 Junnan Zhang
Junnan Zhang Bo Han
Bo Han Yahui Gao
Yahui Gao Dongxiao Sun
Dongxiao Sun- Key Laboratory of Animal Genetics, Breeding and Reproduction of Ministry of Agriculture and Rural Affairs, National Engineering Laboratory for Animal Breeding, Department of Animal Genetics, Breeding and Reproduction, College of Animal Science and Technology, China Agricultural University, Beijing, China
Paratuberculosis in cattle causes substantial economic losses to the dairy industry. Exploring functional genes and corresponding regulatory pathways related to resistance or susceptibility to paratuberculosis is essential to the breeding of disease resistance in cattle. Co-analysis of genome-wide DNA methylation and transcriptome profiles is a critically important approach to understand potential regulatory mechanism underlying the development of diseases. In this study, we characterized the profiles of DNA methylation of jejunum from nine Holstein cows in clinical, subclinical, and healthy groups using whole-genome bisulfite sequencing (WGBS). The average methylation level in functional regions was 29.95% in the promoter, 29.65% in the 5’ untranslated region (UTR), 68.24% in exons, 71.55% in introns, and 72.81% in the 3’ UTR. A total of 3,911, 4,336, and 4,094 differentially methylated genes (DMGs) were detected in clinical vs. subclinical, clinical vs. healthy, and subclinical vs. healthy comparative group, respectively. Gene ontology (GO) and analysis based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) showed that these DMGs were significantly enriched in specific biological processes related to immune response, such as Th1 and Th2 cell differentiation, wnt, TNF, MAPK, ECM-receptor interaction, cellular senescence, calcium, and chemokine signaling pathways (q value <0.05). The integration of information about DMGs, differentially expressed genes (DEGs), and biological functions suggested nine genes CALCRL, TNC, GATA4, CD44, TGM3, CXCL9, CXCL10, PPARG, and NFATC1 as promising candidates related to resistance/susceptibility to Mycobacterium avium subspecies paratuberculosis (MAP). This study reports on the high-resolution DNA methylation landscapes of the jejunum methylome across three conditions (clinical, subclinical, and healthy) in dairy cows. Our investigations integrated different sources of information about DMGs, DEGs, and pathways, enabling us to find nine functional genes that might have potential application in resisting paratuberculosis in dairy cattle.
Introduction
Animal health, as a main factor affecting the development of the animal husbandry economy, is being valued progressively more over time and incorporated into breeding programs. Paratuberculosis is referred to as Johne’s disease and has been reported all over the world bringing huge economic losses to the dairy industry, warranting more attention (Ott et al., 1999). The disease is a chronic debilitating enteritis caused by a Mycobacterium avium subspecies paratuberculosis (MAP) infection (Clarke, 1997), which has a long incubation period. Clinical manifestations contain chronic or intermittent diarrhea, throat and jaw edema, weight loss, and eventually death (Whitlock and Buergelt, 1996). Verschoor et al., 2010 reported that four single nucleotide polymorphisms (SNPs) in the IL10RA gene were significantly associated with paratuberculosis susceptibility in the Canadian Holstein cattle population (Verschoor et al., 2010). Hempel et al., 2016 identified seven genes, FABP6, SLC10A2, MMP13, APOB, IGSF23, GNLY, and FCRLA, related to paratuberculosis through RNA sequencing (RNA-Seq) of ileocecal tissues from Holstein cows (Hempel et al., 2016). Studies have proved that jejunum is the main invasion and residence site of MAP, the infected jejunum wall is diffusely thickened, showing inflammation and tissue edema. The enlargement of mesenteric and other regional lymph nodes are usually apparent (Wentink et al., 1994; Bannantine and Bermudez, 2013; Ibeagha-Awemu et al., 2019). Therefore, we also used jejunum tissue to explore the mechanism of paratuberculosis diseases in our study.
The genome-wide DNA methylation genetic map is an effective strategy to reveal the role of DNA in development and disease. It has been reported that abnormal DNA methylation is related to human diseases, such as cancer and neurodevelopmental diseases (Weksberg et al., 2001; Debaun et al., 2003). Prawitt et al. (2005) found that aberrant methylation at the ICR1 gene led to human Wilms’ tumor and Beckwith–Wiedemann syndrome. Furthermore, a growing number of studies are considering the use of epigenomics to study disease resistance in livestock (Moore et al., 2008; Jin et al., 2011; Hao et al., 2016; Sharifi-Zarchi et al., 2017; Fu et al., 2018). Whole-genome bisulfite sequencing (WGBS) is used to determine the DNA methylation status of single cytosines by treating the DNA with bisulfite before sequencing (Stevens et al., 2013). Compared with the previous methylation sequencing technology, WGBS requires a smaller sample volume and gets a resolution off a single base, which may detect the methylation status of each cytosine.
In our previous studies, we detected the serum antibody levels of MAP based on an enzyme-linked immunosorbent assay (ELISA) for 8,214 Chinese Holstein cows and estimated the heritability of susceptibility to paratuberculosis (0.04–0.11) (Gao et al., 2018a). We further identified 26 genome-wide significant SNPs and 10 functional genes associated with paratuberculosis by performing a genome-wide association study, namely IL-4, IL-5, IL-13, IRF1, MyD88, PACSIN1, DEF6, TDP2, ZAP70, and CSF2 (Gao et al., 2018b). In addition, we conducted RNA sequencing on the jejunum samples from MAP-infected and healthy Holstein cows and identified 134 differentially expressed genes among clinical, subclinical, and healthy groups (unpublished data). However, research related to the regulatory roles of DNA methylation on paratuberculosis in dairy cattle has not been reported until now. In the present study, WGBS was using with the same samples as those for our RNA-Seq study to investigate the regulatory mechanism of DNA methylation on paratuberculosis and identify potentially critical genes for resistance/susceptibility to MAP in order to provide molecular information for a disease-resistance breeding program in dairy cattle.
Methods and Materials
Sample Collection
In our previous studies (Gao et al., 2018a; Gao et al., 2018b), we detected the serum antibody levels for MAP of 8,214 Chinese Holstein cows with the ELISA method, of which 784 cows were identified as positive. Stool samples of each seropositive cow were further collected for quantitative PCR (qPCR) to detect whether MAP was present with an INgene q ParaTB Kit (Ingenasa, Madrid, Spain). Individuals who were positive in both the serum and stool samples and had obvious diarrhea were defined as clinical cows (CC); individuals who were seropositive, but their stools were negative and with no diarrhea, were referred to as subclinical cows (SC); individuals that were negative in both serum and stools samples were treated as healthy cows (HC). Three cows were included in each group (Supplementary Figure S1A). These nine cows were dissected in the same slaughterhouse. The jejunum tissues were collected from each individual within 30 min after slaughtering and placed in liquid nitrogen. The infected part of the jejunum from clinical cows was obviously thickened, inflamed, and edema was present, and that from subclinical cows showed mild inflammation and edema.
Deoxyribonucleic Acid Extraction and Library Preparation
Genomic DNA of the jejunum tissue from the nine cows with three in each group was extracted using a TIANamp Genomic DNA Kit (Tiangen, Beijing, China). The DNA quantity and quality were determined using a NanoDrop2000 spectrophotometer (ThermoFisher, Waltham, MA, United States) and Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, United States), respectively.
First, 5 μg of lambda DNA was added into the genomic DNA of each sample as the negative control. Then, the genomic DNA was randomly broken into 200–300 bp fragments using an S220 focused-ultrasonicator (Covaris, Santa Clara, MA, United States); these fragments were end-repaired, added to the sequencing adapter, and treated with bisulfite. The methylated cytosines were thereby unchanged and the unmethylated cytosines became uracils, which were changed to thymines after PCR amplification (Supplementary Figure S1B). A library quality control was performed with an Agilent 2100 Bioanalyzer.
Bisulfite Sequencing and Data Analysis
With the completion of the library, paired-end sequencing was performed on the Illumina NovaSeq6000 sequencing platform (Illumina, CA, United States). Preliminary quality control of raw reads was carried out with fastqc (v0.11.9, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and these reads were then filtered with fastp software (v0.20) to remove adapters and low quality sequences (Chen et al., 2018). With bowtie2 (v2.4.0), clean reads were deduplicated and aligned against the bovine reference genome (ARS-UCD1.2, https://asia.ensembl.org/index.html), which was bisulfite-converted using bismark (v0.22.1) (Langmead and Salzberg, 2012; Langmead et al., 2018), and the command of “bismark--genome_folder ref.fa -bowtie2 -N 0 -L 20”. Methylation status was determined and methylated CpG sites were marked using the command “bismark_methylation_extractor” (Krueger and Andrews, 2011). The correlation coefficient within each group was calculated using R programing language (v3.6.0).
Identification of Differentially Methylated Regions and Differentially Methylated Genes
The bsseq (v1.24.4) package in the R programing language (v3.6.0) was used to find differentially methylated regions (DMRs) among the three comparative groups based on the information about CpG sites (Hansen et al., 2012). Each DMR had at least five methylated CpG sites where the distance between CpG sites was less than 300 bp and there was a greater than 10% difference in methylation levels between groups. The significant ranking of DMRs was dependent on the absolute value of areaStat that was used to determine the significance threshold. In order to screen differentially methylated genes (DMGs), we retained the genes in which the DMRs were entirely located using the “intersect–f 1” command from bedtools (v2.28.0) (Quinlan and Hall, 2010), eliminating genes that had partial DMRs, all these screened genes were defined as DMGs. The promoter is located kilo bases upstream away from the transcriptional start site in the regulatory sequence controlling gene expression (Hernandez-Garcia and Finer, 2014; Yu et al., 2016). Therefore, we defined 2000 bp upstream of the gene as the promoter region and identified the differentially promoter-methylated genes (DPMGs) with identical screening. Both the files of the gene body and promoter were downloaded from UCSC (https://genome.ucsc.edu/). The genes containing both hypermethylated and hypomethylated regions were considered as hypermethylated and hypomethylated genes.
Gene Ontology and Kyoto Encyclopedia of Genes and Genomes Functional Enrichment Analysis
The clusterProfiler tool (v3.16.0) in R was applied to perform GO and KEGG enrichment analysis on DMGs and DPMGs (Yu et al., 2012). The significant threshold related to enrichment analysis of DMGs and DPMGs was a q value <0.05 and p value <0.05, respectively. The p value is a measure of statistical significance; the q value is a measure of the false discovery rate (FDR) (Benjamini and Hochberg, 1995; Storey, 2003).
Integration Analysis of Whole-Genome Bisulfite Sequencing and Ribonucleic Acid Seq Data
Genes that were both DMGs and DEGs according to the DNA methylation and RNA sequencing data were identified. Further, we detected the methylated status of promoters in these genes and calculated the association between the methylated status and the fragments per kilobase million (FPKM) with Pearson correlation analysis (Schober et al., 2018).
Results
Summary of Methylome Sequencing
By performing WGBS, the raw data of the nine jejunum tissue samples from clinical, subclinical, and healthy groups, each comprising three Holstein cows were completed. After quality control, a total of 2.84 × 109 clean reads were obtained with an average of 3.16 × 108 (average 108.33 G per sample) for each sample, indicating a sequencing depth of 30×. There were 81.03% of non-duplicated clean reads uniquely aligned to the bovine reference genome (ARS-UCD1.2, Supplementary Table S1). Bisulfite conversion efficiency (BCE) reached 99.33% suggesting the reliability of the methylome sequencing in this study (Table 1).
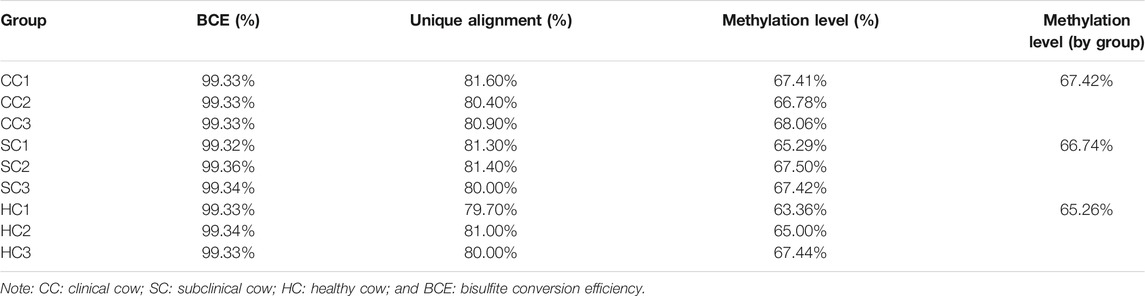
TABLE 1. Basic state of alignment, bisulfite conversion efficiency, and methylation level.
Deoxyribonucleic Acid Methylation Patterns
We found that on average 3.22% of all genomic C sites were methylated across the nine samples. Methylation in advanced mammals generally exists in three sequence contexts: CpG, CHG (where H is A, C, or T), and CHH. Here, we observed the overall genome-wide methylation levels of 70.50% CpG, 0.40% CHG, and 0.38% CHH in the clinical group, 69.60% CpG, 0.39% CHG, and 0.38% CHH methylation in the subclinical group, and 68.03% CpG, 0.39% CHG, and 0.38% CHH methylation in the healthy group (Supplementary Table S1). Further, we calculated the correlation of 0.9 within each group using the methylation level of CpGs.
Deoxyribonucleic Acid Methylation Levels in Different Regions of the Gene
To characterize the distribution of methylation in different genomic elements, we analyzed the average DNA methylation levels in the promoter, 5’ untranslated region (UTR), exons, introns, and the 3’ UTR of each methylated gene, and found the average methylation levels of the three experimental groups were similar in the promoter (29.3–30.48%) and 5’ UTR (29.02–30.29%) but displayed certain differences in partial exons, introns, and 3’UTR (Figure 1). Of note, the methylation levels of partial exons, introns, and the 3’UTR in the clinical group (68.50, 72.40, and 73.35%) were obviously higher than those in the healthy groups (67.90, 70.60, and 71.22%). In addition, the methylation level in the X chromosome (63.45%) was significantly lower than that in the autosomes (69.65%, p value <0.05; Figure 2).
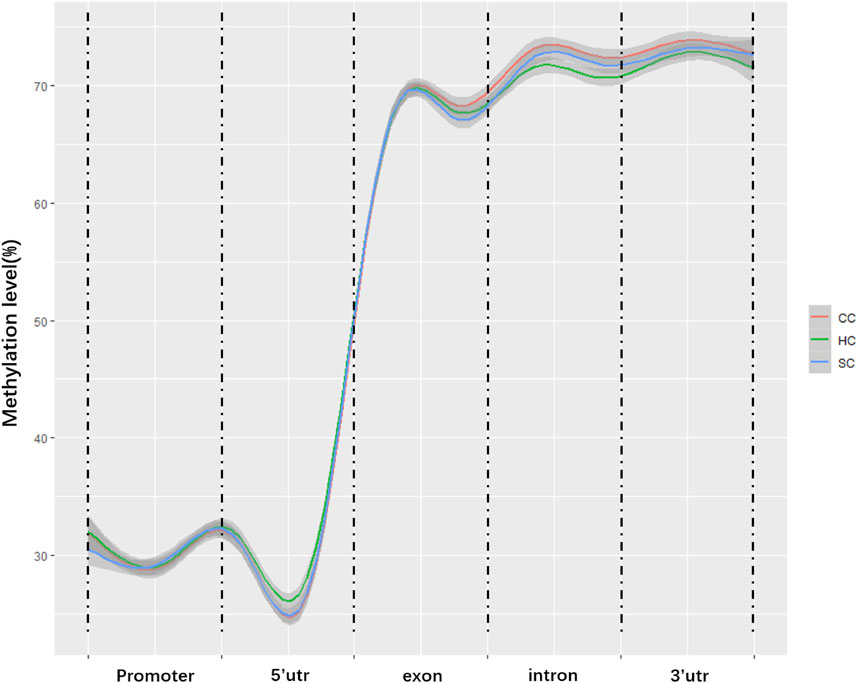
FIGURE 1. Average methylation levels in different genomic regions. The y-axis is the methylation level; the x-axis is the different regions in the genome. CC: clinical cow; SC: subclinical cow; and HC: healthy cow.
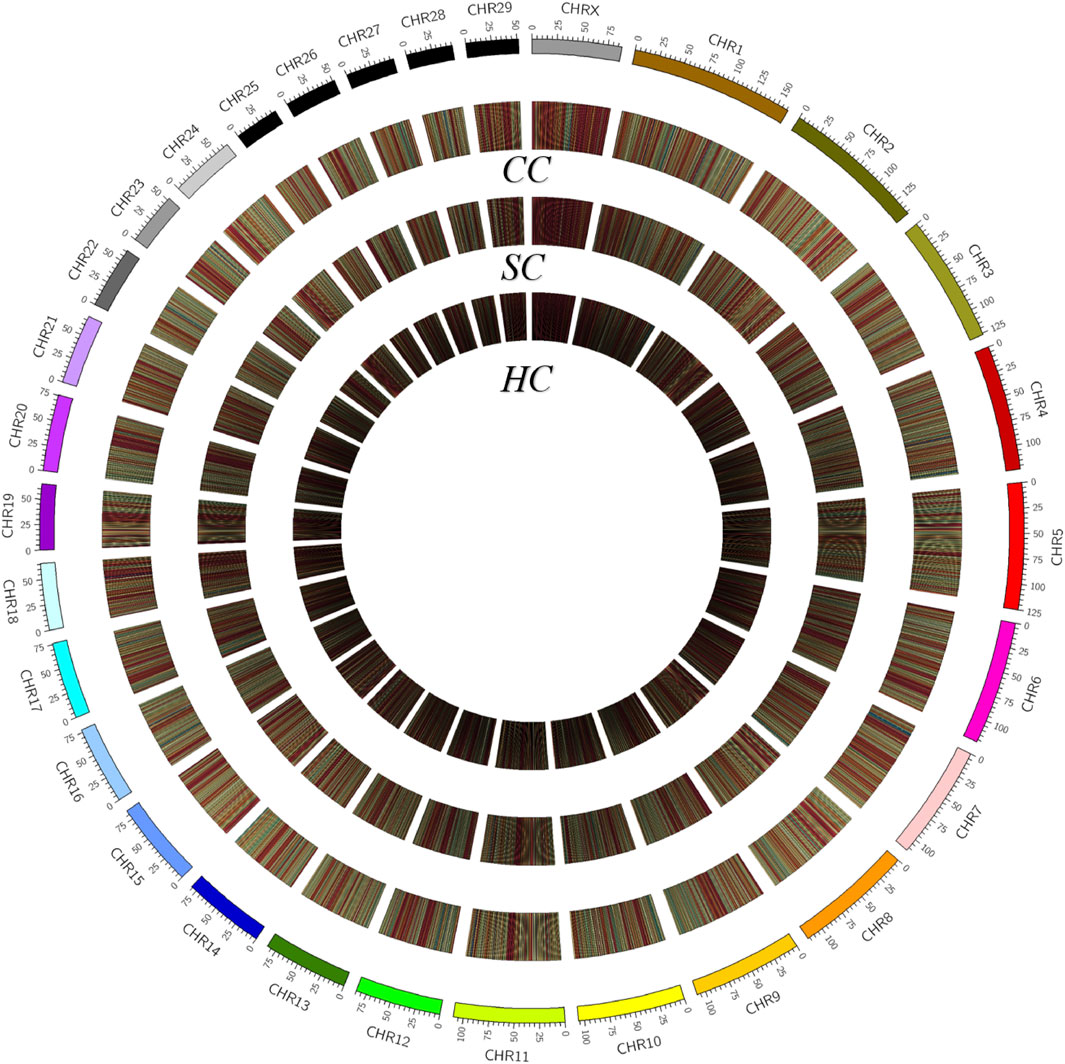
FIGURE 2. Density plot of 5-methylcytosine among various groups. Chromosome numbers and scales are indicated on the periphery, a dark to light color indicates a low to high level of methylation. CC: clinical cow; SC: subclinical cow; and HC: healthy cow.
Differentially Methylated Regions, Differentially Methylated Genes, and Differentially Promoter-Methylated Genes Identification
By comparing the cytosine methylation profiles of MAP-infected and healthy groups, we detected 100,758 differentially methylated regions (DMRs), including 31,750 between the clinical and subclinical groups, 35,082 between the clinical and healthy groups, and 33,926 between the subclinical and healthy groups (Supplementary Table S2). The length of DMRs was 6–2,292 bp with an average of 350 bp, 98.5% of which were less than 1,000 bp (Supplementary Figure S2). Further, a total of 6,653 differentially methylated genes (DMGs) and 1,779 differentially promoter-methylated genes (DPMGs) were identified, i.e., 3,911 DMGs and 661 DPMGs in clinical vs. subclinical groups, 4,336 DMGs and 893 DPMGs in clinical vs. healthy groups, and 4,094 DMGs and 746 DPMGs in subclinical vs. healthy groups (Supplementary Table S2). Details of the top 10 significant DMGs and DPMGs in the three comparisons are described in Table 2 and Table 3, respectively.
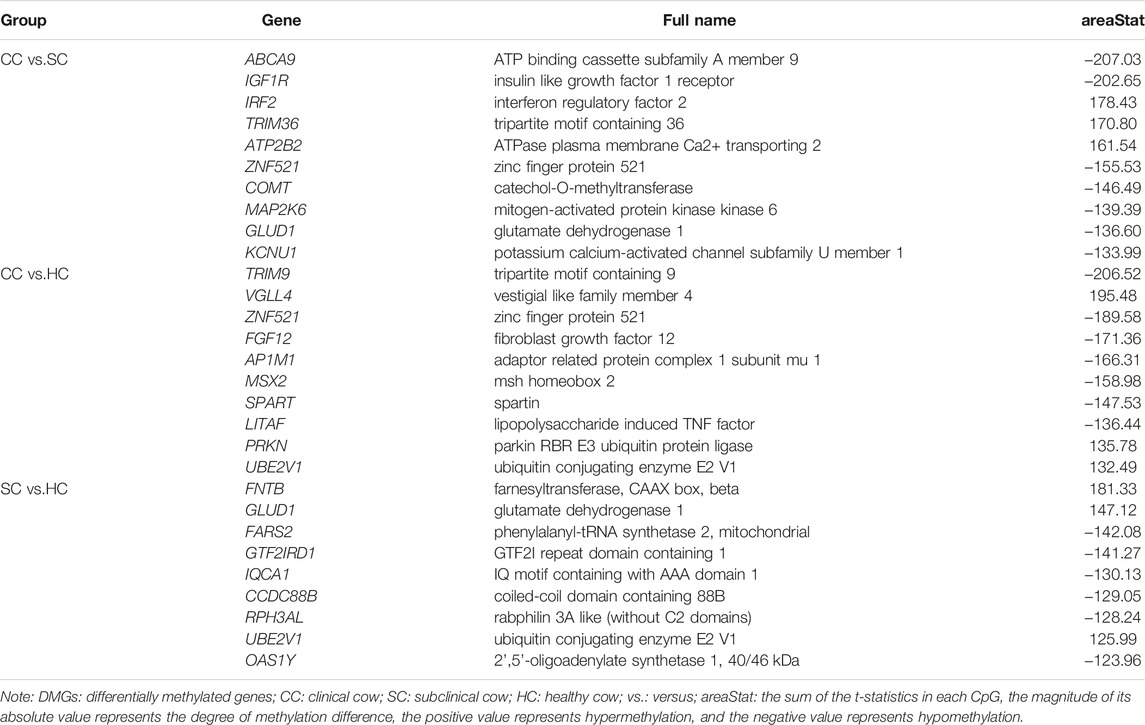
TABLE 2. The top 10 DMGs among clinical, subclinical, and healthy groups.
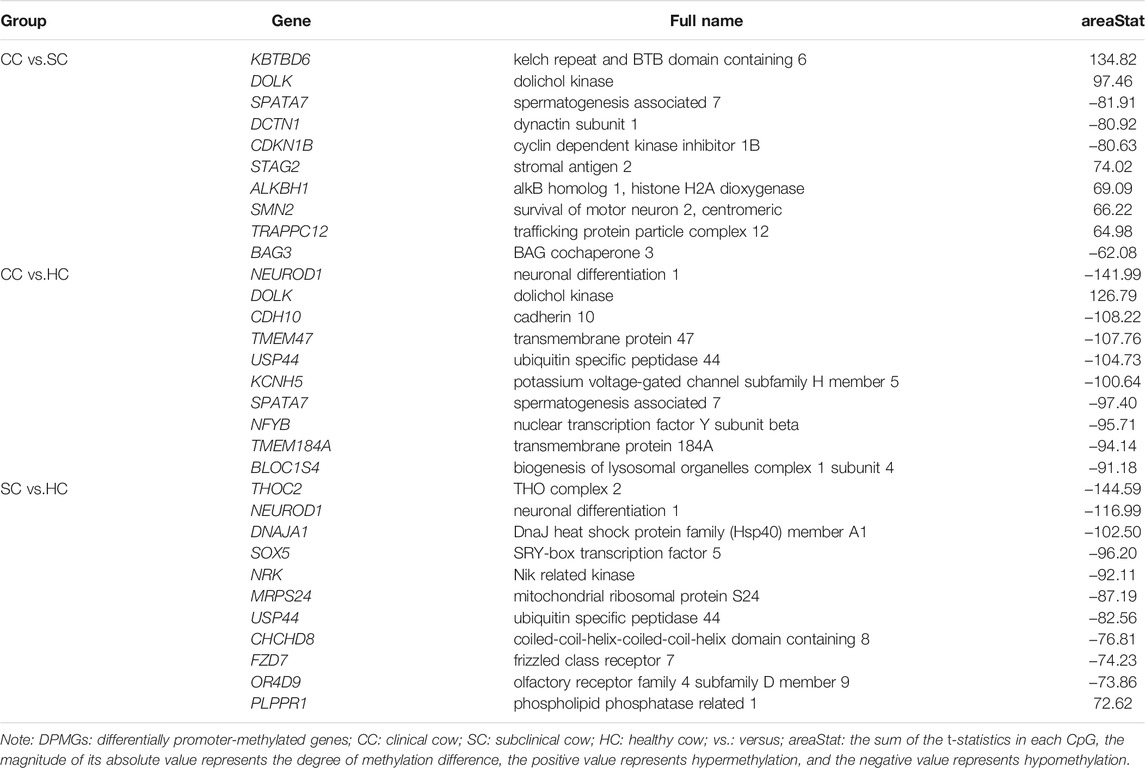
TABLE 3. The top 10 DPMGs among clinical, subclinical, and healthy groups.
Gene Ontology Annotation and Kyoto Encyclopedia of Genes and Genomes Pathways Enrichment on the Differentially Methylated Genes and Differentially Promoter-Methylated Genes
To further investigate the potential associations of the DMGs and DPMGs with paratuberculosis, we implemented functional enrichment analysis with the clusterprofiler package and found that the DMGs were significantly enriched in 77 GO terms (q < 0.05; Figure 3, Supplementary Table S3). From those, the most enriched terms included plasma membrane part (GO:0044459, q = 3.51E-06), phospholipid binding (GO:0005543, q = 3.80E-05), metal ion transmembrane transporter activity (GO:0046873, q = 0.00018), and phosphatidylinositol binding (GO:0035091, q = 0.00223).
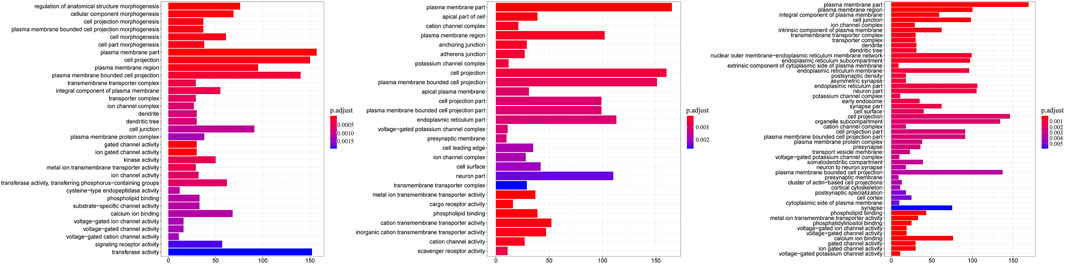
FIGURE 3. Gene ontology categories enriched for DMGs. The y-axis is the name of GO terms; the x-axis is the gene number in terms. CC: clinical cow; SC: subclinical cow; and HC: healthy cow.
According to the KEGG analysis, the DMGs were significantly enriched in 181 pathways (q < 0.05; Supplementary Table S3) with 95 common pathways across all the comparisons (Supplementary Figure S3). These were mainly related to Th1 and Th2 cell differentiation (bta04658, q = 0.03), wnt (bta04310, q = 0.00056), TNF (bta04668, q = 0.0008), MAPK (bta04010, q = 6.98E-08), ECM-receptor interaction (bta04512, q = 0.017), cellular senescence (bta04218, q = 0.03), calcium (bta04020, q = 1.80E-05), and the chemokine signaling pathways (bta04062, q = 0.001).
As for DPMGs, no significant GO terms were enriched (p > 0.05). However, a total of 92 pathways were enriched (p < 0.05, Supplementary Table S3), in which the most significant pathways included oxytocin (bta04921, p = 1.46E-05), influenza A (bta05164, p = 9.65E-05), prostate cancer (bta05215, p = 0.00014), vascular smooth muscle contraction (bta04270, p = 0.00015) and the long-term potentiation signaling pathway (bta04720, p = 0.00022).
Identification of Candidate Genes Related to Paratuberculosis With Integrated Whole-Genome Bisulfite Sequencing, Ribonucleic Acid Seq, and Biological Functional Data
Our previous RNA sequencing on the same jejunum samples of the nine Holstein cows used in the present study detected 134 DEGs including 23 between the clinical and subclinical groups, 71 between the subclinical and healthy groups, and 73 between the clinical and healthy groups (q<0.05; unpublished data). Of these, 31 functional genes were differentially expressed and differentially methylated among the three comparative groups simultaneously (Supplementary Table S4). Eight genes, calcitonin receptor like receptor (CALCRL), tenascin C (TNC), GATA binding protein 4 (GATA4), CD44 molecule (CD44), transglutaminase 3 (TGM3), C-X-C motif chemokine ligand 9 (CXCL9), C-X-C motif chemokine ligand 10 (CXCL10), and peroxisome proliferator activated receptor gamma (PPARG), were significantly enriched in immune-related pathways such as Th1 and Th2 cell differentiation, wnt, TNF, MAPK, ECM-receptor interaction cellular senescence, calcium, and chemokine signaling pathways. Therein, CXCL10 and CALCRL contained two methylated sites in the promoter (Supplementary Figure S4). The Pearson correlation coefficients between the methylated status of the promotor and the mRNA levels (FPKM) of CXCL10 and CALCRL were calculated as -0.07 and -0.56, respectively, indicating downregulation roles of the methylated promoter on gene expression (Supplementary Table S5).
Further, nuclear factor of activated T cells 1 (NFATC1), whose methylation of the intron was shown to affect the expression of IL-2 and IL-4 in the effector T cells in previous studies (Akimzhanov et al., 2008; Huang et al., 2011; Christie and Zhu, 2014), was differentially methylated between the MAP-infected and healthy cows and significantly enriched in the Th1 and Th2 cell differentiation pathway (q = 0.033).
Potential Regulatory Mechanism Underlying Paratuberculosis
Consequently, we inferred the potential of a regulatory mechanism underlying paratuberculosis in dairy cattle. As shown in Figure 4, the differentially methylated NFATC1, TGM3, and GATA4 directly affected the expression of immune factors, including IL-5, IL-2, IL-4, IL-4Ra, IL-2Ra, GATA3, IL-6, IL-12Rb, and IFN-γ secreted in Th1 and Th2 cells, to maintain an inflammatory response. NFATC1 elevated the expression of these immune factors through a cell differentiation pathway. TGM3 activated IL-6 expression via calcium signaling pathways and GATA4 promoted IL-5, IL-4, and IFN-γ through a cellular senescence signaling pathway. PPARG regulated NFATC1 expression through MAPK and calcium signaling pathways thereby modulating immune factors in Th1 and Th2 cells. CXCL9 and CXCL10 stimulated chemokine receptor proteins that promoted the JAK2/3 and STAT pathways, ultimately activating immune factors such as IL-12Rb, IFN-γ, and T-bet. Membrane proteins encoded by TNC and CD44 modulated the expression of IL-5 and IL-4 through the ECM-receptor interaction signaling pathway and another member protein encoded by CALCRL stimulated IL-5 secretion through activation of cAMP in the vascular smooth muscle contraction pathway.
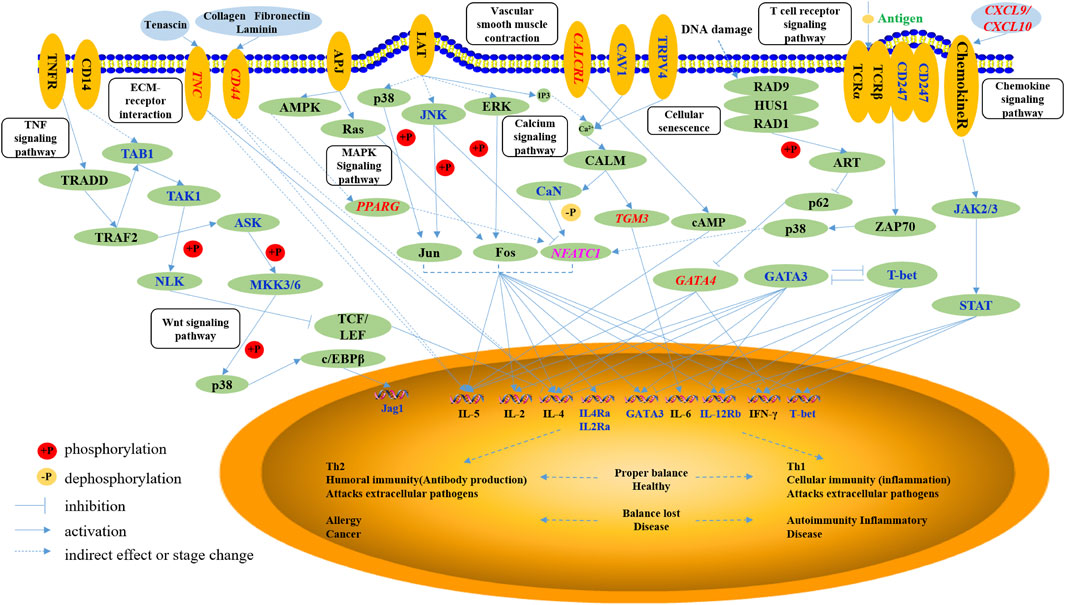
FIGURE 4. The networks of the immune-related pathways to paratuberculosis. The oval with non-italic characters represents the protein, the oval with italic characters represents the gene, and the small circle represents the small molecule. Orange ellipses are the membrane proteins, the blue ellipses are the extracellular matrix, and green ellipses are the intracellular matrix. Blue and pink characters are the differentially methylated genes, and the red characters are the genes with differential methylation and expression.
In sum, invasion of MAP into the animal body induced immune responses whereby the above-mentioned nine functional genes stimulated the secretion of immune factors against MAP, indicating the potential regulatory mechanism underlying paratuberculosis in dairy cattle.
Discussion
In this study, we obtained the integral genome-wide DNA methylation maps of jejunum tissues from clinical, subclinical, and healthy cows with regards to paratuberculosis at the resolution of a single base. We found that the average methylation level was 15–20% in the transcription start site (TSS) and 65–75% in the gene body. These results were similar to the report in humans (TSS: 15–20%, gene body: 60–75%) (Gao et al., 2014), in pigs (TSS: 15–25%, gene body: 65–75%) (Fu et al., 2018; Yang et al., 2021), and even in plants (TSS: 10–20%, gene body: 30–45%) (Song et al., 2013), indicating that a conserved methylation mode existed in trans-species within different species. We observed that most DMRs (46%) were located in the introns region. Del Corvo et al., 2020 also reported that in Italian Simmental, 46% of DMRs in peripheral blood between groups treated by different stress level were located in introns (Del Corvo et al., 2020). This may be because the intron sequence is much longer than the encoding region.
The DNA methylation status of the promoter can affect gene expression via changes in chromatin structure or transcription efficiency (Miller and Grant, 2013). In our study, we found that both CALCRL and CXCL10 genes whose promoters contained two methylated sites showed lower expression levels based on our WGBS and RNA-seq data. This may be due to the methylation state slowing down the progress of RNA polymerase during transcription, given that delay of the transcription extension and appearance can inhibit the abnormal transcription initiation (Barry et al., 1993; Hohn et al., 1996; Rountree and Selker, 1997).
Based on integrated information about the DMGs, DEGs, and their biological functions, we identified nine genes as promising candidates for resistance or susceptibility to paratuberculosis in dairy cattle. They were CALCRL, TNC, GATA4, CD44, TGM3, CXCL9, CXCL10, PPARG, and NFATC1. CALCRL encodes the membrane receptor CRLR, which is associated with diseases of lymphatic malformation and changes contraction of the vascular smooth muscle resulting in an increase in tissue fluid and thus leads to edema (Dong et al., 2005; Garvey, 2018). TNC encodes an extracellular matrix protein, which regulates the expression of immune factors IL-4 and IL-5 and has a critical role in humoral immunity. (Zhou et al., 2019) found that the TNC levels of serum correlated strongly with occurrences of colorectal cancer (Zhou et al., 2019). GATA4 encodes a member of the GATA family of zinc-finger transcription factors that is associated with inflammation induced by DNA damage in mouse embryonic fibroblasts (Reyes et al., 2004; Kang et al., 2015). CD44 participates in a wide variety of cellular functions including lymphocyte activation and tumor metastasis (Dalerba et al., 2007; Schmitt et al., 2015), and CD44 plays important roles in ECM-receptor and wnt signaling pathways related to differentiation of the invasion of tumor cells (Hill et al., 2006; Wan et al., 2019) reported that the CD44 expression influenced susceptibility to colorectal cancer in humans, and that an SNP (rs187115) in CD44 was associated with an increased risk of colorectal cancer (Wan et al., 2019). TGM3 encodes an enzyme that is involved in immune regulation of cancer. (Feng et al., 2020) revealed that TGM3 potentially suppressed cell proliferation through promoting apoptosis and cell cycle regulation and activating phosphorylated AKT serine/threonine kinase to inhibit invasion and metastasis in colorectal cancer cells (Feng et al., 2020). CXCL9 has a critical role in T cell transport and recruitment of mononuclear cells and granulocytes in the host immune response (Liu et al., 2008; Elia and Guglielmi, 2018). Antonelli et al., 2014; Tokunaga et al., 2018 reported that CXCL9 expression in epithelial colonic cells interacted with immune factor IFN-γ in ulcerative colitis (Antonelli et al., 2014; Tokunaga et al., 2018). CXCL10 was associated with chronic inflammation, immune dysfunction, and dissemination of the tumor (Liu et al., 2011). It was reported that the downregulation of CXCL10 expression was connected with tumor metastasis and recurrence of colorectal cancer, and the increasing expression of CXCL10 influenced the recruitment of CD8+ T lymphocytes to tumor sites (Li et al., 2014; Zheng et al., 2016). PPARG decreased the inflammatory response and increased synthesis and release of paraoxonase1, and its activation was shown to inhibit pro-inflammatory genes via trans-repression (Straus and Glass, 2007; Khateeb et al., 2010). PPARG was also associated with beta-catenin, an important molecule in colorectal tumor carcinogenesis and C > T mutation in exon 8 that increased the risk of colorectal cancer (Michalik et al., 2004; Tontonoz and Spiegelman, 2008; Lin et al., 2019). NFATC1 encodes the NFAC1 protein that plays a role in the inducible expression of cytokine genes in T cells. NFATC1 induced IL-2 or IL-4 expression in T cells to regulate the activation differentiation and proliferation of programmed death of T lymphocytes (Huang et al., 2011). In our study, the methylation level of DMRs in CALCRL, TNC, TGM3, and PPARG significantly increased with aggravation of the MAP disease. And the CXCL9, CXCL10, and NFATC1 methylation levels of HC groups was significantly higher than CC and SC groups, indicating that the decrease of their methylation levels may be related to the occurrence of inflammation. The details of methylation level can be found in Supplementary Table S4.
Further, we have proposed the regulatory mechanism by which the immune response occurs after MAP invades the body, and how these candidate genes regulate immune pathways related to paratuberculosis in dairy cattle. Once MAP invades the bovine body, the immune response will control the infection immediately; nevertheless, it cannot completely kill MAP (Arsenault et al., 2014). In the early stage of infection, the Th1 cell-mediated immune response is rapid, with a small number of bacteria excreted from the animal body at this stage. As the cellular immunity mediated by Th1 cells weakens, the humoral immunity that is mediated by Th2 cells is gradually strengthened; the disease has progressed to the clinical stage (Spellberg and Edwards, 2001). At this stage, both the antibody level and the number of bacteria excreted from the body are significantly increased (Nielsen and Toft, 2006). In our study, we found that the nine candidate genes (CALCRL, TNC, GATA4, CD44, TGM3, CXCL9, CXCL10, PPARG, and NFATC1) participated in the main immune-related pathways associated with paratuberculosis, including Th1 and Th2 differentiation, ECM-receptor interaction, wnt, TNF, MAPK, calcium, vascular smooth muscle contraction, T cell receptor, cellular senescence, and chemokine signaling pathways. These pathways have the ability to influence the secretion of immune factors in T cells, involving IL-5, IL-2, IL-4, IL-4Ra, IL-2Ra, GATA3, IL-6, IL-12Rb, and IFN-γ. This implies that they function in transmitting intercellular information, regulating the physiological process of cells and resistance to MAP.
Conclusion
In this study, we obtained genome-wide methylomes with a single-base resolution of jejunum tissue from Holstein cows in clinical, subclinical, and healthy groups for paratuberculosis, and identified 8,432 differentially methylated genes. With integration of information about DMGs, DEGs, and biological functions, we reported nine promising candidate genes associated with resistance/susceptibility to paratuberculosis: CALCRL, TNC, GATA4, CD44, TGM3, CXCL9, CXCL10, PPARG, and NFATC1. Our findings provide new insights into the regulatory mechanism of bovine paratuberculosis and associated molecular information for gene-based improvements to the health of dairy cattle.
Data Availability Statement
We have submitted our sequencing data to the NCBI Sequence Read Archive (SRA) and the data has been reserved under BioProject accession PRJNA745069.
Ethics Statement
The animal study was reviewed and approved by All experiments were carried out in accordance with Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) at China Agricultural University (Beijing, China; permit number: DK996).
Author Contributions
JZ and WZ performed bioinformatics and statistical analysis, JZ was a major contributor to article preparation. SL, HL, and BH performed experiments and sample collection and participated in result interpretation, wrote, revised and approved the article. BH commented the article and were major contribution to article revision. DS conceived and designed the experiments and wrote the article. All authors read and approved the final article.
Funding
This work was financially supported by the National Natural Science Foundation of China (31872330, 31472065) and the Program for Changjiang Scholar and Innovation Research Team in University (IRT_15R62).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We appreciate Professor Jie Cao from the College of Veterinary Medicine, China Agricultural University for providing help with serum and stool sample detection for this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.735147/full#supplementary-material
References
Akimzhanov, A., Krenacs, L., Schlegel, T., Klein-Hessling, S., Bagdi, E., Stelkovics, E., et al. (2008). Epigenetic Changes and Suppression of the Nuclear Factor of Activated T Cell 1 (NFATC1) Promoter in Human Lymphomas with Defects in Immunoreceptor Signaling. Am. J. Pathol. 172, 215–224. doi:10.2353/ajpath.2008.070294
Antonelli, A., Ferrari, S. M., Giuggioli, D., Ferrannini, E., Ferri, C., and Fallahi, P. (2014). Chemokine (C-X-C Motif) Ligand (CXCL)10 in Autoimmune Diseases. Autoimmun. Rev. 13, 272–280. doi:10.1016/j.autrev.2013.10.010
Arsenault, R. J., Maattanen, P., Daigle, J., Potter, A., Griebel, P., and Napper, S. (2014). From Mouth to Macrophage: Mechanisms of Innate Immune Subversion by Mycobacterium avium Subsp. Paratuberculosis. Vet. Res. 45, 54. doi:10.1186/1297-9716-45-54
Bannantine, J. P., and Bermudez, L. E. (2013). No Holes Barred: Invasion of the Intestinal Mucosa by Mycobacterium avium Subsp. Paratuberculosis. Infect. Immun. 81, 3960–3965. doi:10.1128/iai.00575-13
Barry, C., Faugeron, G., and Rossignol, J. L. (1993). Methylation Induced Premeiotically in Ascobolus: Coextension with DNA Repeat Lengths and Effect on Transcript Elongation. Proc. Natl. Acad. Sci. 90, 4557–4561. doi:10.1073/pnas.90.10.4557
Benjamini, Y., and Hochberg, Y. (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodological) 57, 289–300. doi:10.1111/j.2517-6161.1995.tb02031.x
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an Ultra-fast All-In-One FASTQ Preprocessor. Bioinformatics 34, i884–i890. doi:10.1093/bioinformatics/bty560
Christie, D., and Zhu, J. (2014). Transcriptional Regulatory Networks for CD4 T Cell Differentiation. Curr. Top. Microbiol. Immunol. 381, 125–172. doi:10.1007/82_2014_372
Clarke, C. J. (1997). The Pathology and Pathogenesis of Paratuberculosis in Ruminants and Other Species. J. Comp. Pathol. 116, 217–261. doi:10.1016/s0021-9975(97)80001-1
Dalerba, P., Dylla, S. J., Park, I.-K., Liu, R., Wang, X., Cho, R. W., et al. (2007). Phenotypic Characterization of Human Colorectal Cancer Stem Cells. Proc. Natl. Acad. Sci. 104, 10158–10163. doi:10.1073/pnas.0703478104
Debaun, M. R., Niemitz, E. L., and Feinberg, A. P. (2003). Association of In Vitro Fertilization with Beckwith-Wiedemann Syndrome and Epigenetic Alterations of LIT1 and H19. Am. J. Hum. Genet. 72, 156–160. doi:10.1086/346031
Del Corvo, M., Bongiorni, S., Stefanon, B., Sgorlon, S., Valentini, A., Ajmone Marsan, P., et al. (2020). Genome-Wide DNA Methylation and Gene Expression Profiles in Cows Subjected to Different Stress Level as Assessed by Cortisol in Milk. Genes 11, 850. doi:10.3390/genes11080850
Dong, Y.-L., Green, K. E., Vegiragu, S., Hankins, G. D. V., Martin, E., Chauhan, M., et al. (2005). Evidence for Decreased Calcitonin Gene-Related Peptide (CGRP) Receptors and Compromised Responsiveness to CGRP of Fetoplacental Vessels in Preeclamptic Pregnancies. J. Clin. Endocrinol. Metab. 90, 2336–2343. doi:10.1210/jc.2004-1481
Elia, G., and Guglielmi, G. (2018). CXCL9 Chemokine in Ulcerative Colitis. Clin. Ter 169, e235–e241. doi:10.7417/CT.2018.2085
Feng, Y., Ji, D., Huang, Y., Ji, B., Zhang, Y., Li, J., et al. (2020). TGM3 Functions as a Tumor Suppressor by Repressing Epithelial-to-mesenchymal T-ransition and the PI3K/AKT S-ignaling P-athway in C-olorectal C-ancer. Oncol. Rep. 43, 864–876. doi:10.3892/or.2020.7474
Fu, Y., Li, J., Tang, Q., Zou, C., Shen, L., Jin, L., et al. (2018). Integrated Analysis of Methylome, Transcriptome and miRNAome of Three Pig Breeds. Epigenomics 10, 597–612. doi:10.2217/epi-2017-0087
Gao, F., Zhang, J., Jiang, P., Gong, D., Wang, J.-W., Xia, Y., et al. (2014). Marked Methylation Changes in Intestinal Genes during the Perinatal Period of Preterm Neonates. BMC Genomics 15, 716. doi:10.1186/1471-2164-15-716
Gao, Y., Cao, J., Zhang, S., Zhang, Q., and Sun, D. (2018a). Short Communication: Heritability Estimates for Susceptibility to Mycobacterium avium Ssp. Paratuberculosis Infection in Chinese Holstein Cattle. J. Dairy Sci. 101, 7274–7279. doi:10.3168/jds.2017-13264
Gao, Y., Jiang, J., Yang, S., Cao, J., Han, B., Wang, Y., et al. (2018b). Genome-wide Association Study of Mycobacterium avium Subspecies Paratuberculosis Infection in Chinese Holstein. BMC Genomics 19, 972. doi:10.1186/s12864-018-5385-3
Garvey, M. (2018). Mycobacterium avium Subspecies Paratuberculosis: A Possible Causative Agent in Human Morbidity and Risk to Public Health Safety. Open Vet. J. 8, 172–181. doi:10.4314/ovj.v8i2.10
Hansen, K. D., Langmead, B., and Irizarry, R. A. (2012). BSmooth: from Whole Genome Bisulfite Sequencing Reads to Differentially Methylated Regions. Genome Biol. 13, R83. doi:10.1186/gb-2012-13-10-r83
Hao, Y., Cui, Y., and Gu, X. (2016). Genome-wide DNA Methylation Profiles Changes Associated with Constant Heat Stress in Pigs as Measured by Bisulfite Sequencing. Sci. Rep. 6, 27507. doi:10.1038/srep27507
Hempel, R. J., Bannantine, J. P., and Stabel, J. R. (2016). Transcriptional Profiling of Ileocecal Valve of Holstein Dairy Cows Infected with Mycobacterium avium Subsp. Paratuberculosis. PLoS ONE 11, e0153932. doi:10.1371/journal.pone.0153932
Hernandez-Garcia, C. M., and Finer, J. J. (2014). Identification and Validation of Promoters and Cis-Acting Regulatory Elements. Plant Sci. 217-218, 109–119. doi:10.1016/j.plantsci.2013.12.007
Hill, A., Mcfarlane, S., Johnston, P. G., and Waugh, D. J. J. (2006). The Emerging Role of CD44 in Regulating Skeletal Micrometastasis. Cancer Lett. 237, 1–9. doi:10.1016/j.canlet.2005.05.006
Hohn, T., Corsten, S., Rieke, S., Müller, M., and Rothnie, H. (1996). Methylation of Coding Region Alone Inhibits Gene Expression in Plant Protoplasts. Proc. Natl. Acad. Sci. 93, 8334–8339. doi:10.1073/pnas.93.16.8334
Huang, T., Xie, Z., Wang, J., Li, M., Jing, N., and Li, L. (2011). Nuclear Factor of Activated T Cells (NFAT) Proteins Repress Canonical Wnt Signaling via its Interaction with Dishevelled (Dvl) Protein and Participate in Regulating Neural Progenitor Cell Proliferation and Differentiation. J. Biol. Chem. 286, 37399–37405. doi:10.1074/jbc.m111.251165
Ibeagha-Awemu, E. M., Wang, M., Bissonnette, N., Griebel, P., Dudemaine, P.-L., Do, D. N., et al. (2019). 23 Differential microRNA Expression in Jejunal Tissue and Jejunal Lymph Nodes Following Naturally Occurring Mycobacterium avium Subspecies Paratuberculosis Infection in Holstein Cows. J. Anim. Sci. 97, 20–21. doi:10.1093/jas/skz258.038
Jin, B., Li, Y., and Robertson, K. D. (2011). DNA Methylation: superior or Subordinate in the Epigenetic Hierarchy? Genes & Cancer 2, 607–617. doi:10.1177/1947601910393957
Kang, C., Xu, Q., Martin, T. D., Li, M. Z., Demaria, M., Aron, L., et al. (2015). The DNA Damage Response Induces Inflammation and Senescence by Inhibiting Autophagy of GATA4. Science 349, aaa5612. doi:10.1126/science.aaa5612
Khateeb, J., Gantman, A., Kreitenberg, A. J., Aviram, M., and Fuhrman, B. (2010). Paraoxonase 1 (PON1) Expression in Hepatocytes Is Upregulated by Pomegranate Polyphenols: A Role for PPAR-γ Pathway. Atherosclerosis 208, 119–125. doi:10.1016/j.atherosclerosis.2009.08.051
Krueger, F., and Andrews, S. R. (2011). Bismark: a Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinformatics 27, 1571–1572. doi:10.1093/bioinformatics/btr167
Langmead, B., and Salzberg, S. L. (2012). Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 9, 357–359. doi:10.1038/nmeth.1923
Langmead, B., Wilks, C., Antonescu, V., and Charles, R. (2018). Scaling Read Aligners to Hundreds of Threads on General-Purpose Processors. Bioinformatics 35, 421–432. doi:10.1093/bioinformatics/bty648
Li, C., Wang, Z., Liu, F., Zhu, J., Yang, L., Cai, G., et al. (2014). CXCL10 mRNA Expression Predicts Response to Neoadjuvant Chemoradiotherapy in Rectal Cancer Patients. Tumor Biol. 35, 9683–9691. doi:10.1007/s13277-014-2234-0
Lin, J., Chen, Y., Tang, W. F., Liu, C., Zhang, S., Guo, Z. Q., et al. (2019). PPARG Rs3856806 C>T Polymorphism Increased the Risk of Colorectal Cancer: A Case-Control Study in Eastern Chinese Han Population. Front. Oncol. 9, 63. doi:10.3389/fonc.2019.00063
Liu, C., Papewalis, C., Domberg, J., Scherbaum, W., and Schott, M. (2008). Chemokines and Autoimmune Thyroid Diseases. Horm. Metab. Res. 40, 361–368. doi:10.1055/s-2008-1073153
Liu, M., Guo, S., and Stiles, J. K. (2011). The Emerging Role of CXCL10 in Cancer (Review). Oncol. Lett. 2, 583–589. doi:10.3892/ol.2011.300
Michalik, L., Desvergne, B., and Wahli, W. (2004). Peroxisome-proliferator-activated Receptors and Cancers: Complex Stories. Nat. Rev. Cancer 4, 61–70. doi:10.1038/nrc1254
Miller, J. L., and Grant, P. A. (2013). The Role of DNA Methylation and Histone Modifications in Transcriptional Regulation in Humans. Subcell Biochem. 61, 289–317. doi:10.1007/978-94-007-4525-4_13
Moore, L. E., Pfeiffer, R. M., Poscablo, C., Real, F. X., Kogevinas, M., Silverman, D., et al. (2008). Genomic DNA Hypomethylation as a Biomarker for Bladder Cancer Susceptibility in the Spanish Bladder Cancer Study: a Case-Control Study. Lancet Oncol. 9, 359–366. doi:10.1016/s1470-2045(08)70038-x
Nielsen, S. S., and Toft, N. (2006). Age-specific Characteristics of ELISA and Fecal Culture for Purpose-specific Testing for Paratuberculosis. J. Dairy Sci. 89, 569–579. doi:10.3168/jds.s0022-0302(06)72120-8
Ott, S. L., Wells, S. J., and Wagner, B. A. (1999). Herd-level Economic Losses Associated with Johne's Disease on US Dairy Operations. Prev. Vet. Med. 40, 179–192. doi:10.1016/s0167-5877(99)00037-9
Prawitt, D., Enklaar, T., Gärtner-Rupprecht, B., Spangenberg, C., Oswald, M., Lausch, E., et al. (2005). Microdeletion of Target Sites for Insulator Protein CTCF in a Chromosome 11p15 Imprinting center in Beckwith-Wiedemann Syndrome and Wilms' Tumor. Proc. Natl. Acad. Sci. 102, 4085–4090. doi:10.1073/pnas.0500037102
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 26, 841–842. doi:10.1093/bioinformatics/btq033
Reyes, J. C., Muro-Pastor, M. I., and Florencio, F. J. (2004). The GATA Family of Transcription Factors in Arabidopsis and rice. Plant Physiol. 134, 1718–1732. doi:10.1104/pp.103.037788
Rountree, M. R., and Selker, E. U. (1997). DNA Methylation Inhibits Elongation but Not Initiation of Transcription in Neurospora Crassa. Genes Development 11, 2383–2395. doi:10.1101/gad.11.18.2383
Schmitt, M., Metzger, M., Gradl, D., Davidson, G., and Orian-Rousseau, V. (2015). CD44 Functions in Wnt Signaling by Regulating LRP6 Localization and Activation. Cell Death Differ 22, 677–689. doi:10.1038/cdd.2014.156
Schober, P., Boer, C., and Schwarte, L. A. (2018). Correlation Coefficients. Anesth. Analgesia 126, 1763–1768. doi:10.1213/ane.0000000000002864
Sharifi-Zarchi, A., Gerovska, D., Adachi, K., Totonchi, M., Pezeshk, H., Taft, R. J., et al. (2017). DNA Methylation Regulates Discrimination of Enhancers from Promoters through a H3K4me1-H3K4me3 Seesaw Mechanism. BMC Genomics 18, 964. doi:10.1186/s12864-017-4353-7
Song, Q.-X., Lu, X., Li, Q.-T., Chen, H., Hu, X.-Y., Ma, B., et al. (2013). Genome-wide Analysis of DNA Methylation in Soybean. Mol. Plant 6, 1961–1974. doi:10.1093/mp/sst123
Spellberg, B., and Edwards, J. E. (2001). Type 1/Type 2 Immunity in Infectious Diseases. Clin. Infect. Dis. 32, 76–102. doi:10.1086/317537
Stevens, M., Cheng, J. B., Li, D., Xie, M., Hong, C., Maire, C. L., et al. (2013). Estimating Absolute Methylation Levels at Single-CpG Resolution from Methylation Enrichment and Restriction Enzyme Sequencing Methods. Genome Res. 23, 1541–1553. doi:10.1101/gr.152231.112
Storey, J. D. (2003). The Positive False Discovery Rate: A Bayesian Interpretation and the Q-Value. Ann. Stat. 31, 2013–2035. doi:10.1214/aos/1074290335
Straus, D. S., and Glass, C. K. (2007). Anti-inflammatory Actions of PPAR Ligands: New Insights on Cellular and Molecular Mechanisms. Trends Immunol. 28, 551–558. doi:10.1016/j.it.2007.09.003
Tokunaga, R., Zhang, W., Naseem, M., Puccini, A., Berger, M. D., Soni, S., et al. (2018). CXCL9, CXCL10, CXCL11/CXCR3 axis for Immune Activation - A Target for Novel Cancer Therapy. Cancer Treat. Rev. 63, 40–47. doi:10.1016/j.ctrv.2017.11.007
Tontonoz, P., and Spiegelman, B. M. (2008). Fat and beyond: The Diverse Biology of PPARγ. Annu. Rev. Biochem. 77, 289–312. doi:10.1146/annurev.biochem.77.061307.091829
Verschoor, C. P., Pant, S. D., You, Q., Schenkel, F. S., Kelton, D. F., and Karrow, N. A. (2010). Polymorphisms in the Gene Encoding Bovine Interleukin-10 Receptor Alpha Are Associated with Mycobacterium avium Ssp. Paratuberculosis Infection Status. BMC Genet. 11, 23. doi:10.1186/1471-2156-11-23
Wan, Q., Zhang, D., Zhou, Q., Li, M., Wang, Y., Song, Y., et al. (2019). Association of CD44 Gene Rs187115 Polymorphism with Colorectal Cancer Risk and Prognosis in Chinese Han Population: a Case-Control Study. Aging 11, 9616–9625. doi:10.18632/aging.102408
Weksberg, R., Nishikawa, J., Caluseriu, O., Fei, Y. L., Shuman, C., Wei, C., et al. (2001). Tumor Development in the Beckwith-Wiedemann Syndrome Is Associated with a Variety of Constitutional Molecular 11p15 Alterations Including Imprinting Defects of KCNQ1OT1. Hum. Mol. Genet. 10, 2989–3000. doi:10.1093/hmg/10.26.2989
Wentink, G. H., Bongers, J. H., Zeeuwen, A. A. P. A., and Jaartsveld, F. H. J. (1994). Incidence of Paratuberculosis after Vaccination against M. Paratuberculosis in Two Infected Dairy Herds. Zentralbl Veterinarmed B 41, 517–522. doi:10.1111/j.1439-0450.1994.tb00258.x
Whitlock, R. H., and Buergelt, C. (1996). Preclinical and Clinical Manifestations of Paratuberculosis (Including Pathology). Vet. Clin. North America: Food Anim. Pract. 12, 345–356. doi:10.1016/s0749-0720(15)30410-2
Yang, Y., Fan, X., Yan, J., Chen, M., Zhu, M., Tang, Y., et al. (2021). A Comprehensive Epigenome Atlas Reveals DNA Methylation Regulating Skeletal Muscle Development. Nucleic Acids Res. 49, 1313–1329. doi:10.1093/nar/gkaa1203
Yu, C.-P., Lin, J.-J., and Li, W.-H. (2016). Positional Distribution of Transcription Factor Binding Sites in Arabidopsis thaliana. Sci. Rep. 6, 25164. doi:10.1038/srep25164
Yu, G., Wang, L.-G., Han, Y., and He, Q.-Y. (2012). clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS: A J. Integr. Biol. 16, 284–287. doi:10.1089/omi.2011.0118
Zheng, H., Zhao, W., Yan, C., Watson, C. C., Massengill, M., Xie, M., et al. (2016). HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 22, 4119–4132. doi:10.1158/1078-0432.ccr-15-2584
Keywords: paratuberculosis, DNA methylation, candidate gene, regulatory mechanism, whole-genome bisulfite sequencing, dairy cattle
Citation: Zhang J, Han B, Zheng W, Lin S, Li H, Gao Y and Sun D (2021) Genome-Wide DNA Methylation Profile in Jejunum Reveals the Potential Genes Associated With Paratuberculosis in Dairy Cattle. Front. Genet. 12:735147. doi: 10.3389/fgene.2021.735147
Received: 02 July 2021; Accepted: 23 September 2021;
Published: 15 October 2021.
Edited by:
Alaguraj Veluchamy, St. Jude Children’s Research Hospital, United StatesReviewed by:
Tang Zhonglin, Agricultural Genomics Institute at Shenzhen (CAAS), ChinaBohao Zhao, Yangzhou University, China
Copyright © 2021 Zhang, Han, Zheng, Lin, Li, Gao and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongxiao Sun, c3VuZHhAY2F1LmVkdS5jbg==