and Qubit fluorometer (Thermo Fisher Scientific). Library preparation was performed using the reagents provided in the Clinical Exome sequencing panel kit, version 2 (Sophia Genetics SA, Saint Sulpice, Switzerland), according to the manufacturer’s protocol. Sequencing was performed on NextSeq Instrument (Illumina, San Diego, CA, United States). Sequencing data analysis and variant annotation were performed with the Sophia DDM® software version 5.9.1.1 (Sophia Genetics SA). A bioinformatic filter was constructed, including all the genes previously reported to be related to CDG.
The HSFpro software (Genomnis; Desmet et al., 2009) was used to predict the effect of variants on splicing. All predictions were made with the DYSF transcript ENST00000268261.
Total mRNA from the patient’s fibroblasts was obtained using TRIzol reagent (Life Technologies) and cDNA was synthesized using M-MLV Reverse Transcriptase (Life Technologies). The cDNA-based PCR product corresponding to the coding sequence of ALG1 was obtained using forward primer ALG1s 5′-TGACTGCTGCGGGCCAG-3′ and reverse primer ALG1as 5′-CACTGGGAGGTGCTGCTCG-3′. In the case of the patient, the amplicon was isolated in low melting point agarose gel, purified, subcloned, and screened for alternative splicing.
The inheritance of variants was determined by analyzing the patient’s and parents’ gDNA using primer ALG1s and reverse primer ALG1gas 5′-CTAAAGGAGCACTTCCGCC-3′ for the c.208 + 25G > T pathogenic variant and forward primer ALG1-E13s 5′-CAGGCAATGAGGTAAGCTCTG-3′ and reverse primer ALG1-E13as 5′-CAATTCTTTTACCAGGCAGTACC-3′ for the c.1312C > T pathogenic variant. Sequencing was performed by an ABI Prism 3130xl autoanalyzer (Applied Biosystems, Foster City, CA, United States), and results were visualized using SnapGene Viewer 2.2.2 (GSL Biotech LLC, Chicago, IL, United States).
The novel pathogenic variant observed in this study has been deposited in the ClinVar database with the accession SCV001761655.
The studies involving human participants were reviewed and approved by the Ethics Committee of the Sociedad Latinoamericana de Glicobiología. Written informed consent to participate in this study was provided by the participants’ parents.
IM-D and CG-D conceived and designed the study. CG-D, MF-R, SG-C, AK-A, MH, ED, KR, MA-G, SM-H, AG-J, RS-M, CM-G, KC-S, LF-L, MJ-O, AM-R, MC-M, MR-G, HH-F, and CA-V contributed to data acquisition and analysis. IM-D, CG-D, CA-V, HH-F, and HM-M contributed to curation and interpretation of data. HM-M and IM-D: provided supervision. IM-D wrote the original draft. CG-D, MF-R, SG-C, A-KA, MH, ED, KR, M-AG, SM-H, AG-J, RS-M, CM-G, KC-S, LF-L, MJ-O, AM-R, MC-M, MR-G, HM-M, HH-F, CA-V, and IM-D revised, edited, and approved the final version of the manuscript.
IM-D and CG-D were supported by grants 293399 Red Temática Glicociencia en Salud – CONACyT and the Sociedad Latinoamericana de Glicobiología, A.C. CG-D is recipient of a scholarship 957250 from CONACYT. HH-F was supported by The Rocket Fund and National Institutes of Health Grant R01DK99551.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Felipe Olvera Rodríguez and Maricela Olvera Rodríguez for aiding in the production of rabbit anti-transferrin serum for IEF.
Aebi, M. (2013). N-linked protein glycosylation in the ER. Biochim. Biophys. Acta Mol. Cell Res. 1833, 2430–2437. doi: 10.1016/j.bbamcr.2013.04.001
Bahena-Bahena, D., López-Valdez, J., Raymond, K., Salinas-Marín, R., Ortega-García, A., Ng, B. G., et al. (2014). ATP6V0A2 mutations present in two Mexican Mestizo children with an autosomal recessive cutis laxa syndrome type IIA. Mol. Genet. Metab. Rep. 1, 203–212.
Barba, C., Darra, F., Cusmai, R., Procopio, E., Dionisi Vici, C., Keldermans, L., et al. (2016). Congenital disorders of glycosylation presenting as epileptic encephalopathy with migrating partial seizures in infancy. Dev. Med. Child Neurol. 58, 1085–1091. doi: 10.1111/dmcn.13141
Bengtson, P., Ng, B. G., Jaeken, J., Matthijs, G., Freeze, H. H., and Eklund, E. A. (2016). Serum transferrin carrying the xeno-tetrasaccharide NeuAc-Gal-GlcNAc2 is a biomarker of ALG1-CDG. J. Inherit. Metab. Dis. 39, 107–114. doi: 10.1007/s10545-015-9884-y
Bogdañska, A., Lipiński, P., Szymańska-Rożek, P., Jezela-Stanek, A., Rokicki, D., Socha, P., et al. (2021). Clinical, biochemical and molecular phenotype of congenital disorders of glycosylation: long-term follow-up. Orphanet J. Rare Dis. 16:17. doi: 10.1186/s13023-020-01657-5
Bowling, K. M., Thompson, M. L., Amaral, M. D., Finnila, C. R., Hiatt, S. M., Engel, K. L., et al. (2017). Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 9:43. doi: 10.1186/s13073-017-0433-1
Chen, J., Li, X., Edmondson, A., Meyers, G. D., Izumi, K., Ackermann, A. M., et al. (2019). Increased clinical sensitivity and specificity of plasma protein N-glycan profiling for diagnosing congenital disorders of glycosylation by use of flow injection-electrospray ionization-quadrupole time-of-flight mass spectrometry. Clin. Chem. 65, 653–663. doi: 10.1373/CLINCHEM.2018.296780
Couto, J. R., Huffaker, T. C., and Robbins, P. W. (1984). Cloning and expression in Escherichia coli of a yeast mannosyltransferase from the asparagine-linked glycosylation pathway. J. Biol. Chem. 259, 378–382. doi: 10.1016/s0021-9258(17)43670-2
Desmet, F. O., Hamroun, D., Lalande, M., Collod-Bëroud, G., Claustres, M., and Béroud, C. (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37:e67. doi: 10.1093/nar/gkp215
Dupré, T., Vuillaumier-Barrot, S., Chantret, I., Yayé, H. S., Le Bizec, C., Afenjar, A., et al. (2010). Guanosine diphosphate-mannose: GlcNAc2-PP-dolichol mannosyltransferase deficiency (congenital disorders of glycosylation type Ik): five new patients and seven novel mutations. J. Med. Genet. 47, 729–735. doi: 10.1136/jmg.2009.072504
Gao, X. D., Nishikawa, A., and Dean, N. (2004). Physical interactions between the Alg1, Alg2, and Alg11 mannosyltransferases of the endoplasmic reticulum. Glycobiology 14, 559–570. doi: 10.1093/glycob/cwh072
González-Domínguez, C. A., Raya-Trigueros, A., Manrique-Hernández, S., González Jaimes, A., Salinas-Marín, R., Molina-Garay, C., et al. (2020). Identification through exome sequencing of the first PMM2-CDG individual of Mexican mestizo origin. Mol. Genet. Metab. Rep. 25:100637. doi: 10.1016/j.ymgmr.2020.100637
González-Domínguez, C. A., Villarroel, C. E., Rodríguez-Morales, M., Manrique-Hernández, S., González-Jaimes, A., and Olvera-Rodriguez, F. (2021). Non-functional alternative splicing caused by a Latino pathogenic variant in a case of PMM2-CDG. Mol. Genet. Metab. Rep. 28:100781. doi: 10.1016/j.ymgmr.2021.100781
Grubenmann, C. E., Frank, C. G., Hülsmeier, A. J., Schollen, E., Matthijs, G., Mayatepek, E., et al. (2004). Deficiency of the first mannosylation step in the N-glycosylation pathway causes congenital disorder of glycosylation type Ik. Hum. Mol. Genet. 13, 535–542. doi: 10.1093/hmg/ddh050
Harshman, L. A., Ng, B. G., Freeze, H. H., Trapane, P., Dolezal, A., Brophy, P. D., et al. (2016). Congenital nephrotic syndrome in an infant with ALG1-congenital disorder of glycosylation. Pediatr. Int. 58, 785–788. doi: 10.1111/ped.12988
Jones, M. A., Rhodenizer, D., da Silva, C., Huff, I. J., Keong, L., Bean, L. J. H., et al. (2013). Molecular diagnostic testing for congenital disorders of glycosylation (CDG): detection rate for single gene testing and next generation sequencing panel testing. Mole. Genet. Metabol. 110, 78–85. https://doi.org/10.1016/j.ymgme.2013.05.012
Köhler, S., Gargano, M., Matentzoglu, N., Carmody, L. C., Lewis-Smith, D., Vasilevsky, N. A., et al. (2021). The human phenotype ontology in 2021. Nucleic Acids Res. 49, D1207–D1217. doi: 10.1093/nar/gkaa1043
Kranz, C., Denecke, J., Lehle, L., Sohlbach, K., Jeske, S., Meinhardt, F., et al. (2004). Congenital Disorder of Glycosylation Type Ik (CDG-Ik): a Defect of Mannosyltransferase I. Am. J. Hum Genet. 74, 545–551. doi: 10.1086/382493
Lei, Y. L., Zhen, L., Xu, L. L., Yang, Y. D., and Li, D. Z. (2020). Foetal phenotype of ALG1-CDG caused by paternal uniparental disomy 16. J. Obstet. Gynaecol. 41, 828–830. doi: 10.1080/01443615.2020.1786031
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Manickam, K., Mcclain, M. R., Demmer, L. A., Biswas, S., Kearney, H. M., Malinowski, J., et al. (2021). Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. [Epub Online ahead of print]. doi: 10.1038/s41436-021-01242-6
Marques-da-Silva, D., dos Reis Ferreira, V., Monticelli, M., Janeiro, P., Videira, P. A., Witters, P., et al. (2017). Liver involvement in congenital disorders of glycosylation (CDG). A systematic review of the literature. J. Inherit. Metab. Dis. 40, 195–207. doi: 10.1007/s10545-016-0012-4
Medrano, C., Vega, A., Navarrete, R., Ecay, M. J., Calvo, R., Pascual, S. I., et al. (2019). Clinical and molecular diagnosis of non-phosphomannomutase 2 N-linked congenital disorders of glycosylation in Spain. Clin. Genet. 95, 615–626. doi: 10.1111/cge.13508
Morava, E., Vodopiutz, J., Lefeber, D. J., Janecke, A. R., Schmidt, W. M., Lechner, S., et al. (2012). Defining the phenotype in congenital disorder of glycosylation due to ALG1 mutations. Pediatrics 130, e1034–e1039. doi: 10.1542/peds.2011-2711
Ng, B. G., Shiryaev, S. A., Rymen, D., Eklund, E. A., Raymond, K., Kircher, M., et al. (2016). ALG1-CDG: clinical and Molecular Characterization of 39 Unreported Patients. Hum. Mutat. 37, 653–660. doi: 10.1002/humu.22983
Ondruskova, N., Cechova, A., Hansikova, H., Honzik, T., and Jaeken, J. (2021). Congenital disorders of glycosylation: still “hot” in 2020. Biochim. Biophys. Acta Gen. Subj. 1865:129751. doi: 10.1016/j.bbagen.2020.129751
O’Reilly, M. K., Zhang, G., and Imperiali, B. (2006). In vitro evidence for the dual function of Alg2 and Alg11: essential mannosyltransferases in N-linked glycoprotein biosynthesis. Biochemistry 45, 9593–9603. doi: 10.1021/bi060878o
Pérez-Cerdá, C., Girós, M. L., Serrano, M., Ecay, M. J., Gort, L., Pérez Dueñas, B., et al. (2017). A Population-Based Study on Congenital Disorders of Protein N- and Combined with O-Glycosylation Experience in Clinical and Genetic Diagnosis. J. Pediatr. 183, 170–177.e1. doi: 10.1016/j.jpeds.2016.12.060
Quelhas, D., Martins, E., Azevedo, L., Bandeira, A., Diogo, L., Garcia, P., et al. (2021). Congenital Disorders of Glycosylation in Portugal—Two Decades of Experience. J. Pediatr. 231, 148–156. doi: 10.1016/j.jpeds.2020.12.026
Rohlfing, A. K., Rust, S., Reunert, J., Tirre, M., du Chesne, I., Wemhoff, S., et al. (2014). ALG1-CDG: a new case with early fatal outcome. Gene 534, 345–351. doi: 10.1016/j.gene.2013.10.013
Schwarz, M., Thiel, C., Lübbehusen, J., Dorland, B., de Koning, T., von Figura, K., et al. (2004). Deficiency of GDP-Man:GlcNAc2-PP-Dolichol Mannosyltransferase Causes Congenital Disorder of Glycosylation Type Ik. Am. J. Hum. Genet. 74, 472–481. doi: 10.1086/382492
Zhang, W., James, P. M., Ng, B. G., Li, X., Xia, B., Rong, J., et al. (2016). A novel N-tetrasaccharide in patients with congenital disorders of glycosylation, including asparagine-linked glycosylation protein 1, phosphomannomutase 2, and mannose phosphate isomerase deficiencies. Clin. Chem. 62, 208–217. doi: 10.1373/clinchem.2015.243279
Keywords: CDG, splicing, metabolic, glycosylation, ALG1, mutation, tetrasaccharide
Citation: González-Domínguez CA, Fiesco-Roa MO, Gómez-Carmona S, Kleinert-Altamirano API, He M, Daniel EJP, Raymond KM, Abreu-González M, Manrique-Hernández S, González-Jaimes A, Salinas-Marín R, Molina-Garay C, Carrillo-Sánchez K, Flores-Lagunes LL, Jiménez-Olivares M, Muñoz-Rivas A, Cruz-Muñoz ME, Ruíz-García M, Freeze HH, Mora-Montes HM, Alaez-Verson C and Martínez-Duncker I (2021) ALG1-CDG Caused by Non-functional Alternative Splicing Involving a Novel Pathogenic Complex Allele. Front. Genet. 12:744884. doi: 10.3389/fgene.2021.744884
Received: 21 July 2021; Accepted: 05 August 2021;
Published: 09 September 2021.
Edited by:
Karolina Stepien, Salford Royal NHS Foundation Trust, United KingdomReviewed by:
Domenico Garozzo, Italian National Research Council, ItalyCopyright © 2021 González-Domínguez, Fiesco-Roa, Gómez-Carmona, Kleinert-Altamirano, He, Daniel, Raymond, Abreu-González, Manrique-Hernández, González-Jaimes, Salinas-Marín, Molina-Garay, Carrillo-Sánchez, Flores-Lagunes, Jiménez-Olivares, Muñoz-Rivas, Cruz-Muñoz, Ruíz-García, Freeze, Mora-Montes, Alaez-Verson and Martínez-Duncker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iván Martínez-Duncker, ZHVuY2tlckB1YWVtLm14
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.