 Mohd Amir
Mohd Amir Saleem Javed
Saleem Javed
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 22 October 2021
Sec. Epigenomics and Epigenetics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.742802
This article is part of the Research Topic Epigenetic and Transcriptional Dysregulations in Cancer and Therapeutic Opportunities View all 19 articles
Chronic myeloid leukemia is a malignancy of bone marrow that affects white blood cells. There is strong evidence that disease progression, treatment responses, and overall clinical outcomes of CML patients are influenced by the accumulation of other genetic and epigenetic abnormalities, rather than only the BCR/ABL1 oncoprotein. Both genetic and epigenetic factors influence the efficacy of CML treatment strategies. Targeted medicines known as tyrosine-kinase inhibitors have dramatically improved long-term survival rates in CML patients during the previous 2 decades. When compared to earlier chemotherapy treatments, these drugs have revolutionized CML treatment and allowed most people to live longer lives. Although epigenetic inhibitors’ activity is disrupted in many cancers, including CML, but when combined with TKI, they may offer potential therapeutic strategies for the treatment of CML cells. The epigenetics of tyrosine kinase inhibitors and resistance to them is being studied, with a particular focus on imatinib, which is used to treat CML. In addition, the use of epigenetic drugs in conjunction with TKIs has been discussed. Resistance to TKIs is still a problem in curing the disease, necessitating the development of new therapies. This study focused on epigenetic pathways involved in CML pathogenesis and tumor cell resistance to TKIs, both of which contribute to leukemic clone breakout and proliferation.
The malignancy of white blood cells in chronic myeloid leukemia (CML), often referred to as chronic myelogenous leukemia. CML is a clonal myeloproliferative disease that affects over 15% of adults and the average age for the outbreak of this disease is 57 years (Hochhaus et al., 2020; Jabbour and Kantarjian, 2020). CML is more prevalent in men than in women (ratio 1.4:1), accounting for 15–25% of all juvenile leukemias and 14% of all leukemias in the countries lying towards the west. The Philadelphia chromosome (Ph), which occurs between chromosomes 9 and 22, is the characteristic feature of CML and has been regarded to be the first malignancy related to genetic abnormalities. This translocation results in the formation of the BCR-ABL1 hybrid gene, which expresses a constitutively active oncokinase protein. The ABL kinase domain that spans around 300 amino acids, has a bilobed conformation with a C-terminal and an N-terminal lobe, with ATP binding in a cleft flanked by these lobes. Because of the flexibility of C-terminal loop, which contains a “DFG”-motif (Asp381-Phe382-Gly383), the kinase can be in an active (open) or inactive (closed) conformation (Reddy and Aggarwal, 2012; Balabanov et al., 2014). CML is classified into three phases namely chronic phase (CP), accelerated phase (AP) and blast crisis (BC). The presence of one of the various hematological (like persistent thrombocytopenia) or provisional (like hematological resistance to the drug) criteria for responding to tyrosine kinase inhibitor (TKI) defines AP phase, as per WHO criteria (Arber et al., 2016).
TKIs are small molecules that are used to treat a wide range of malignancies, including leukemia and solid tumors (Amir et al., 2020). TKIs have been developed as a targeted treatment for BCR-ABL1 kinase activity suppression (Cilloni and Saglio, 2012). TKI therapy for CML patients has resulted in a substantial improvement in disease outcomes and a near-normal lifespan. CML is a well-studied TK-dependent alteration model disease. As a consequence of fundamental and clinical research in clinical practice, TKIs are currently the benchmark in the treatment of CML patients and an effective example of targeted therapy (Balabanov et al., 2014). Imatinib, for example, is one of the first TKIs to be certified for the treatment of CML and is presently used as a first-line treatment, while nilotinib and dasatinib are the next discoveries (Özgür Yurttaş and Eşkazan, 2020).
The first generation TKI binds to an inactive form of BCR-ABL conformation, while second generation TKIs show binding affinity for both the active and inactive forms. The TKIs of second generation (dasatinib, nilotinib, bosutinib) are approximately 1,000 times more sensitive than imatinib in terms of efficacy against BCR-ABL resistant mutations (Kantarjian et al., 2010; Saglio et al., 2010). The binding of imatinib with BCR-ABL kinase in its ATP-binding pocket is hampered by T315I mutation, which is the result of a substitution of isoleucine for threonine at position 315 of the amino acid chain. Approximately 12 percent of adult patients with BCR-ABL leukemia have seen this mutation, and is more common in people with severe conditions. However, dasatinib and nilotinib against T315I are not effective (Soverini et al., 2006; Soverini et al., 2007).
The survival has improved significantly to the point the survival in patients with CML is approaching that of the normal population with access to TKI therapy. Patients with newly diagnosed CML-CP had an OS rate that is only marginally lower than the normal population when treated with TKIs. Patients who obtained CCyR or better, on the other hand, have an OS rate that is comparable to the general population. This highlights the need of ensuring the best possible care for CML patients of all ages to maximize the benefits of these life-changing treatments (Sasaki et al., 2015). The long-term response of patients treated with different TKI therapies and their outcomes were examined by Jain et al. using Kaplan-Meier method. Patients treated with imatinib 800, dasatinib, or nilotinib had better long-term responses and outcomes than those treated with imatinib 400. Transformation to AP and BC phases, as well as mortality from CML, are now uncommon with current TKI therapy methods when well managed. However, discontinuation of treatment was more prevalent in imatinib patients than in TKIs of the second generation (Jain et al., 2015).
In the era of TKIs, CML-CP patients who lived for a certain number of years had good clinical results. Patients may, however, quit medication for some reason throughout time, affecting OS. It’s worth noting that the good conditional long-term outcomes predicted are based on rigorous ongoing monitoring of patients for CML and other comorbidities, as well as side events related to TKI usage, as done in clinical studies. It is not reasonable to believe that this may be generalized to situations where patients are treated less strictly (Sasaki et al., 2016). A clinical study showed that patients with the best long-term ELN responses have the best long-term results, and patients who received imatinib 800, dasatinib, or nilotinib have a greater chance of attaining optimum response at various periods (Jain et al., 2016).
According to Issa et al. during TKI therapy, if non-Y CCA/Ph¯ (clonal chromosomal abnormalities in Philadelphia chromosome-negative) is detected, physicians should be alerted to the possibility of poor survival outcomes, which may need careful monitoring. For most indices of response and long-term survival, patients with CCA/Ph¯ exhibited a trend toward inferiority. Early response assessment, such as BCR-ABL, was found to have a substantial predictive influence on the multivariate analysis. Patients with CCA/Ph¯ were less likely to get this reaction (74 vs. 81%). For individuals with monosomy 7, the emergence of nondeletion Y CCA/Ph¯ following therapy with different TKI is linked to reduced FFS, EFS, TFS, and OS, as well as a low but substantial chance of developing MDS or AML (Issa et al., 2017; Sasaki et al., 2018).
COVID-19 patients have been compared to an age, sex, and condition matched control group to determine the therapeutic effects of TKIs on their outcome (Başcı et al., 2020). Neither CP CML nor BCR-ABL TKIs induces clinically significant immune suppression, and there is no evidence that CP CML patients are at greater risk of complications by SARS-CoV-2 than the normal community. The clinical diagnosis of COVID-19 is not worse in TKI-treated CML patients than it is in the non-TKI-treated individuals. TKIs should be studied in wide-scale prospective and randomized studies to see if they improve COVID-19 outcomes (Başcı et al., 2020).
The clinical parameters of various TKI drugs used to treat CML are shown in Table 1 (Gleevec, 2008; Sprycel, 2010; Iclusig, 2016; Bosulif, 2017; Tasigna, 2017).
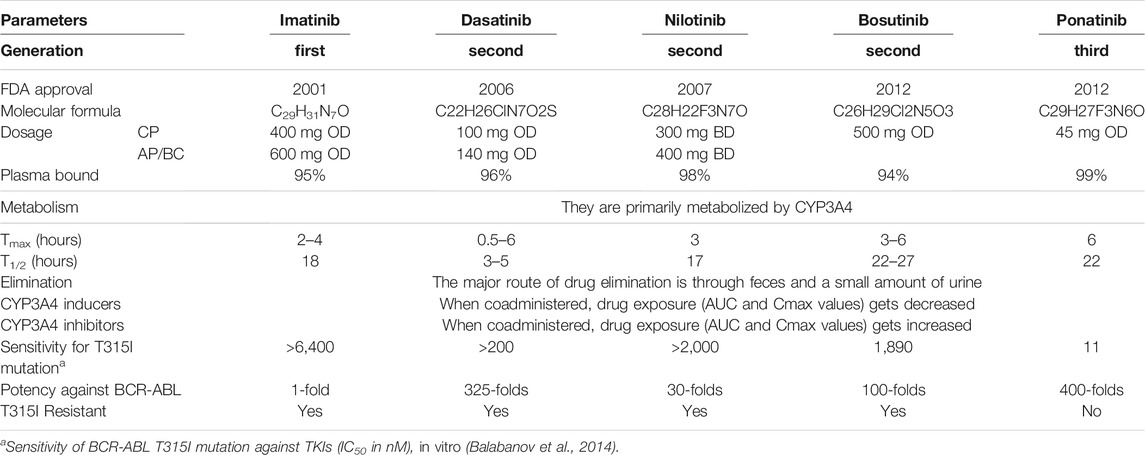
TABLE 1. Clinical parameters of different TKIs approved by FDA to treat CML.
Imatinib is generally well absorbed, with far fewer complications than traditional drug treatment. The percentage of individuals with CML in CP that had neutropenia and thrombocytopenia of grade 3/4, is approximately 13 and 7%, respectively. In children, 27 percent of patients had neutropenia, 5 percent had thrombocytopenia, and 2.5 percent had anemia, all of grade 3 or 4. By temporarily halting imatinib, the cytopenia is usually fairly controllable. Gastrointestinal disorders like diarrhea, nausea, and vomiting are examples of nonhematologic toxicity. Bone pain, edema, rashes, myalgia and elevated levels of ALT and AST, etc., are the further side effects (Millot et al., 2011).
Although TKIs are found to be one of the effective targeted chemotherapy for treatment of individuals with CML. But every drug has some toxic responses against the body mechanism or metabolism that has to be overcome either by drug dose modifications or, in some cases, by permanently discontinuing the drug. Similar challenges and actions that have to be taken under treatment of CML using TKI are depicted in Table 2 (Gleevec, 2008; Sprycel, 2010; Iclusig, 2016; Bosulif, 2017; Tasigna, 2017).
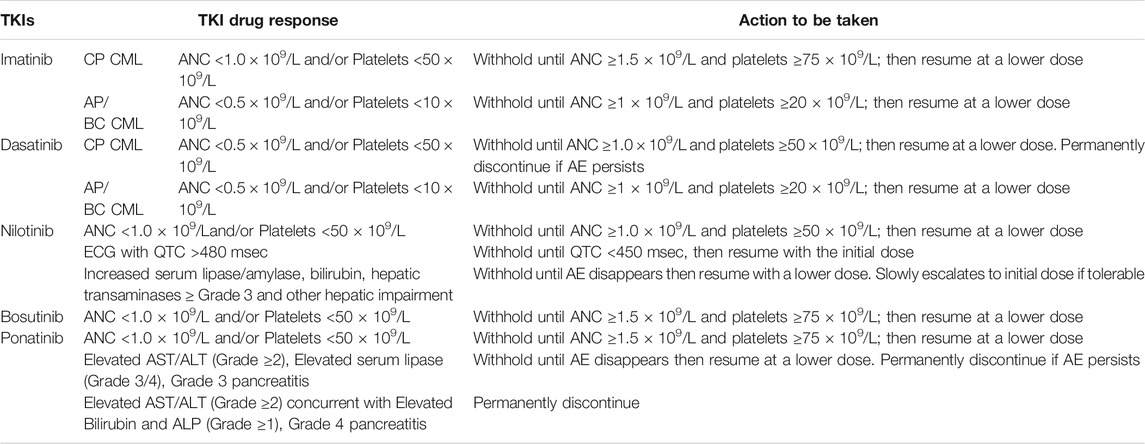
TABLE 2. The adverse conditions and dose modifications of TKIs during CML treatment.
Even with more effective second-and third-generation TKIs, many CML individuals have developed primary (5–10%) or secondary (20–30%) intolerance and thus become resistant to TKI therapy. Secondary resistance is most commonly caused by acquired Abl-kinase domain (Abl-KD) alterations, which can be noticed in roughly 60% of instances; nevertheless, the most of primary resistance pathways are still unknown (Bower et al., 2016; Russo et al., 2020). The 6-year review of the IRIS study revealed that imatinib therapy is ineffective in around one-third of patients because of 1° or 2° resistance. Approximately 40–90 percent of individuals who are resistant to imatinib have BCR-ABL mutation, based on the cancer stage and detection equipment’s sensitivity. T315I is an especially important mutation as it is quite prevalent (15–20%) and causes tolerance to almost all TKIs currently used in clinical practice, except ponatinib. It is found in ABL kinase domain’s ATP-binding region and provides one of imatinib’s six binding sites. This binding site is destroyed when threonine is replaced by isoleucine, as well as a structural impediment that prevents imatinib, nilotinib, dasatinib, and bosutinib from reaching it (Hughes et al., 2014). Imatinib resistance can be caused by either primary or secondary causes. Pharmacokinetic changes, such as anomalies in transport or drug efflux, may cause primary resistance. Because of BCR-ABL mutations or the involvement of salvage mechanisms, patients are more prone to develop secondary or acquired resistance. The T315I mutation is the most dreaded mutation since it is resistant to all the TKIs that have been approved by FDA for this disease. Several drugs, such as ponatinib, are being tested in clinical studies to block kinase activity linked to T315I (O’Hare et al., 2009). According to prior studies, TKI can restore the levels of specific miRNAs, and this mechanism can mediate the effects of TKIs on CML cells (Bugler et al., 2019).
CML is divided into three stages, referred to as CP, AP, and BP. With the introduction of TKIs and the subsequent success of TKI cessation, a fourth-phase, treatment-free remission (TFR-CML), has developed. In the absence of CML-directed treatment, this phase begins after TKI therapy is stopped and is defined by sustained remission with undetectable (or very low detectable) illness by the most sensitive technique of testing. This phase differs from CP since there is no additional molecular, cytogenetic, or hematological development. A clinical study revealed that patients were treated with TKIs for 11 years, and MMR and MR4.5 were maintained for 10 and 5.5 years, respectively, until therapy was discontinued. As a result, the rapid transition to BP CML was not preceded by CP development, and this sudden transformation had been reported in fewer than 1% of the CML individuals undergoing imatinib therapy. Cellular immunity, in the form of natural killer cells and cytotoxic T lymphocytes, may have a role in TFR-CML, according to one theory. This rapid blastic change might be the outcome of a lack of immune monitoring or a disease breakout (Alfayez et al., 2019). But it might also be related to some genetic and epigenetic abnormalities that require further clinical research for validation.
Clinical study data show that epigenetic medication treatment can produce objective responses in CML patients, even in later stages and amid imatinib resistance. Though, these reactions are rarely long-lasting, necessitating the pursuit of alternative recovery methods. Not only are many TKI options available (e.g., Imatinib, nilotinib, dasatinib, ponatinib, and bosutinib), but novel agents (e.g., ABL001) are being investigated, and for individuals with chronic or non-responsive illness, allogeneic transplantation remains a surgical option.
Given below are the possible reasons/mechanisms for TKI resistance (Balabanov et al., 2014).
1. BCR-ABL Dependent
i. Overexpression of BCR-ABL kinases as a result of amplification leads to a TKI-resistant scenario.
ii. Mutations in BCR-ABL resist TKI binding as a result of conformational change in ABL.
2. BCR-ABL Independent
i. Initiation of other compensatory intracellular signaling pathways (like LYN, HCK), due to which, despite successful suppression of the primary oncogenic driver kinase, cells continue to proliferate and thus results in TKI resistance.
ii. The overexpression of efflux transporters, like ABCB1 (MRD-1, for which imatinib acts as a substrate), results in a decrease of TKI levels in cells.
iii. The downregulation of influx transporters, like OCT1 (in the case of imatinib), also results in reduced efficacy of TKI and finally leads to TKI resistance.
The inhibition of the HOXA4 protein may interfere with the normal growth and proliferation of myeloid progenitors. Imatinib resistance in CML patients may be caused by an epigenetic mechanism in the BCR-ABL-independent pathway. The hypermethylation of HOXA4 gene appears to be inhibiting the clinical response to IM, and as a result, is acting as a significant inhibitor of normal leukemogenesis. Inhibition of this process might be a superior treatment option, necessitating hypomethylating drugs in CML patients with this epigenetic resistance mechanism. As a result, apart from BCR-ABL gene mutation analyses, the hypermethylation profile of the HOXA4 gene might be used as an epigenetic biomarker in CML patients for predicting reaction to imatinib treatment (Elias et al., 2013).
According to recent research, the long noncoding RNA (lncRNA) HOTAIR (HOX transcript antisense RNA) is involved in the development of both solid tumors like breast cancer and NSCLC, as well as hematopoietic malignancies like AML. The epigenetic regulatory mechanisms of HOTAIR in advanced CML, on the other hand, remain unknown. Li et al. suggested that demethylation drugs may provide a novel remedy for CML-BC, as HOTAIR is a possible therapeutic target (Li and Luo, 2018).
In the age of TKIs, the incidence of second malignancies in CML patients has increased. Sasaki et al. (2019), reported data of newly diagnosed CML patients in 2001–2014, and it was found that 5.6% of the cancer patients had more than one malignancy after the CML diagnosis. The most prevalent sites of second cancers were the male genital system, digestive system and respiratory system. CML-CP had a slightly higher relative incidence of total second malignancies than the general population, with a modest rise in the AER (Sasaki et al., 2019).
There have been reports of cardiovascular or arteriothrombotic adverse events (CV- or AT-AEs) in CML patients receiving TKI therapy. The second or third-generation TKIs, such as ponatinib, significantly increase the risk of CV-AEs (excluding hypertension) and AT-AEs in CML patients (Jain et al., 2019). Myelopoeisis epigenetic regulation is controlled by the TET2 gene, which codes for the protein TET2 methylcytosine dioxygenase. Through the oxidation of methyl-cytosine, TET proteins play a role in DNA alteration, also in normal and malignant growth. A DNA epigenetic modification termed as hydroxymethylation is catalyzed by Tet2. As a result, it had been postulated that Tet2 regulated gene expression in macrophages exposed to excessive cholesterol. As a result, somatic mutations in hematopoietic cells have a role in the onset of atherosclerosis in humans. It was suggested that clonal hematopoiesis might be a controllable risk factor, either by using cholesterol-lowering medicines or targeting certain inflammatory processes (Jaiswal et al., 2017).
Patients with severe CML are more likely to have additional cytogenetic abnormalities (ACAs) in addition to the Philadelphia chromosome. Long-term survival may be improved by combining TKI treatment with systemic chemotherapy. No matter what the blast count is, the existence of CBF translocations determines whether or not a patient has AML, because CBF rearrangements are seldom diagnosed as ACAs. In individuals with CML, CBF rearrangements as ACAs might be regarded as high-risk characteristics (Morita et al., 2021a).
There is significant evidence that the accumulation of various genetic and epigenetic abnormalities, in addition to the BCR/ABL1 oncoprotein, influences disease progression, treatment response, and overall clinical prognosis in CML patients. The development of a variety of illnesses has been related to DNA hydroxymethylation. Disease development, drug reaction, and clinical outcome of different diseases are all influenced by the DNA hydroxymethylation of gene promoters (Jelinek et al., 2011). The previous results of the clinical studies suggest that in CML patients undergoing imatinib treatment, hydroxymethylation of gene promoters of cell cycle regulators and autophagy genes may be a significant predictor of tumor growth, identifies inefficient imatinib responders, and indicates a relatively poor treatment outcome. As a result, the findings encouraged the use of demethylating agents in conjunction with TKI treatment to improve clinical consequences (Jelinek et al., 2011). The previous studies linking hypermethylation of SOCS1 gene to downregulation of expression suggested that the lack of negative cytokine signaling caused by SOCS1 protein epigenetic suppression might contribute to CML development. Small molecules with two src homology domains make up SOCS proteins. As a result, SOCS1 expression may be a new and valuable marker for CML therapeutic follow-up, as well as a new therapeutic strategy for developing successful CML target therapies (Liu et al., 2003).
Over the last 20 years, the invention of TKIs to counteract the BCR-ABL protein’s constitutive tyrosine kinase function has greatly increased disease control and clinical outcomes. However, most patients are not healed, and designing treatment strategies that target epigenetic pathways is a potential way of increasing cure rates. During the formation and growth of CML, various epigenetic pathways are changed or reprogrammed, leading to changes in histone modifications, DNA methylation, and transcriptional dysregulation (Bugler et al., 2019). Consequently, with the advent of TKIs as the primary mode of therapy, the occurrence of CML has risen, rendering it a manageable, chronic condition. Although TKI therapy has revolutionized CML treatment, 25–30 percent of patients with CP-CML have failed TKI treatment, half had mutations in the BCR–ABL1 kinase domain and 50 percent of patients are unknown for the failure of this therapy (Rohrbacher and Hasford, 2009; Baccarani et al., 2013). Potentially harmful somatic mutations of epigenetic modifiers are common in CP-CML at the time of diagnosis, and when combined with other factors associated, they can be useful predictive indicators to identify the best TKI for every patient (Nteliopoulos et al., 2019). The chemotherapy resistance during treatment might occur at any stage, and thus the emergence of drug resistance remains a major issue.
In 1983, after the discovery of the first cancer-epigenetic reprogramming relations, emerging evidence indicates that genetic and epigenetic changes cause cancers and that some of them occurring before profound leukemia commences preleukemia (Feinberg et al., 2006; Feinberg and Vogelstein, 1983). The BCR-ABL1 mutation induces epigenetic reprogramming, which is important in CML, as well as converting HSCs to LSCs. Polycomb-group (PcG) proteins are one kind of epigenetic regulator that is believed to be disrupted in CML LSC (Di Carlo et al., 2019). There are two complexes in PcG proteins, namely PRC1 and PRC2 (Polycomb Repressive Complex). Given the number of studies that PRC2 plays a role in cancer (including solid tumors and multiple myeloma), it’s no surprise that a variety of therapeutics targeting this complex have appeared and are under clinical studies of Phase I and II (Fioravanti et al., 2018; Gulati et al., 2018). In comparison to TKI therapy alone, Scott et al. demonstrated significant targeting of CML stem cells using the EZH2 (a core component of PRC2) inhibitor Tazemetostat in conjunction with TKI. As a result, combined therapy may be a new clinical option for CML treatment, as shown in Figure 1 (Scott et al., 2016).
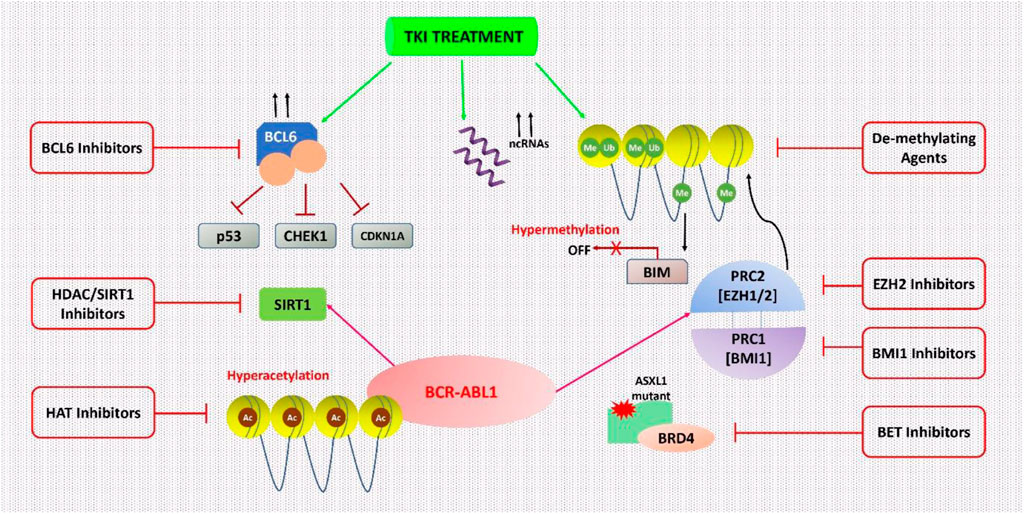
FIGURE 1. Epigenetic processes in CML cells are being targeted. CML cells are epigenetically reprogrammed via several mechanisms. This dysregulation can occur only in the presence of TKI therapy in various cases. With this understanding, many epigenetic treatments were suggested to inhibit these mechanisms and to result in the complete destruction of CML cells especially in combination with TKI therapy.
Sirtuin 1 (SIRT1) is a NAD-dependent HDAC that acts on the nonhistone target p53 in CD34+ CML cells and has been associated with leukemogenesis and the persistence of CML LSCs. After imatinib therapy, SIRT1 expression significantly decreased but not completely depleted in CML cell lines, paving the way for effective SIRT1 inhibition. The treatment with the SIRT1 inhibitor (e.g., Selisistat or EX-527) via activation of p53 signaling, improved the effect of TKI treatment (Chen and Bhatia, 2013; Li et al., 2012; Yuan et al., 2012). SIRT1’s function is one of the most prevalent causes of treatment resistance in several malignancies, including CML (S et al., 2021).
BCL6 is a transcription factor that can epigenetically control a variety of its targets by altering the accessibility of chromatin at promoter and enhancer sites. It is often mutated in lymphoma cells (Hatzi et al., 2013). BCL6 expression is modest in TKI-naive CML cells, but it is substantially increased in CML cell lines and primary CD34+ cells after TKI therapy, although its function in CML is unknown (Hurtz et al., 2011). In Ph+ ALL patients, the BCL6 peptide inhibitor, RI-BPI was coupled with imatinib to avoid TKI resistance and to enhance imatinib’s efficacy (Duy et al., 2011). In CD34+38- CML LSCs, the use of RI-BPI increased the effectiveness of imatinib treatment, and decreased the ability to form CML cell colony, thus eliminated CD34+38- LSCs by enhancing programmed cell death. Other BCL6 small molecule inhibitors, such as FX1, significantly reduced CML CD34+ cells’ colony-forming ability, in combination with TKI (Madapura et al., 2017).
It had been suggested by San José-Eneriz et al. (2009), that the concomitant use of imatinib and a de-methylating drug such as 5-aza-2′-deoxycytidine (Decitabine) might result in better outcomes in individuals with reduced expression of the pro-apoptotic BCL-2-interacting mediator (BIM). This downregulation is epigenetically mediated by BIM methylation in most CML individuals and has a negative impact on health (San José-Eneriz et al., 2009). TKI-resistant leukemic stem cells (LSCs) are still a big problem in CML, and finding ways to get rid of them is a major unsatisfied clinical need. Scott et al. (2016) showed that for the existence of LSC, EZH2 and H3K27me3 reprogramming is essential, nevertheless, it also makes LSCs vulnerable to the mutual effects of EZH2 inhibitor and TKI. That becomes a novel strategy for more efficiently targeting LSCs in TKI receiving patients (Scott et al., 2016).
DNMT inhibitors are cytosine analogs that bind to DNA and inactivate genes, inhibiting the methyltransferase enzyme reversibly. Azacytidine and decitabine are DNMT inhibitors that had been approved by the FDA for the treatment of myelodysplastic syndrome (MDS). Low doses of these inhibitors are effective, but high doses are lethal (Yang et al., 2006). Decitabine is a cytosine derivative that is used as an intravenous antineoplastic agent in the treatment of MDS. It receives FDA approval in 2006 for the treatment of MDS and was also approved by the EU for the treatment of AML. The strategy of concomitant use of TKIs with epigenetic drugs for CML therapy was initiated after the approval of the first tinib drug (imatinib) in 2001 (Figure 2).
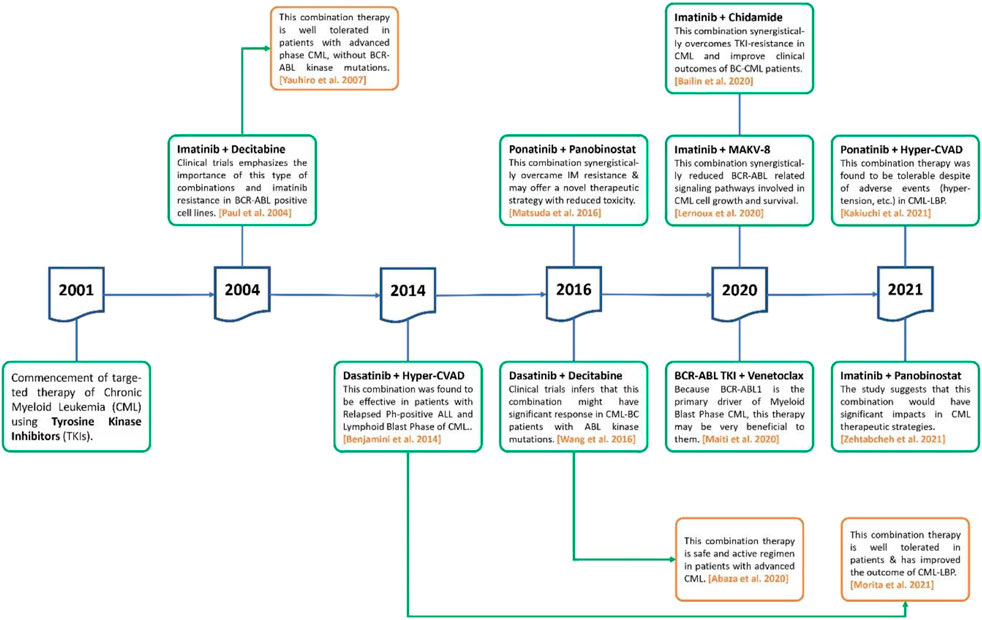
FIGURE 2. The role of TKIs in combination with epigenetic drugs to treat CML during last 2 decades.
In CML patients with AP or BC phase, a Phase II trial of low-dose decitabine in conjunction with an active metabolite of imatinib (i.e., imatinib mesylate, IM) was performed. Imatinib 600 mg OD and decitabine 15 mg/m2 intravenously (5 days a week) were given to patients for 2 weeks. The major and modest cytogenetic responses were reported which includes: 32% patients with complete and 4% patients had moderate hematologic responses, while 7% patients had hematological development. The individuals who lack BCR-ABL kinase mutations had a greater hematologic response rate (53%) than those with mutations (14%). The median hematologic response lasted 18 weeks. The most common side effect was myelosuppression, which resulted in neutropenic fever in 32% of individuals. So, in AP CML without BCR-ABL kinase mutations, it was concluded that decitabine with imatinib in combination treatment, is well tolerated and effective (Oki et al., 2007).
In recent years, the reprogramming of mitochondrial metabolism has been identified as a hallmark of cancer, including CML, and could be used for medicinal purposes. The impacts of various drugs on mitochondrial activity in the CML cell line K562 were explored, and it was shown that decitabine in combination with 5-aminoimidazole-4-carboxamide ribotide (AICAR) might efficiently boost ATP content and mitochondrial biogenesis. Furthermore, it has been shown that AICAR-decitabine therapy increased the sensitivity of the K562 cells for imatinib. Based on these findings, it is clear that TKIs coupled with mitochondrial modulation may give treatment strategies for CML treatment (Zhu et al., 2020).
Treatment for AP CML still lacks, as single-agent TKIs exhibit weak and short-lived action. A phase I/II study was undertaken in patients with advanced CML to see if the concomitant use of dasatinib and decitabine was safe and effective. Two distinct dosing regimens were tested with a beginning decitabine dose of either 10 or 20 mg/m2 OD for 10 days with dasatinib 100 mg OD. Decitabine 10 or 20 mg/m2 OD for 10 days, with dasatinib 140 mg OD, was the target dosage. There was no dose-limiting toxicity identified with either regimen at the initial dosages. In 93% of individuals, treatment-emergent hematological adverse effects of grade 3 were observed. Meanwhile, 48% of patients had a major hematopoietic response, whereas 22% had a weak hematologic reaction, with 44% of them having a large cytogenetic reaction and 33% having a significant biomolecular response. Decitabine with dasatinib is a safe, innovative, and effective treatment for advanced CML, with a life expectancy among patients that looks to be higher than with either agent alone (Abaza et al., 2020).
As a BCL2 inhibitor, venetoclax is authorized for use in aged or “unsuitable” individuals with newly diagnosed AML, together with low-dose cytarabine or HMA. Venetoclax has demonstrated preclinical efficacy against TKI-resistant CML cells and synergy with BCR-ABL TKI in eliminating leukemic stem cells in advanced CML In CML-MBP, dasatinib is effective as a single treatment; however, the venetoclax-TKI combination may be more effective than dasatinib alone. Ponatinib has shown promising results in advanced Ph+ leukemias and relapsed/refractory pre-B ALL whether used alone or in conjunction with nonchemotherapeutic treatments. Ponatinib synergizes with venetoclax in Ph+ ALL samples by inducing LYN-mediated pro-apoptotic BIM, reducing anti-apoptotic MCL1, and perhaps removing venetoclax resistance. The combination of venetoclax and ponatinib in this patient population is therefore supported by clinical trials. Thus, treatment of advanced Ph+ myeloid leukemias with venetoclax and TKI-based combination regimens is a viable option. Because BCR-ABL1 is the primary factor of CML-MBP, this combination could be very beneficial to them (Maiti et al., 2020).
Crawford et al. (2009) observed that a rise in proteasome proteolytic activity is linked to BCR/ABL genetic and epigenetic alterations, and inhibiting the proteasome might cause extremely higher cell apoptosis in BCR/ABL positive cells than in BCR/ABL negative cells. The combined inhibition of HDACs and the proteasome leads to a significant decrease in both efficacy and metabolism of CML-derived BCR/ABL-expressing K562 cells than either drug alone, indicating that treatment with concomitant use of HDAC and proteasome inhibitors could be a successful approach in CML therapy. According to Jagannath et al. (2010) in relapsed/refractory multiple myeloma patients, the clinical therapy with the combined use of HDAC and proteasome inhibitors showed promising anticancer activity (Jagannath et al., 2010).
It has been demonstrated that TKI + IC/HMA (intensive chemotherapy/hypomethylating agent) therapy provides greater response rates, reduced recurrence risk, and superior 5-years EFS/OS than TKI alone, suggesting that rather than TKI alone, the combination therapy with IC or HMA + TKI (especially received decitabine) should be taken into account for the appropriate frontline therapy for such patients. Long-term survival rates were similar between IC + TKI and HMA + TKI, with a long-term survival rate of around 30% and improved outcomes for patients who were able to undergo ASCT (allogeneic stem cell transplant). However, while being less intensive than an IC method, the combination of HMA and second/third generation TKI is extremely successful (Saxena et al., 2021). As a result of their poor prognosis, patients with CML-BP require novel treatment options (Jain et al., 2017). It was found that the use of hyper-CVAD (cyclophosphamide, vincristine sulfate, doxorubicin hydrochloride/adriamycin, and dexamethasone) with dasatinib therapy, the outcome of CML-lymphoid BP has improved, and the survival rate is equivalent to that of Ph+ ALL. Using new TKIs and targeted agents, further improvements may be made in the future (Morita et al., 2021b).
HDAC inhibitors are chemical compounds that inhibit histone deacetylases. They have a long tradition of use as mood stabilizers and anti-epileptics in psychology and neurology. They’ve recently been studied as potential cancer, viral, and infectious disease therapies (Miller et al., 2003; Blanchard and Chipoy, 2005; Mwakwari et al., 2010; Patil et al., 2010). HDAC inhibitors are derived from natural and synthetic compounds of varying target specificity and actions. HDAC inhibitors are divided into four categories: hydroxamic acids (or hydroxamates), cyclic tetrapeptides, benzamides and short-chain fatty acids. Due to the concept that BCR/ABL signaling and epigenetic changes might act as two major reasons for disease and tolerance, the combined effects of BCR/ABL inhibition utilizing imatinib with panobinostat (a multi-HDAC inhibitor) in K562 cells were studied (Housman et al., 2014). In CML-derived K562 cells, the combination of panobinostat with imatinib showed synergistic antineoplastic responses and improved therapeutic effectiveness, as predicted (Zehtabcheh et al., 2021).
MAKV-8 has been first reported for its IC50 of 2 pM, in vitro, against HDAC3 and HDAC6 and its anti-proliferative effect for pancreatic tumor cells, which was analogous to SAHA. It comprises the 6-methylene linker and a CAP group containing arylisoxazole. The chemical MAKV-8 is a potent pan-HDAC inhibitor in vitro and also in several cell lines of CML. In addition, in imatinib-sensitive/resistant BCR-ABL-positive CML cells, MAKV-8 in combination with imatinib displayed substantial anti-tumor properties, while healthy cell types exposed to this co-therapy showed relatively minimal impact. BCR-ABL expression and phosphorylation, as well as downstream targets expression important in CML growth and survival, were all inhibited by MAKV-8-imatinib combination (Lernoux et al., 2020).
Investigation of the combined effects of BCR/ABL inhibition with Imatinib and the multi-HDAC inhibitor panobinostat was performed in K562 cells The combination of panobinostat with imatinib was reported to have synergistic antileukemic effects and increased treatment effectiveness in K562 cells generated from CML patients (Zehtabcheh et al., 2021).
The concomitant use of panobinostat with ponatinib to treat IM resistance which was caused by either gene amplification or T315I mutation in BCR-ABL was found to be effective. This synergetic impact could just be because of the kinase activity suppression of ABL accompanied by BCR-ABL protein degradation. The concomitant use of these drugs could lead to a novel treatment approach with outstanding anti-CML efficacy and few side effects. The XTT test was used to assess the growth inhibitory effects of IM, panobinostat, and ponatinib alone or in combination on K562 cells, K562/IM-R1 cells, Ba/F3 cells, and Ba/F3/T315I cells. K562/IM-R1 cells 12 times, while Ba/F3/T315I cells were shown to be 13 times more IM-resistant than parental counterparts in this experiment. Importantly, as compared to either drug alone, the panobinostat and ponatinib combination demonstrated a significantly greater inhibitory growth impact on all cell lines (Table 3) (Matsuda et al., 2016).
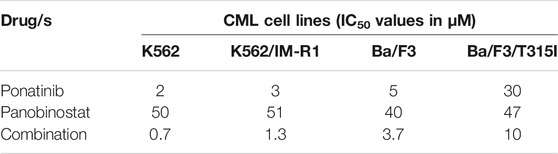
TABLE 3. This data has been extracted from Matsuda et al. (2016). For 72 h, cells were exposed to the same concentration of ponatinib, panobinostat, or their combination, before being tested using XTT assay.
Chidamide is a new selective HDAC inhibitor that has demonstrated its efficacy to treat hematological cancers. Chidamide reduced the development of the CML cell, causing apoptosis and arrest of the cell cycle when used alone. Furthermore, in the CML cell line KBM5, as well as IM-resistant CML cells KBM5 bearing T315I mutations, chidamide in conjunction with imatinib induced synergistic fatality, with a significant decrease in kinase activity of BCR-ABL and expression of acetyl-histone H3. In IM-resistant cells, the combined therapy significantly reduced the fundamental activity of β-catenin signaling and eliminated the mesenchymal stromal cells (MSCs) shielding effects on CML cells. As a result, this combination therapy is likely to be a feasible way to treat TKI-resistant CML and improve treatment strategies with BC-CML (Table 4) (He et al., 2020).
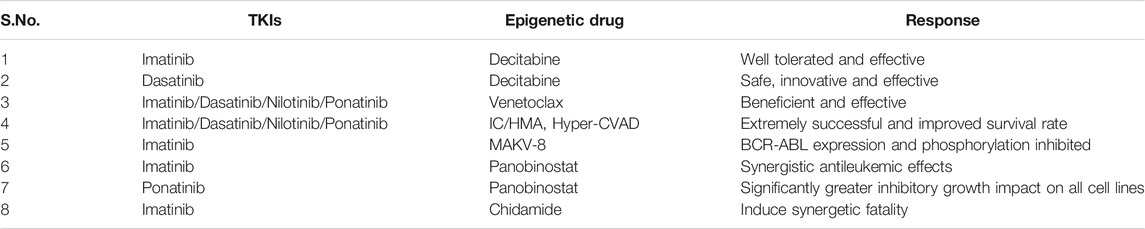
TABLE 4. The combination therapy for CML using TKIs with epigenetic drugs.
Today, one of the finest examples of effective targeted therapy in CML is that the survival rate of CML patients treated with TKIs seems to be relatively close to the healthy individuals. Since the development of first and second-line TKIs, substantial progress in CML therapy has been accomplished. Despite the TKI treatment’s excellent efficacy, some patients developed resistance owing to BCR-ABL kinase domain alterations, resulting in therapeutic failure. The development of CML and its resistance to treatments are both influenced by abnormal epigenetic control of key genes. Because epigenetic alterations may be controlled, novel medications that target distinct epigenetic pathways, such as demethylating agents and HDAC inhibitors, have been evaluated for CML treatment, particularly in individuals who have developed imatinib resistance. Finally, recent advances in biotechnology and bioinformatics have created new potential techniques for de novo epigenetic factor characterization and greater knowledge of epigenetic pathways, which can further develop tailored CML therapies in the future.
MA prepared the manuscript and SJ reviewed and edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
MA is thankful to UGC for providing fellowship.
Abaza, Y., Kantarjian, H., Alwash, Y., Borthakur, G., Champlin, R., Kadia, T., et al. (2020). Phase I/II Study of Dasatinib in Combination with Decitabine in Patients with Accelerated or Blast Phase Chronic Myeloid Leukemia. Am. J. Hematol. 95 (11), 1288–1295. doi:10.1002/ajh.25939
Alfayez, M., Richard‐Carpentier, G., Jabbour, E., Vishnu, P., Naqvi, K., Sasaki, K., et al. (2019). Sudden Blastic Transformation in Treatment‐free Remission Chronic Myeloid Leukaemia. Br. J. Haematol. 187 (4), 543–545. doi:10.1111/bjh.16245
Amir, M., Qureshi, M. A., and Javed, S. (2020). Biomolecular Interactions and Binding Dynamics of Tyrosine Kinase Inhibitor Erdafitinib, with Human Serum Albumin. J. Biomol. Struct. Dyn. 39 (11), 3934–3947. doi:10.1080/07391102.2020.1772880
Arber, D. A., Orazi, A., Hasserjian, R., Thiele, J., Borowitz, M. J., Le Beau, M. M., et al. (2016). The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 127 (20), 2391–2405. doi:10.1182/blood-2016-03-643544
Baccarani, M., Deininger, M. W., Rosti, G., Hochhaus, A., Soverini, S., Apperley, J. F., et al. (2013). European LeukemiaNet Recommendations for the Management of Chronic Myeloid Leukemia: 2013. Blood 122 (6), 872–884. doi:10.1182/blood-2013-05-501569
Balabanov, S., Braig, M., and Brümmendorf, T. H. (2014). Current Aspects in Resistance against Tyrosine Kinase Inhibitors in Chronic Myelogenous Leukemia. Drug Discov. Today Tech. 11 (1), 89–99. doi:10.1016/j.ddtec.2014.03.003
Başcı, S., Ata, N., Altuntaş, F., Yiğenoğlu, T. N., Dal, M. S., Korkmaz, S., et al. (2020). Outcome of COVID-19 in Patients with Chronic Myeloid Leukemia Receiving Tyrosine Kinase Inhibitors. J. Oncol. Pharm. Pract. 26 (7), 1676–1682. doi:10.1177/1078155220953198
Blanchard, F., and Chipoy, C. (2005). Histone Deacetylase Inhibitors: New Drugs for the Treatment of Inflammatory Diseases?. Drug Discov. Today 10 (3), 197–204. doi:10.1016/S1359-6446(04)03309-4
Bosulif (2017). Bosutinib. US FDA, 1–17. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203341s009lbl.pdf (Accessed September 10, 2021).
Bower, H., Björkholm, M., Dickman, P. W., Höglund, M., Lambert, P. C., and Andersson, T. M.-L. (2016). Life Expectancy of Patients with Chronic Myeloid Leukemia Approaches the Life Expectancy of the General Population. Jco 34 (24), 2851–2857. doi:10.1200/JCO.2015.66.2866
Bugler, J., Kinstrie, R., Scott, M. T., and Vetrie, D. (2019). Epigenetic Reprogramming and Emerging Epigenetic Therapies in CML. Front. Cel Dev. Biol. 7 (July), 1–14. doi:10.3389/fcell.2019.00136
Chen, W., and Bhatia, R. (2013). Roles of SIRT1 in Leukemogenesis. Curr. Opin. Hematol. 20 (Issue 4), 308308–313313. doi:10.1097/MOH.0b013e328360ab64
Cilloni, D., and Saglio, G. (2012). Molecular Pathways: BCR-ABL. Clin. Cancer Res. 18 (Issue 4), 930–937. doi:10.1158/1078-0432.CCR-10-1613
Crawford, L. J., Windrum, P., Magill, L., Melo, J. V., McCallum, L., McMullin, M. F., et al. (2009). Proteasome Proteolytic Profile is Linked to Bcr-Abl Expression. Exp. Hematol. 37, 357–366. doi:10.1016/j.exphem.2008.11.004
Di Carlo, V., Mocavini, I., and Di Croce, L. (2019). Polycomb Complexes in normal and Malignant Hematopoiesis, Rockefeller University Press. J. Cel Biol. 218 (Issue 1), 55–69. doi:10.1083/jcb.201808028
Duy, C., Hurtz, C., Shojaee, S., Cerchietti, L., Geng, H., Swaminathan, S., et al. (2011). BCL6 Enables Ph+ Acute Lymphoblastic Leukaemia Cells to Survive BCR-ABL1 Kinase Inhibition. Nature 473 (7347), 384–388. doi:10.1038/nature09883
Elias, M. H., Baba, A. A., Husin, A., Sulong, S., Hassan, R., Sim, G. A., et al. (2013). HOXA4Gene Promoter Hypermethylation as an Epigenetic Mechanism Mediating Resistance to Imatinib Mesylate in Chronic Myeloid Leukemia Patients. Biomed. Res. Int. 2013, 1–7. doi:10.1155/2013/129715
Feinberg, A. P., Ohlsson, R., and Henikoff, S. (2006). The Epigenetic Progenitor Origin of Human Cancer. Nat. Rev. Genet. 7 (Issue 1), 21–33. doi:10.1038/nrg1748
Feinberg, A. P., and Vogelstein, B. (1983). Hypomethylation Distinguishes Genes of Some Human Cancers from Their normal Counterparts. Nature 301 (5895), 89–92. doi:10.1038/301089a0
Fioravanti, R., Stazi, G., Zwergel, C., Valente, S., and Mai, A. (2018). Six Years (2012-2018) of Researches on Catalytic EZH2 Inhibitors: The Boom of the 2‐Pyridone Compounds, John Wiley and Sons Inc. Chem. Rec. 18 (Issue 12), 1818–1832. doi:10.1002/tcr.201800091
Gleevec (2008). Gleevec (Imatinib Mesylate) FDA US. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021588s024lbl.pdf (Accessed September 10, 2021).
Gulati, N., Béguelin, W., and Giulino-Roth, L. (2018). Enhancer of Zeste Homolog 2 (EZH2) Inhibitors, Taylor and Francis Ltd. Leuk. Lymphoma. 59 (7), 1574–1585. doi:10.1080/10428194.2018.1430795
Hatzi, K., Jiang, Y., Huang, C., Garrett-Bakelman, F., Gearhart, M. D., Giannopoulou, E. G., et al. (2013). A Hybrid Mechanism of Action for BCL6 in B Cells Defined by Formation of Functionally Distinct Complexes at Enhancers and Promoters. Cel Rep. 4 (3), 578–588. doi:10.1016/j.celrep.2013.06.016
He, B., Wang, Q., Liu, X., Lu, Z., Han, J., Pan, C., et al. (2020). A Novel HDAC Inhibitor Chidamide Combined with Imatinib Synergistically Targets Tyrosine Kinase Inhibitor Resistant Chronic Myeloid Leukemia Cells. Biomed. Pharmacother. 129, 110390. doi:10.1016/j.biopha.2020.110390
Hochhaus, A., Baccarani, M., Silver, R. T., Schiffer, C., Apperley, J. F., Cervantes, F., et al. (2020). European LeukemiaNet 2020 Recommendations for Treating Chronic Myeloid Leukemia, Springer Nature. Leukemia 34 (4), 966–984. doi:10.1038/s41375-020-0776-2
Housman, G., Byler, S., Heerboth, S., Lapinska, K., Longacre, M., Snyder, N., et al. (2014). Drug Resistance in Cancer: An Overview. CancersMDPI AG 6 (3), 1769–1792. doi:10.3390/cancers6031769
Hughes, T. P., Saglio, G., Kantarjian, H. M., Guilhot, F., Niederwieser, D., Rosti, G., et al. (2014). Early Molecular Response Predicts Outcomes in Patients with Chronic Myeloid Leukemia in Chronic Phase Treated with Frontline Nilotinib or Imatinib. Blood 123 (9), 1353–1360. doi:10.1182/blood-2013-06-510396
Hurtz, C., Hatzi, K., Cerchietti, L., Braig, M., Park, E., Kim, Y.-m., et al. (2011). BCL6-mediated Repression of P53 Is Critical for Leukemia Stem Cell Survival in Chronic Myeloid Leukemia. J. Exp. Med. 208 (11), 2163–2174. doi:10.1084/jem.20110304
Iclusig (2016). Ponatinib. FDA US, 3. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203469lbl.pdf (Accessed September 10, 2021).
Issa, G. C., Kantarjian, H. M., Gonzalez, G. N., Borthakur, G., Tang, G., Wierda, W., et al. (2017). Clonal Chromosomal Abnormalities Appearing in Philadelphia Chromosome-Negative Metaphases during CML Treatment. Blood 130 (19), 2084–2091. doi:10.1182/blood-2017-07-792143
Jabbour, E., and Kantarjian, H. (2020). Chronic Myeloid Leukemia: 2020 Update on Diagnosis, Therapy and Monitoring. Am. J. Hematol. 95 (6), 691–709. doi:10.1002/ajh.25792
Jagannath, S., Dimopoulos, M. A., and Lonial, S. (2010). Combined Proteasome and Histone Deacetylase Inhibition: A Promising Synergy for Patients with Relapsed/refractory Multiple Myeloma. Leuk. Res. 34 (Issue 9), 1111–1118. doi:10.1016/j.leukres.2010.04.001
Jain, P., Kantarjian, H., Alattar, M. L., Jabbour, E., Sasaki, K., Gonzalez, G. N., et al. (2015). Analysis of Long Term Responses and Their Impact on Outcomes in Patients with Chronic Phase CML Treated with Four Different TKI Modalities – Analysis of 5 Prospective Clinical Trials. Lancet Haematol. 2 (3), 118–128. doi:10.1016/S2352-3026(15)00021-6
Jain, P., Kantarjian, H., Boddu, P. C., Nogueras-González, G. M., Verstovsek, S., Garcia-Manero, G., et al. (2019). Analysis of Cardiovascular and Arteriothrombotic Adverse Events in Chronic-phase CML Patients after Frontline TKIs. Blood Adv. 3 (6), 851–861. doi:10.1182/bloodadvances.2018025874
Jain, P., Kantarjian, H. M., Ghorab, A., Sasaki, K., Jabbour, E. J., Nogueras Gonzalez, G., et al. (2017). Prognostic Factors and Survival Outcomes in Patients with Chronic Myeloid Leukemia in Blast Phase in the Tyrosine Kinase Inhibitor Era: Cohort Study of 477 Patients. Cancer 123 (22), 4391–4402. doi:10.1002/cncr.30864
Jain, P., Kantarjian, H., Sasaki, K., Jabbour, E., Dasarathula, J., Nogueras Gonzalez, G., et al. (2016). Analysis of 2013 European LeukaemiaNet (ELN) Responses in Chronic Phase CML across Four Frontline TKI Modalities and Impact on Clinical Outcomes. Br. J. Haematol. 173 (1), 114–126. doi:10.1111/bjh.13936
Jaiswal, S., Natarajan, P., Silver, A. J., Gibson, C. J., Bick, A. G., Shvartz, E., et al. (2017). Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 377 (2), 111–121. doi:10.1056/nejmoa1701719
Jelinek, J., Gharibyan, V., Estecio, M. R., Kondo, K., He, R., Chung, W., et al. (2011). Aberrant DNA Methylation is Associated with Disease Progression, Resistance to Imatinib and Shortened Survival in Chronic Myelogenous Leukemia. PLoS One 6 (7), 22110. doi:10.1371/journal.pone.0022110
Kantarjian, H., Shah, N. P., Hochhaus, A., Cortes, J., Shah, S., Ayala, M., et al. (2010). Dasatinib versus Imatinib in Newly Diagnosed Chronic-phase Chronic Myeloid Leukemia. N. Engl. J. Med. 362 (24), 2260–2270. doi:10.1056/NEJMoa1002315
Lernoux, M., Schnekenburger, M., Losson, H., Vermeulen, K., Hahn, H., Gérard, D., et al. (2020). Novel HDAC Inhibitor MAKV-8 and Imatinib Synergistically Kill Chronic Myeloid Leukemia Cells via Inhibition of BCR-ABL/MYC-signaling: Effect on Imatinib Resistance and Stem Cells. Clin. Epigenet. 12 (1), 1–26. doi:10.1186/s13148-020-00839-z
Li, L., Wang, L., Li, L., Wang, Z., Ho, Y., McDonald, T., et al. (2012). Activation of P53 by SIRT1 Inhibition Enhances Elimination of CML Leukemia Stem Cells in Combination with Imatinib. Cancer Cell. 21 (2), 266–281. doi:10.1016/j.ccr.2011.12.020
Li, Z., and Luo, J. (2018). Epigenetic Regulation of HOTAIR in Advanced Chronic Myeloid Leukemia. Cmar 10, 5349–5362. doi:10.2147/CMAR.S166859
Liu, T.-C., Lin, S.-F., Chang, J.-G., Yang, M.-Y., Hung, S.-Y., and Chang, C.-S. (2003). Epigenetic Alteration of theSOCS1gene in Chronic Myeloid Leukaemia. Br. J. Haematol. 123 (4), 654–661. doi:10.1046/j.1365-2141.2003.04660.x
Madapura, H. S., Nagy, N., Ujvari, D., Kallas, T., Kröhnke, M. C. L., Amu, S., et al. (2017). Interferon γ Is a STAT1-dependent Direct Inducer of BCL6 Expression in Imatinib-Treated Chronic Myeloid Leukemia Cells. Oncogene 36 (32), 4619–4628. doi:10.1038/onc.2017.85
Maiti, A., Franquiz, M. J., Ravandi, F., Cortes, J. E., Jabbour, E. J., Sasaki, K., et al. (2020). Venetoclax and BCR-ABL Tyrosine Kinase Inhibitor Combinations: Outcome in Patients with Philadelphia Chromosome-Positive Advanced Myeloid Leukemias. Acta Haematol. 143 (6), 567–573. doi:10.1159/000506346
Matsuda, Y., Yamauchi, T., Hosono, N., Uzui, K., Negoro, E., Morinaga, K., et al. (2016). Combination of Panobinostat with Ponatinib Synergistically Overcomes Imatinib‐resistant CML Cells. Cancer Sci. 107 (7), 1029–1038. doi:10.1111/cas.12965
Miller, T. A., Witter, D. J., and Belvedere, S. (2003). Histone Deacetylase Inhibitors. J. Med. Chem. 46 (24), 5097–5116. doi:10.1021/jm0303094
Millot, F., Baruchel, A., Guilhot, J., Petit, A., Leblanc, T., Bertrand, Y., et al. (2011). Imatinib Is Effective in Children with Previously Untreated Chronic Myelogenous Leukemia in Early Chronic Phase: Results of the French National Phase IV Trial. Jco 29 (20), 2827–2832. doi:10.1200/JCO.2010.32.7114
Morita, K., Jabbour, E., Ravandi, F., Borthakur, G., Khoury, J. D., Hu, S., et al. (2021a). Clinical Outcomes of Patients with Chronic Myeloid Leukemia with Concurrent Core Binding Factor Rearrangement and Philadelphia Chromosome. Clin. Lymphoma Myeloma Leuk. 21 (5), 338–344. doi:10.1016/j.clml.2020.12.025
Morita, K., Kantarjian, H. M., Sasaki, K., Issa, G. C., Jain, N., Konopleva, M., et al. (2021b). Outcome of Patients with Chronic Myeloid Leukemia in Lymphoid Blastic Phase and Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia Treated with hyper‐CVAD and Dasatinib. Cancer 127 (15), 2641–2647. doi:10.1002/cncr.33539
Mwakwari, S. C., Patil, V., Guerrant, W., and Oyelere, A. K. (2010). Macrocyclic Histone Deacetylase Inhibitors. Curr. Top. Med. Chem. 10 (14), 1423–1440. doi:10.2174/156802610792232079
Nteliopoulos, G., Bazeos, A., Claudiani, S., Gerrard, G., Curry, E., Szydlo, R., et al. (2019). Somatic Variants in Epigenetic Modifiers Can Predict Failure of Response to Imatinib but Not to Second-Generation Tyrosine Kinase Inhibitors. Haematologica 104 (12), 2400–2409. doi:10.3324/haematol.2018.200220
O'Hare, T., Shakespeare, W. C., Zhu, X., Eide, C. A., Rivera, V. M., Wang, F., et al. (2009). AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell. 16 (5), 401–412. doi:10.1016/j.ccr.2009.09.028
Oki, Y., Kantarjian, H. M., Gharibyan, V., Jones, D., O'Brien, S., Verstovsek, S., et al. (2007). Phase II Study of Low-Dose Decitabine in Combination with Imatinib Mesylate in Patients with Accelerated or Myeloid Blastic Phase of Chronic Myelogenous Leukemia. Cancer 109 (5), 899–906. doi:10.1002/cncr.22470
Özgür Yurttaş, N., and Eşkazan, A. E. (2020). Novel Therapeutic Approaches in Chronic Myeloid Leukemia. Leuk. Res. 91, 106337. doi:10.1016/J.LEUKRES.2020.106337
Patil, V., Guerrant, W., Chen, P. C., Gryder, B., Benicewicz, D. B., Khan, S. I., et al. (2010). Antimalarial and Antileishmanial Activities of Histone Deacetylase Inhibitors with Triazole-Linked Cap Group. Bioorg. Med. Chem. 18 (1), 415–425. doi:10.1016/j.bmc.2009.10.042
Reddy, E. P., and Aggarwal, A. K. (2012). The Ins and Outs of Bcr-Abl Inhibition. Genes. Cancer 3 (5–6), 447–454. doi:10.1177/1947601912462126
Rohrbacher, M., and Hasford, J. (2009). Epidemiology of Chronic Myeloid Leukaemia (CML). Best Pract. Res. Clin. Haematol. 22 (3), 295–302. doi:10.1016/j.beha.2009.07.007
Russo, D., Garcia-Gutierrez, J. V., Soverini, S., and Baccarani, M. (2020). Chronic Myeloid Leukemia Prognosis and Therapy: Criticisms and Perspectives. Jcm 9 (6), 1709. doi:10.3390/jcm9061709
Saglio, G., Kim, D.-W., Issaragrisil, S., le Coutre, P., Etienne, G., Lobo, C., et al. (2010). Nilotinib versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia. New Engl. J. Med. 362 (24), 2251–2259. doi:10.1056/nejmoa0912614
San José-Eneriz, E., Agirre, X., Jiménez-Velasco, A., Cordeu, L., Martín, V., Arqueros, V., et al. (2009). Epigenetic Down-Regulation of BIM Expression Is Associated with Reduced Optimal Responses to Imatinib Treatment in Chronic Myeloid Leukaemia. Eur. J. Cancer. 45 (10), 1877–1889. doi:10.1016/j.ejca.2009.04.005
Sasaki, K., Kantarjian, H. M., Jain, P., Jabbour, E. J., Ravandi, F., Konopleva, M., et al. (2016). Conditional Survival in Patients with Chronic Myeloid Leukemia in Chronic Phase in the Era of Tyrosine Kinase Inhibitors. Cancer 122 (2), 238–248. doi:10.1002/cncr.29745
Sasaki, K., Kantarjian, H. M., O’Brien, S., Ravandi, F., Konopleva, M., Borthakur, G., et al. (2019). Incidence of Second Malignancies in Patients with Chronic Myeloid Leukemia in the Era of Tyrosine Kinase Inhibitors. Int. J. Hematol. 109 (5), 545–552. doi:10.1007/s12185-019-02620-2
Sasaki, K., Kantarjian, H., O’Brien, S., Ravandi, F., Konopleva, M., Borthakur, G., et al. (2018). Prediction for Sustained Deep Molecular Response of BCR-ABL1 Levels in Patients with Chronic Myeloid Leukemia in Chronic Phase. Cancer 124 (6), 1160–1168. doi:10.1002/cncr.31187
Sasaki, K., Strom, S. S., O’brien, S., Jabbour, E., Ravandi, F., Konopleva, M., et al. (2015). Prospective Analysis: Relative Survival in Patients with Chronic Myeloid Leukemia in Chronic Phase in the Era of Tyrosine Kinase Inhibitors. Lancet Haematol. 2 (5), 186–193. doi:10.1016/S2352-3026(15)00048-4
Saxena, K., Jabbour, E., Issa, G., Sasaki, K., Ravandi, F., Maiti, A., et al. (2021). Impact of Frontline Treatment Approach on Outcomes of Myeloid Blast Phase CML. J. Hematol. Oncol. 14 (1), 1–10. doi:10.1186/s13045-021-01106-1
Scott, M. T., Korfi, K., Saffrey, P., Hopcroft, L. E. M., Kinstrie, R., Pellicano, F., et al. (2016). Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov. 6 (11), 1248–1257. doi:10.1158/2159-8290.CD-16-0263
Soverini, S., Colarossi, S., Gnani, A., Rosti, G., Castagnetti, F., Poerio, A., et al. (2006). Contribution of ABL Kinase Domain Mutations to Imatinib Resistance in Different Subsets of Philadelphia-positive Patients: By the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin. Cancer Res. 12 (24), 7374–7379. doi:10.1158/1078-0432.CCR-06-1516
Soverini, S., Iacobucci, I., Baccarani, M., and Martinelli, G. (2007). Targeted Therapy and the T315I Mutation in Philadelphia-positive Leukemias. Haematologica 92 (4), 437–439. doi:10.3324/haematol.11248
Sprycel (2010). Dasatinib. FDA US, 1–37. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021986s7s8lbl.pdf (Accessed September 10, 2021).
Tasigna (2017). Nilotinib. US FDA. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022068s004s005lbl.pdf (Accessed September 10, 2021).
Yang, A. S., Doshi, K. D., Choi, S. W., Mason, J. B., Mannari, R. K., Gharybian, V., et al. (2006). DNA Methylation Changes After 5-aza-2′-deoxycytidine Therapy in Patients with Leukemia. Cancer Res. 66 (10), 5495–5503. doi:10.1158/0008-5472.CAN-05-2385
Yuan, H., Wang, Z., Li, L., Zhang, H., Modi, H., Horne, D., et al. (2012). Activation of Stress Response Gene SIRT1 by BCR-ABL Promotes Leukemogenesis. Blood 119 (8), 1904–1914. doi:10.1182/blood-2011-06-361691
Zehtabcheh, S., Yousefi, A.-M., Salari, S., Safa, M., Momeny, M., Ghaffari, S. H., et al. (2021). Abrogation of Histone Deacetylases (HDACs) Decreases Survival of Chronic Myeloid Leukemia Cells: New Insight into Attenuating Effects of the PI3K/c-Myc axis on Panobinostat Cytotoxicity. Cel Biol. Int. 45 (5), 1111–1121. doi:10.1002/CBIN.11557
Zhu, X.-Y., Liu, W., Liang, H.-T., Tang, L., Zou, P., You, Y., et al. (2020). AICAR and Decitabine Enhance the Sensitivity of K562 Cells to Imatinib by Promoting Mitochondrial Activity. Curr. Med. Sci. 40 (5), 2020. doi:10.1007/s11596-020-2266-1
ACAs Additional cytogenetic abnormalities
AE Adverse effects
AER Absolute excess risk
ALL Acute lymphoblastic leukemia
ALT Alanine transaminase
AML Acute myeloid leukemia
ANC Absolute neutrophil count
AP Accelerated phase
ASCT Allogeneic stem cell transplant
AST Aspartate aminotransferase
AUC Area under the curve that represents the total drug exposure across time
BC Blast crisis
BCR Breakpoint cluster region
BD Bis in die (two times daily)
BP Blast Phase
CBF Core binding factor
CCyR Complete cytogenetic response
CHR Complete hematologic remission
Cmax Maximum serum concentration that a drug achieves after administration
CML Chronic myeloid leukemia
COVID-19 Coronavirus Disease 2019
CP Chronic phase
DNMT DNA methylation
EFS Event-free survival
ELN European LeukemiaNet
EMR Major molecular remission
FDA Food and drug administration
FFS Failure-free survival
HDAC Histone deacetylase
HMA Hypomethylating agent
HOTAIR HOX transcript antisense RNA
IC Intensive chemotherapy
IC50 Half maximal inhibitory concentration
LSC Leukemic stem cell
MDS Myelodysplastic Syndrome
OD Omne in die (once a day)
OS Overall survival
PcG Polycomb-group
PCyR Partial cytogenetic response
PRC Polycomb repressive complex
QTc QT corrected for heart rate
SAHA Suberoylanilide hydroxamic acid
SARS CoV-2 Severe acute respiratory syndrome coronavirus 2
SIRT Sirtuin
SOCS Suppressor of cytokine signaling
T1/2 Elimination half-life of a drug
TFS Transformation-free survival
TKI Tyrosine kinase inhibitor
Tmax Time at which the Cmax is observed
WHO World Health Organization
Keywords: chronic myeloid leukemia, tyrosine kinase inhibitors, imatinib, epigenetic, DNMT inhibitors, HDAC inhibitors, TKI resistance
Citation: Amir M and Javed S (2021) A Review on the Therapeutic Role of TKIs in Case of CML in Combination With Epigenetic Drugs. Front. Genet. 12:742802. doi: 10.3389/fgene.2021.742802
Received: 16 July 2021; Accepted: 30 September 2021;
Published: 22 October 2021.
Edited by:
Rais Ahmad Ansari, Nova Southeastern University, United StatesReviewed by:
Ahmet Emre Eskazan, Istanbul University-Cerrahpasa, TurkeyCopyright © 2021 Amir and Javed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saleem Javed, c2FsZWVtamF2ZWQ3MEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.