 Xiaojing Gu
Xiaojing Gu Yongping Chen
Yongping Chen Qianqian Wei
Qianqian Wei Yanbing HouBei Cao
Yanbing HouBei Cao Lingyu ZhangRuwei Ou
Lingyu ZhangRuwei Ou Junyu Lin
Junyu Lin Kuncheng LiuBi Zhao
Kuncheng LiuBi Zhao Huifang Shang*
Huifang Shang*- Department of Neurology, Laboratory of Neurodegenerative Disorders, Rare Disease Center, West China Hospital, Sichuan University, Chengdu, China
Background: CYLD Lysine 63 Deubiquitinase gene (CYLD) was recently identified to be a novel causative gene for frontal temporal dementia (FTD)-amyotrophic lateral sclerosis (ALS). In the current study, we aimed to (1) systematically screen the mutations of CYLD in a large cohort of Chinese ALS patients, (2) study the genotype–phenotype correlation, and (3) explore the role of CYLD in ALS via rare variants burden analysis.
Methods: A total of 978 Chinese sporadic ALS (sALS) patients and 46 familial ALS (fALS) patients were sequenced with whole-exome sequencing and analyzed rare variants in CYLD with minor allele frequency <0.1%.
Results: In total, seven rare missense variants in CYLD have been identified in 7 (0.72%) patients among 978 sALS patients. Two (4.3%) rare missense variants were identified among the 46 fALS cases, in which one patient was diagnosed as having comorbidity of ALS and progressive supranuclear palsy (PSP). Moreover, the burden analysis indicated no enrichment of rare variants in CYLD among patients with ALS.
Conclusion: In conclusion, our study extended the genotype and phenotype of CYLD in ALS, but the pathogenicity of these variants needs to be further verified. Moreover, burden analysis argued against the role of CYLD in the pathogenesis of ALS. More studies from different ethnicities would be needed.
Introduction
Recently, a heterozygous missense variant in CYLD Lysine 63 Deubiquitinase gene (CYLD) was found to co-segregate with the FTD-ALS in an autosomal dominant inherited European Australian family (Dobson-stone et al., 2020). Further functional studies revealed that mutated CYLD can lead to an increased lysine 63 deubiquitinase activity and gave rise to the pathogenesis of ALS and FTD (Dobson-stone et al., 2020). A subsequent study on a cohort of 65 Portuguese FTD patients identified two rare CYLD variants in 2 FTD patients (Tábuas-Pereira et al., 2020). ALS and FTD are considered as two related diseases in a continuous clinical spectrum that share not only clinical manifestations, but also genetic features and neuropathological abnormalilities (Murphy et al., 2007), while differences in genetic background have been described among ALS patients from different ethnicities (Wei et al., 2017). Therefore, the current study aimed to study the role of CYLD in Chinese patients with ALS.
Methods
We recruited 978 Chinese sporadic ALS (sALS) patients and 46 familial ALS (fALS) probands admitted to the Department of Neurology, West China Hospital of Sichuan University. All the ALS patients were diagnosed based on the El Escorial revised criteria for definite or probable ALS (Brooks et al., 2000). Written and signed informed consent were obtained from all the participants. The study was approved by the Ethics Committee of West China Hospital, Sichuan University.
Genomic DNA was collected from peripheral blood leukocytes and underwent whole-exome sequencing (WES). Procedures of WES and variants annotation were performed as previously described (Gu et al., 2020). C9orf72 was sequenced using Repeat-primed PCR and gel electrophoresis methods as previously described (Chen et al., 2015). Patients with mutations in known ALS causative genes such as SOD1, FUS, C9orf72, TARDBP, and TBK1 were excluded (Gregory et al., 2020). The average depth for CYLD was over 100×. Considering the autosomal dominant-inherited model for CYLD, we selected the variants according to the minor allele frequency (MAF) less than 0.1% in public databases, and the pathogenicity was based on multiple lines of in silico prediction tools including SIFT, Polyphen2, LRT, and Mutation Taster. Sanger sequencing was performed to confirm the variants.
Regarding burden analysis, controls were from the gnomAD East Asian population non-neuro dataset (https://gnomad.broadinstitute.org/). Combined Annotation Dependent Depletion (CADD) integrates predictions from numerous bioinformatic algorithms into a single “C-score” and ranks all possible nucleotide changes in the genome based on potential to disrupt gene/protein function (Uitterlinden et al., 2017). In accordance with the previous study, we defined a stringent CADD C-score ≥12.37 as likely damaging variants, representing the top 2% most damaging of all possible nucleotide changes in the genome—this subset is enriched for known pathogenic alleles (Uitterlinden et al., 2018). Moreover, the in-frame deletion/insertion variants lead to the change of the protein length and are classified as moderately strong evidence for pathogenicity (Richards et al., 2015). Therefore, in-frame deletion/insertion variants were also considered as damaging variants in the current study. Five different algorithms, including Comprehensive Approach to Analyzing Rare Genetic Variants (CARV), Sum of Squared Score (SSU), Sum Test (SUM), Cumulative Minor Allele Test (CMAT), and Bayesian Score Test (BST) were used for burden analysis with AssotesteR Package in the rare variants level and rare deleterious variants level (Lee et al., 2014).
Results
The demographic characteristics of all patients are listed in Supplementary Table S1. In total, seven rare missense variants (p. K33R and p. R156K in the exon 4, p. E203V in the exon 5, p. D328H in the exon 8, p. I377N in the exon 9, and p. R397S and p. V454I in the exon 10) in CYLD were identified in 7 (0.72%) patients from 978 sALS patients, and two (4.3%) rare missense variants (p. D190E in the exon 4 and p. Q443K in the exon 10) were identified in 2 patients from the 46 fALS cases (Table 1 and Figure 1). All the nine variants had a very low frequency or were even absent in the public databases of East Asia controls (PM2), and multiple lines of in silico prediction tools supported a deleterious effect (PP3). Therefore, all of them were classified into variants of uncertain significance (VUS) (Richards et al., 2015). No variant was found in the deubiquitinase domain of CYLD (amino acids 593–948), which was revealed to have a significant enrichment in FTD cases by the previous study (Dobson-stone et al., 2020).
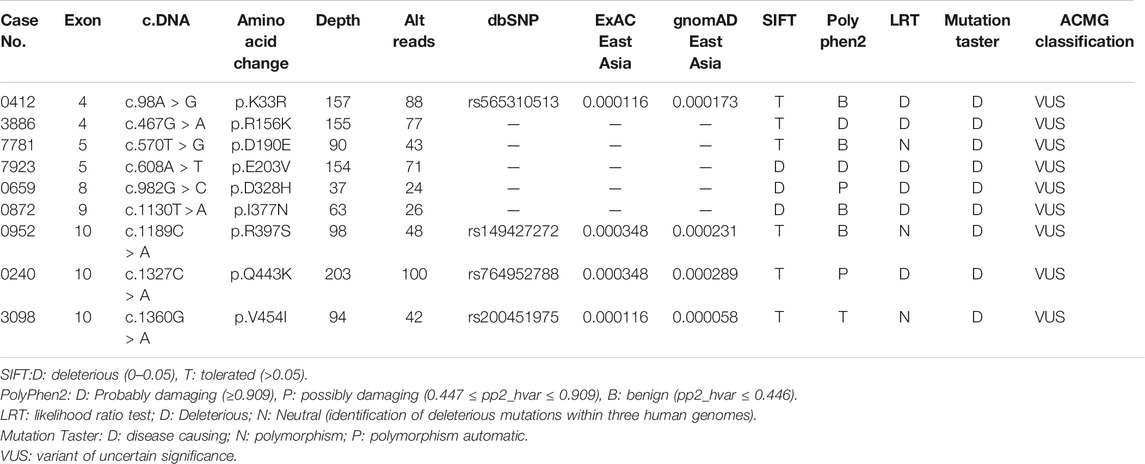
TABLE 1. Detailed description of the observed rare variants in CYLD.
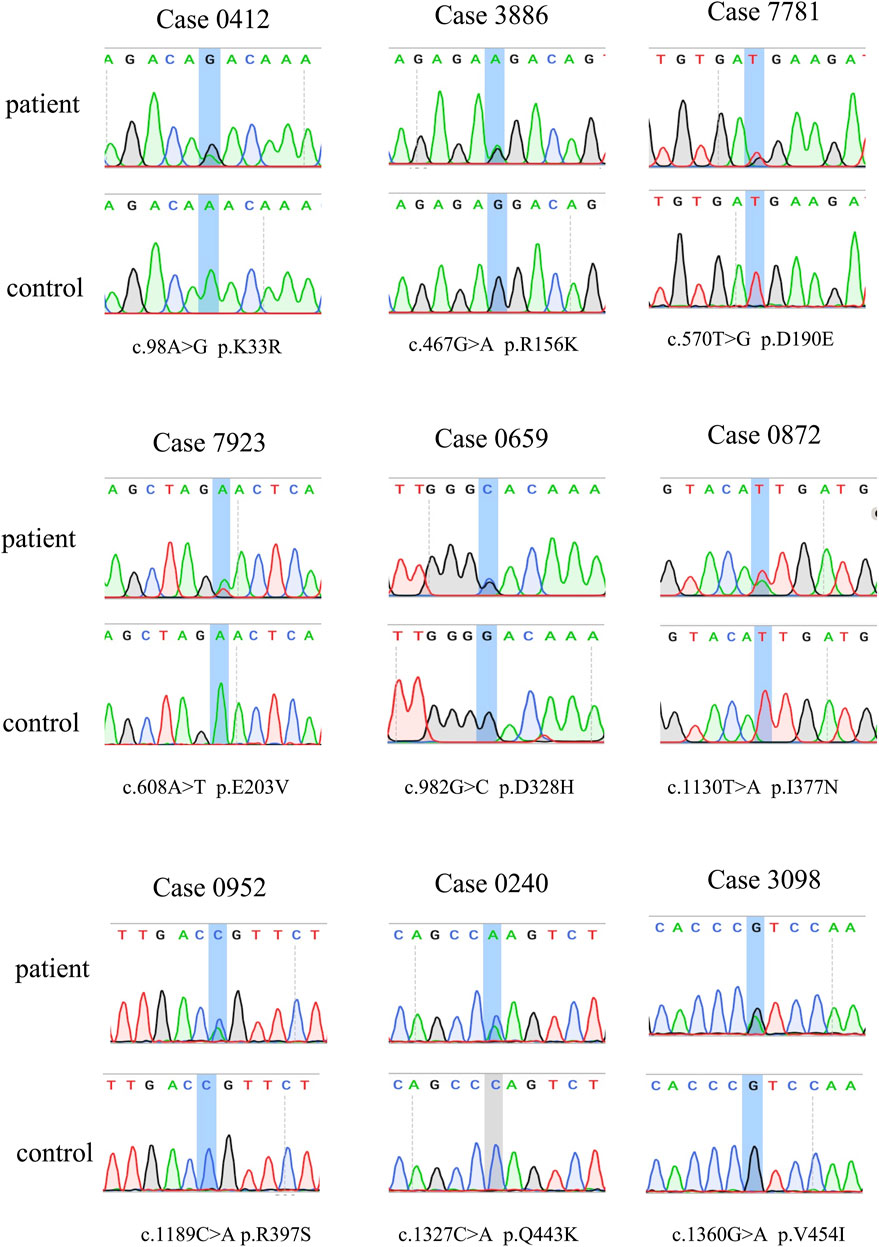
FIGURE 1. Sanger sequencing for the observed variants in CYLD.
The clinical features of the nine patients carrying rare variants in CYLD are listed in Supplementary Table S2. Most of the patients (8/9) had limb onset. The disease progression and survival time varied greatly among patients, but most of the patients were likely to have a relatively slow progression and long survival time. Two patients from six patients whose cognition were detailed assessed showed global cognitive impairment and frontal lobe dysfunction but did not present the typical features of FTD such as changes in behavior, personality, and aphasia.
Interestingly, among the nine patients, case 0659 had atypical ALS. He developed rigidity, bradykinesia, and weakness in both upper limbs at the age of 63. Then, he developed dysarthria 3 months later, and developed rigidity and weakness of both lower limbs 6 months later. He was diagnosed as having progressive supranuclear palsy (PSP) at the local hospital and was treated with levodopa but had no response. At the disease duration of 1 year since onset, he came to our clinic and was found to have difficulty in shifting his gaze horizontally and vertically, muscle atrophy of both hands, and increased muscle tone of limbs. Muscle strength of the proximal and distal part of lower limbs was grade 4+ and grade 3+, respectively, whereas muscle strength of lower limbs was grade 5. Pyramidal signs were found. The electromyographic examination indicated diffused neurogenic degeneration in the cervical, thoracic, and lumbosacral spinal cord segments. Therefore, the patient was diagnosed to have a comorbidity of ALS and PSP. The patient had a faster progression, and the muscle strength of extremities decreased to grade 2 after 3 months. The patient died of respiratory failure at the disease duration at 20.8 months. The patient’s mother developed cognitive impairment at the age of 74, progressed to have muscle weakness, and died within 2 years (Supplementary Figure S1). Moreover, two younger brothers of the proband’s mother both developed muscle weakness around 50 years old and died within several years since onset. Backtracking the previous generation, the patient’s grandmother on his mother’s side was recalled having dementia; however, the detailed medical history could not be obtained. Unfortunately, all the affected patients in the pedigree had died, so the co-segregation of the variant was not available.
To further evaluate the accumulated association of the rare variants in CYLD with ALS, we did gene-level rare variant burden analyses. There was no significant enrichment of putative pathogenic variants in our study when compared with the gnomAD East Asian control (Supplementary Table S3). Furthermore, when the variants were divided into “damaging” and “benign” based on the cutoff value at 12.37 of CADD scores, there were also no differences between patients and controls (Supplementary Table S3).
Discussion
CYLD is a deubiquitinating enzyme that targets Lys63-linked ubiquitin chains and regulates biological pathways such as the NF-ĸB signaling pathway and autophagy (Kovalenko et al., 2003; Yamashita et al., 2019). Loss-of-function mutations in CYLD have been shown to cause skin diseases, while very recently, missense mutations in CYLD were found to cause ALS-FTD via gain-of-function mechanism (Dobson-stone et al., 2020).
In the current study, nine patients were found to carry rare missense variants in CYLD with the MAF <0.1%. In total, seven rare missense variants in CYLD have been identified in 7 (0.72%) patients among 978 sALS patients, and two (4.3%) rare missense variants were identified among the 46 fALS cases, where familial cases had a relatively higher variation frequency. This phenomenon could be due to the following: firstly, as found in our previous study, familial cases are much more likely be caused by genetic causes than sporadic cases (40.6% in fALS vs. 8.6% in sALS) (Chen et al., 2021); secondly, the sample size of familial cases was relatively small. It is interesting to note that, besides the classical phenotype of ALS and ALS with cognitive impairment, our current study found one familial case presenting with a comorbidity of ALS and PSP who had rapid progression and short survival. The previous study on the large ALS-FTD pedigree reported that one patient had clinical diagnosis of FTD-ALS, Paget’s disease, and parkinsonism (Dobson-stone et al., 2020), whose pathological findings met neuropathological criteria for both cortical basal degeneration (CBD) FTLD with tau pathology (CBD FTLD-tau) and FTLD with type B TDP-43 pathology (type B FTLD-TDP). Moreover, TDP-43 pathology has also been reported in PSP (Koga et al., 2017). Therefore, our study further expanded the phenotype for CYLD mutation carriers. Moreover, two patients in our cohort (Patient 3886 and Patient 0952) had global cognitive impairments, frontal lobe dysfunction, and irritative symptoms but without FTD symptoms including behavior changes and aphasia.
CYLD is composed of three N-terminal cytoskeleton-associated protein-glycine conserved domains (CAP-Gly), which are responsible for binding to microtubules and other proteins, and a C-terminal ubiquitin-specific protease domain (USP), which is functioning for the disassembly of Lys63-linked ubiquitin polymers (Komander et al., 2008). Most of the variants identified in our study were not located in the functional domain, while two variants (p. D190E and p. E203V) were located in the CAP-Gly domain. However, two variants reported by previous studies (p. S615F and p. M791V) were located in the USP domain (Dobson-stone et al., 2020; Tábuas-Pereira et al., 2020) (Figure 2). This evidence emphasized the genetic heterogeneity among different ethnicities.
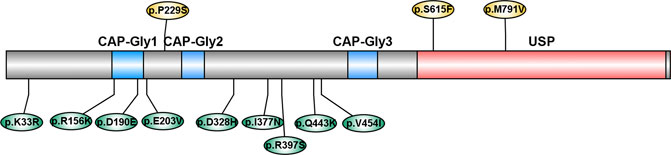
FIGURE 2. Schematic diagram of variants in CYLD. CAP-Gly: cytoskeleton-associated protein-glycine conserved domain; USP: ubiquitin-specific protease domain. Variants reported by previous studies were labeled above, and variants reported in our study were labeled below.
For the rare variants burden analysis, we did not find a significant enrichment of rare variants of CYLD in Chinese patients with ALS, which was consistent with the finding of the original study, which only detected a significant enrichment of rare variants in the deubiquitinase domain of CYLD (amino acids 593–948) in FTD patients, but not in ALS either in the whole gene level or the deubiquitinase domain (Dobson-stone et al., 2020). These results indicated that rare variants in the deubiquitinase domain of CYLD may contribute to the risk for FTD but not ALS.
However, some limitations of our study should be acknowledged. First, the sample size of familial cases was relatively small and the pedigree validation of the familial case was not available, which compromised the evidence to support pathogenicity of these variants. Second, there are genetic heterogeneity across races and rare variants can be (sub) population specific; therefore, using public databases as control for burden analysis will cause some unavoidable bias. Although we used the East Asian population in gnomAD as our control, which could decrease the heterogeneity to some extent, it would be better to enroll controls from the same region. Thirdly, no functional study has been performed to confirm the pathogenicity of CYLD variants.
Conclusion
In conclusion, the current study on a large cohort of Chinese patients with ALS suggested that the frequency of variant in CYLD is relatively uncommon in Chinese patients with sALS (0.72%) and fALS (4.3%), while the pathogenicity of the variants needs to be further verified. Our findings of ALS-PSP phenotype further expanded the phenotypic spectrum of the CYLD in ALS. Moreover, burden analysis argued against the role of CYLD for the risk of ALS. More studies from different ethnicities are needed.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of West China Hospital, Sichuan University. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
1. Research project: A: Conception and design; B: Acquisition of data; C: Analysis and interpretation of data; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing of the first draft, B. Review and Critique. XG: 1A, 1B, 1C, 2B, 3A; YC: 1B, 1C, 2B, 2C, 3B; QW: 1C, 2A, 2C, 3B; YH: 1B, 2B; BC: 1B, 2B; LZ: 1B, 2B; RO: 1B, 2B; JL: 1B, 2B; KL: 1B, 2B; BZ: 1B, 2B; HS: 1A, 1B, 2A, 2C, 3B.
Funding
This work was supported by the National Key Research and Development Program of China (Grant No. 2016YFC0901504), the 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYJC18038 and ZY2016203), and the Science Foundation of Chengdu Science and Technology Bureau (2019-YF05-00307-SN).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank all the patients and family members for their participation in the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.740052/full#supplementary-material
References
Brooks, B. R., Miller, R. G., Swash, M., and Munsat, T. L. (2000). El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 293–299. doi:10.1080/146608200300079536
Chen, Y.-P., Yu, S.-H., Wei, Q.-Q., Cao, B., Gu, X.-J., Chen, X.-P., et al. (2021). Role of Genetics in Amyotrophic Lateral Sclerosis: a Large Cohort Study in Chinese mainland Population. J. Med. Genet., jmedgenet–2021. doi:10.1136/jmedgenet-2021-107965
Chen, Y., Lin, Z., Chen, X., Cao, B., Wei, Q., Ou, R., et al. (2016). Large C9orf72 Repeat Expansions Are Seen in Chinese Patients with Sporadic Amyotrophic Lateral Sclerosis. Neurobiol. Aging 38, e15. doi:10.1016/j.neurobiolaging.2015.11.016
Dobson-stone, C., Hallupp, M., Shahheydari, H., Ragagnin, A. M. G., Chatterton, Z., Carew-jones, F., et al. (2020). CYLD Is a Causative Gene for Frontotemporal Dementia - Amyotrophic Lateral Sclerosis. Brain 143, 783–799. doi:10.1093/brain/awaa039
Gregory, J. M., Fagegaltier, D., Phatnani, H., and Harms, M. B. (2020). Genetics of Amyotrophic Lateral Sclerosis. Curr. Genet. Med. Rep. 8, 121–131. doi:10.1024/1661-8157/a00315310.1007/s40142-020-00194-8
Gu, X., Li, C., Chen, Y., Ou, R., Cao, B., Wei, Q., et al. (2020). Mutation Screening and burden Analysis of VPS13C in Chinese Patients with Early-Onset Parkinson's Disease. Neurobiol. Aging 94, e1. doi:10.1016/j.neurobiolaging.2020.05.005
Koga, S., Sanchez-contreras, M., Josephs, K. A., Uitti, R. J., Graff-radford, N., van Gerpen, J. A., et al. (2017). Distribution and Characteristics of Transactive Response DNA Binding Protein 43 kDa Pathology in Progressive Supranuclear Palsy. Mov Disord. 32, 246–255. doi:10.1002/mds.26809.Distribution
Komander, D., Lord, C. J., Scheel, H., Swift, S., Hofmann, K., Ashworth, A., et al. (2008). The Structure of the CYLD USP Domain Explains its Specificity for Lys63-Linked Polyubiquitin and Reveals a B Box Module. Mol. Cel 29, 451–464. doi:10.1016/j.molcel.2007.12.018
Kovalenko, A., Chable-Bessia, C., Cantarella, G., Israël, A., Wallach, D., and Courtois, G. (2003). The Tumour Suppressor CYLD Negatively Regulates NF-Κb Signalling by Deubiquitination. Nature 424, 801–805. doi:10.1038/nature01802
Lee, S., Abecasis, G. R., Boehnke, M., and Lin, X. (2014). Rare-Variant Association Analysis: Study Designs and Statistical Tests. Am. J. Hum. Genet. 95, 5–23. doi:10.1016/j.ajhg.2014.06.009
Murphy, J., Henry, R., and Lomen-hoerth, C. (2007). Establishing Subtypes of the Continuum of Frontal Lobe Impairment in Amyotrophic Lateral Sclerosis. Arch. Neurol. 64, 330–334. doi:10.1001/archneur.64.3.330
Richards, S., Aziz, N., Aziz, N., Bale, S., Bick, D., Das, S., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423. doi:10.1038/gim.2015.30
Robak, L. A., Jansen, I. E., van Rooij, J., Uitterlinden, A. G., Kraaij, R., Jankovic, J., et al. (2017). Excessive burden of Lysosomal Storage Disorder Gene Variants in Parkinson's Disease. Brain 140, 3191–3203. doi:10.1093/brain/awx285
Tábuas-Pereira, M., Santana, I., Kun-Rodrigues, C., Bras, J., and Guerreiro, R. (2020). CYLD Variants in Frontotemporal Dementia Associated with Severe Memory Impairment in a Portuguese Cohort. Brain 143, e67. doi:10.1093/brain/awaa183
Uitterlinden, G., Robak, L. A., Jansen, I. E., Rooij, J. V., and Kraaij, R. (2018). Excessive burden of Lysosomal Storage Disorder Gene Variants in Parkinson ’ S Disease. Brain 140, 3191–3203. doi:10.1093/brain/awx285
Wei, Q., Zhou, Q., Chen, Y., Ou, R., Cao, B., Xu, Y., et al. (2017). Analysis of SOD1 Mutations in a Chinese Population with Amyotrophic Lateral Sclerosis: a Case-Control Study and Literature Review. Sci. Rep. 7, 44606. doi:10.1038/srep44606
Keywords: CYLD lysine 63 deubiquitinase gene, amyotrophic lateral sclerosis, mutation screening, phenotype, burden analysis
Citation: Gu X, Chen Y, Wei Q, Hou Y, Cao B, Zhang L, Ou R, Lin J, Liu K, Zhao B and Shang H (2021) Rare CYLD Variants in Chinese Patients With Amyotrophic Lateral Sclerosis. Front. Genet. 12:740052. doi: 10.3389/fgene.2021.740052
Received: 13 July 2021; Accepted: 14 October 2021;
Published: 12 November 2021.
Edited by:
Nicola Ticozzi, University of Milan, ItalyReviewed by:
Qifei Li, Boston Children’s Hospital and Harvard Medical School, United StatesSheng Wang, University of California, San Francisco, United States
Copyright © 2021 Gu, Chen, Wei, Hou, Cao, Zhang, Ou, Lin, Liu, Zhao and Shang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huifang Shang, aGZzaGFuZzIwMDJAMTI2LmNvbQ==