 Youjia Zhang
Youjia Zhang Xueli Chen1
Xueli Chen1 Li Wang
Li Wang Xinghuai Sun
Xinghuai Sun Yuhong Chen
Yuhong Chen- 1Department of Ophthalmology and Visual Science, Eye and ENT Hospital, Shanghai Medical College, Fudan University, Shanghai, China
- 2NHC Key Laboratory of Myopia, Chinese Academy of Medical Sciences, and Shanghai Key Laboratory of Visual Impairment and Restoration, Fudan University, Shanghai, China
- 3State Key Laboratory of Medical Neurobiology and MOE Frontiers Center for Brain Science, Institutes of Brain Science, Fudan University, Shanghai, China
Axenfeld–Rieger Syndrome (ARS) is a rare disease with a wide spectrum of ocular and systemic manifestations. The genetic spectrum of Chinese patients with ARS and genotype-phenotype correlations have yet to be described. To explore the molecular and clinical features in Chinese patients, fifty-five patients clinically diagnosed with ARS from independent families were recruited. Complete ophthalmic examinations and next generation sequencing of anterior segment dysgenesis associated genes were performed in all patients, and segregation in available relatives was verified using Sanger sequencing. 18 FOXC1 variants, 13 PITX2 variants, and two gross deletions spanning FOXC1 were detected in 35 out of 55 (63.6%) patients. 12 FOXC1 variants, 9 PITX2 variants, and two gross deletions were novel. There was a wide range of variability and severity in ocular and systemic manifestations displayed in our patients. Patients with FOXC1 variants were diagnosed at a younger age and had a lower prevalence of systemic manifestations than patients harboring PITX2 variants and those without variants. To our best knowledge, this is the largest study of Chinese patients with ARS to date. Our findings expand the genetic spectrum of ARS and reveal genotype-phenotype correlations in Chinese patients with ARS. Genetic and clinical heterogeneity were present in our patients. Awareness of the extensive characterization may aid in the clinical management and genetic counseling of patients with this rare disease.
Introduction
Axenfeld–Rieger Syndrome (ARS; OMIM: 180500, 601499, 602482) is an autosomal dominant developmental disorder that has both clinical and genetic heterogeneity, and primarily affects the anterior segment structure of the eye (Strungaru et al., 2007; Reis et al., 2012). The prevalence of ARS has been estimated to be one in 200,000 individuals (Lewis et al., 2017). ARS comprises a subgroup of anterior segment dysgenesis (ASD) and refers to a constellation of ocular abnormalities, including posterior embryotoxon, iris hypoplasia, corectopia, polycoria, and iridocorneal adhesions (Strungaru et al., 2007; Souzeau et al., 2017). Because of its anomalous anterior segment features, glaucoma is the most serious consequence of ARS, which can lead to irreversible vision loss or even complete blindness (Lewis et al., 2017; Souzeau et al., 2017). Approximately 50% of patients with ARS will develop glaucoma, and treating these patients is difficult (Strungaru et al., 2007). Aside from ocular anomalies, systemic problems also often exist, which typically include dental anomalies, craniofacial abnormalities, and redundant periumbilical skin (Tumer and Bach-Holm 2009; Seifi and Walter 2018). Hearing loss, heart defects, developmental delay, and other variable manifestations have also been reported in patients with ARS (Reis et al., 2012; Seifi and Walter 2018).
FOXC1 and PITX2 are two major causative genes of ARS. They are estimated to explain disease pathogenesis in approximately 40–63% of patients with ARS (D’Haene et al., 2011; Reis et al., 2012; Wang et al., 2018; Souzeau et al., 2017). In addition, candidate loci at 13q14 and 16q24 have also been found to be associated with ARS, but no underlying genes have been detected (Chang et al., 2012). FOXC1 is a member of the large forkhead box (FOX) transcription factor family, whereas PITX2 is a member of bicoid-like homeodomain transcription factor family. FOXC1 and PITX2 are both transcription factors that are coexpressed in the periocular mesenchyme and play key roles in the regulation of embryonic development (Berry et al., 2006).
To date, only limited studies with small sample sizes have been reported in Chinese individuals with ARS. The genotype and phenotype spectrums of Chinese patients with ARS are yet to be described. In this study, we performed a clinical and genetic investigation of 55 Chinese patients with ARS from independent families. We described their clinical ophthalmologic features, presented novel variants in FOXC1 and PITX2 genes, and established the genetic and clinical heterogeneity in the present Chinese cohort of patients with ARS.
Materials and Methods
Patients
We consecutively recruited 55 patients from independent families that were diagnosed with ARS at the Ophthalmology Department of the Eye and ENT Hospital of Fudan University between December 2004 and June 2020. Both sporadic and familial patients were included. This study was approved by the Institutional Review Board of the Eye and ENT Hospital of Fudan University, and written informed consent was obtained from all patients or their legal guardians. ARS was diagnosed as a group of distinctive ocular features, including malformations of the anterior chamber angle, posterior embryotoxon, iridocorneal adhesions traversing the anterior chamber, corectopia, polycoria, and iris hypoplasia (Alward 2000; Lines et al., 2002). Glaucoma was diagnosed as the presence of at least two of the following criteria: high intraocular pressure (≥ 22 mmHg), glaucomatous optic disc damage, or glaucomatous visual fields defects (Strungaru et al., 2007).
Clinical Investigation
The family histories and detailed medical histories of the patients were recorded, including the onset and diagnosis ages of ARS and glaucoma, ocular and systemic manifestations, and treatments and their effects. All patients underwent a complete ophthalmologic examination, including visual acuity examination, slit-lamp biomicroscopy, ophthalmoscopy, color fundus photography, gonioscopy, B-mode ultrasonography, A-mode ultrasonography, intraocular pressure (IOP) measurement (Goldmann Applanation Tonometer or Tono-PEN; Reichert, Depew, NY, United States), and ultrasound biomicroscopy (UBM, MD-300L; MEDA Co., Tianjin, China). Perimetry was performed in cooperative children and adults using the Octopus 101 (Haag-Streit, Inc., Köniz, Switzerland) or Humphrey Visual Field Analyzer 750 (Zeiss Humphrey Systems, Dublin, CA, United States).
Genetic Analysis
Whole blood samples of all patients and available relatives were collected for genomic DNA extraction by Gentra PureGene blood kits (Qiagen, Valencia, CA, United States) according to the manufacturer’s instructions. Genetic testing was performed in 55 patients using next generation sequencing between October 2016 and June 2020. A panel of 289 genes associated with ASD disorders was sequenced by the Illumina Miseq platform (Illumina, San Diego, CA, United States) with the 2 × 300 bp paired-end read module. The average depth was 100x, and 90% of the target region was covered above 40x. Low quality bases (< Q20) were removed using SolexaQA (Cox et al., 2010).
Whole genome sequencing was performed in two patients. In brief, a total of 0.2 μg DNA per sample was fragmented using sonication to a size of 350 bp. The DNA fragments then underwent end-repairing and A-tailing, and ligation was performed with the full-length adapters for Illumina sequencing, followed by polymerase chain reaction amplification and purification. After library quality assessment, clustering of the index-coded samples was performed on the cBot Cluster Generation System using the Illumina PE Cluster Kit (Illumina), and DNA libraries were sequenced on the Illumina platform and 150 bp paired-end reads were generated.
Reads were aligned to the hg19 human reference genome using the Burrows–Wheeler Aligner (BWA; ver. 0.7.11) (Li and Durbin 2009). The detected variants were annotated using ANNOVAR (Wang et al., 2010) and the following databases: the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php), Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/), and 1,000 Genomes Project (https://www.internationalgenome.org/). The copy number variants were analyzed by calculating the sequencing depth of each region covered by probes. The ExomeDepth Package (Plagnol et al., 2012) was also used to find potential copy number variants. The reference sequences of NM_001453.2 (FOXC1) and NM_153427.2 (PITX2) were used for mutation nomenclature. Novel variants were classified into five categories according to the American College of Medical Genetics and Genomics (ACMG) guidelines: pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, and benign (Richards et al., 2015). Conservation of the novel variant sites was evaluated using Clustal Omega (Sievers et al., 2011). Polymorphism Phenotyping 2 (PolyPhen2) (Adzhubei et al., 2010), Sorting Intolerant from Tolerant (SIFT) (Kumar et al., 2009), and Provean (Choi and Chan 2015) were applied for the assessment of the pathogenicity of detected missense variants. All detected variants were confirmed using Sanger sequencing, and the segregation on available family members was also verified using Sanger sequencing.
Statistical Analysis
Statistical analyses were performed using SPSS version 20.0 (IBM-SPSS, Chicago, IL, United States). Age is presented as the median (range). Patients were classified into three groups according to their genotypes. Differences in age with non-normal distributions were assessed using the Kruskal-Wallis test with Dunn’s post-hoc test. Differences between categorical variables among the three groups were assessed using chi-square test or Fisher’s exact test with Bonferroni correction for post-hoc multiple comparisons. The statistical significance was set at p-value of 0.05.
Results
Patients
Fifty-five Chinese patients with ARS from independent families were enrolled in our study. These included 22 females (40.0%) and 33 males (60.0%). The median age at diagnosis was 14.0 (0.1–64.0) years. 25 patients (45.5%) had family histories of ARS or glaucoma. 53 patients (96.4%) were diagnosed with glaucoma and 36 patients (65.5%) had systemic features.
Genetic Analysis
In total, 33 variants were identified in 35 patients, including 18 variants in FOXC1, 13 variants in PITX2 and two gross deletions of 6p25 (Figures 1, 2). Among them, 12 FOXC1 variants, 9 PITX2 variants, and two gross deletions were novel. All detected variants were heterozygous. The variant detection rate was 63.6% (35/55), with 19 patients carrying the FOXC1 variants, 14 patients carrying the PITX2 variants, and two patients carrying gross deletions of 6p25. No variants were found in the remaining 20 patients. Among the 35 patients in whom variants were detected, six patients carried de novo variants, 11 patients carried heterozygous variants inherited from their parents, and data of parents of 18 patients were unavailable.
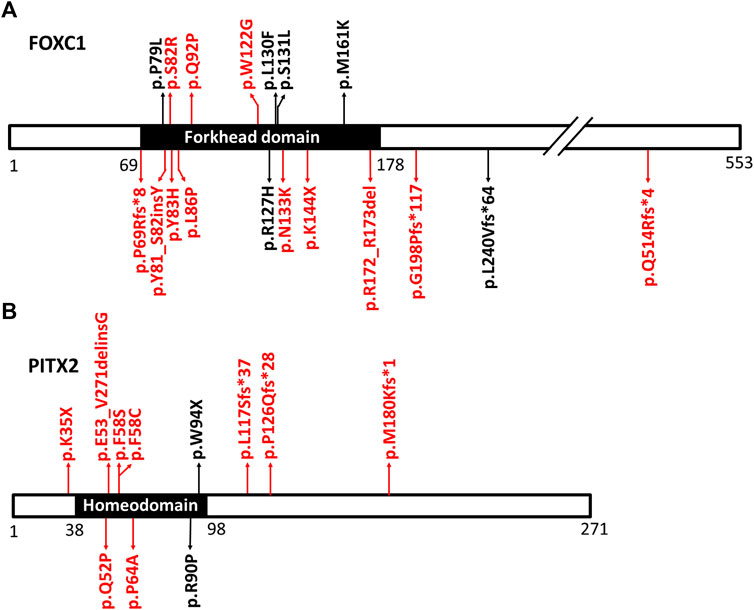
FIGURE 1. The positions of the detected variants in FOXC1 and PITX2. (A) is the diagram of FOXC1 protein and its variants. (B) is the diagram of PITX2 protein and its variants. The main function domains (forkhead domain in FOXC1 protein and homeodomain in PITX2 protein) are shown in black. The positions of the variants in FOXC1 and PITX2 are indicated by arrows. The novel variants are shown in red. Gross deletions spanning FOXC1 and splicing variants in PITX2 were not shown in Figure 1.
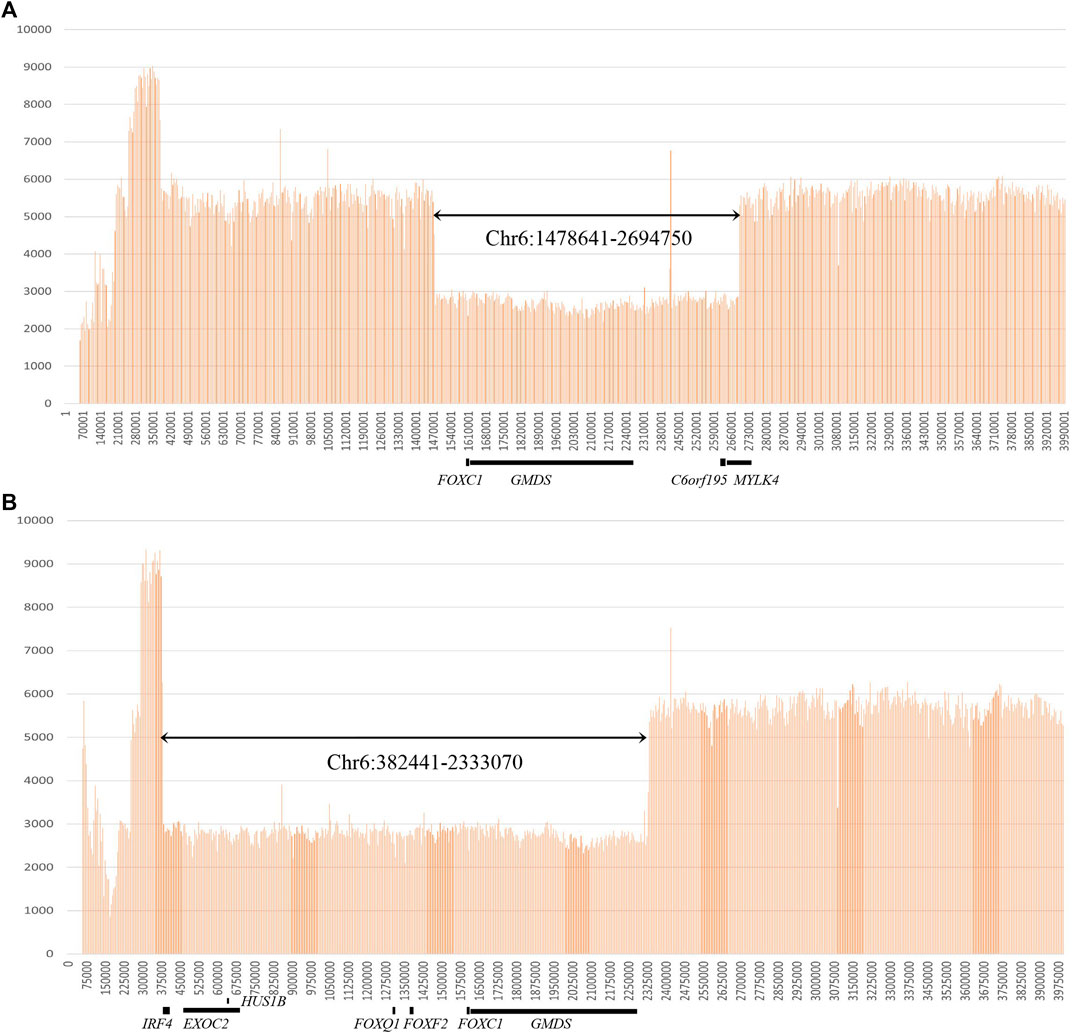
FIGURE 2. Gross deletions identified by whole-genome sequencing. (A) Genome sequencing read depth at the gross deletion (chr6:1478641-2694750) for patient No. 20. (B) Genome sequencing read depth at the gross deletion (chr6: 382441-2333070) for patient No. 21.
Eighteen FOXC1 mutations and two gross deletions of 6p25 spanning FOXC1 were detected in 21 patients (21/55, 38.2%) (Table 1). Most of the FOXC1 variants (15/18) were located in the forkhead domain, which is the DNA interaction function domain of FOXC1 (Figure 1). Six of eighteen FOXC1 variants have been previously described (Nishimura et al., 1998; Kawase et al., 2001; Nishimura et al., 2001; Panicker et al., 2002; Cella et al., 2006; Strungaru et al., 2007). Other 12 FOXC1 variants were novel variants, including p.P69Rfs*8, p.Y81_S82insY, p.S82R, p.Y83H, p.L86P, p.Q92P, p.W122G, p.N133K, p.K144X, p.R172_R173del, p.G198Pfs*117, and p.Q514Rfs*4. Ten of them were classified as pathogenic or likely pathogenic and two of them were classified as VUS according to the ACMG classification. All novel missense mutations were predicted to be highly deleterious to the structure and function of FOXC1 by PolyPhen2, SIFT, and/or Provean (Table 1), located at the evolutionary conserved positions of FOXC1 corresponding to multiple sequence alignment across species. Heterozygous deletions spanning whole FOXC1 gene were detected in two patients (No. 20 and No. 21) who presented with both ocular and systemic disorders with very early onset ages. To further determine the exact range of gross deletions in these two patients, whole-genome sequencing was used. By checking the depth of coverage across the whole genome, a large deletion (Chr 6:1478641-2694750) spanning FOXC1, GMDS, C6orf195, and MYLK4 was detected in patient No. 20, and a large deletion (Chr 6:382441-2333070) spanning IRF4, EXOC2, HUS1B, FOXQ1, FOXF2, FOXC1, and GMDS was detected in patient No. 21 (Figure 2).
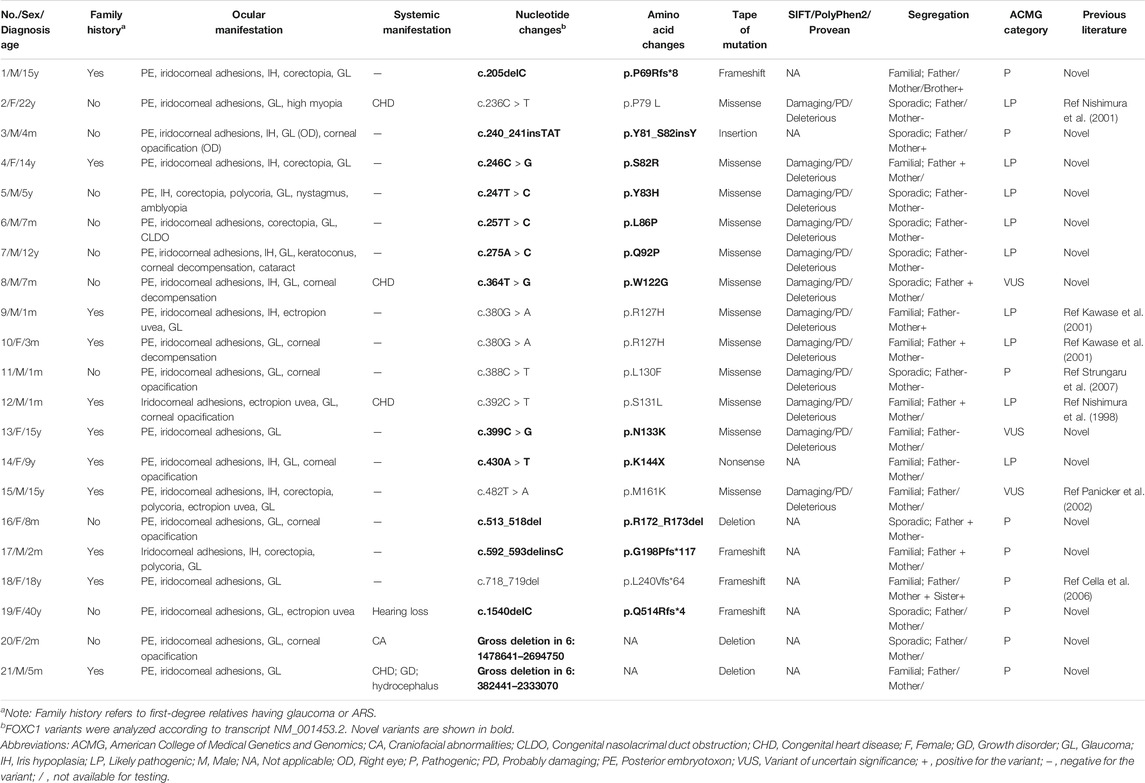
TABLE 1. The genotype and phenotype of ARS patients with FOXC1 mutations or gross deletions of 6p25.
We also identified 13 PITX2 variants in 14 patients (25.5%, 14/55) (Table 2), including five missense mutations, three nonsense mutations, two splice site mutations, and three deletions. Four of these have been previously reported (D’Haene et al., 2011; Phillips 2002; Reis et al., 2012; Semina et al., 1996). Most of the variants were located in the homeodomain, which is the DNA binding domain of PITX2 (Figure 1). Nine of these PITX2 variants were novel variants, including p.K35X, p.Q52P, p.E53_V271delinsG, p.F58C, p.F58S, p.P64A, p.L117Sfs*37, p.P126Qfs*28, and p.M180Kfs*1. All of them were classified as pathogenic or likely pathogenic according to the ACMG guidelines. Four novel missense mutations were located in highly conserved residues and were predicted to be damaging using Provean, PolyPhen2, and/or SIFT.
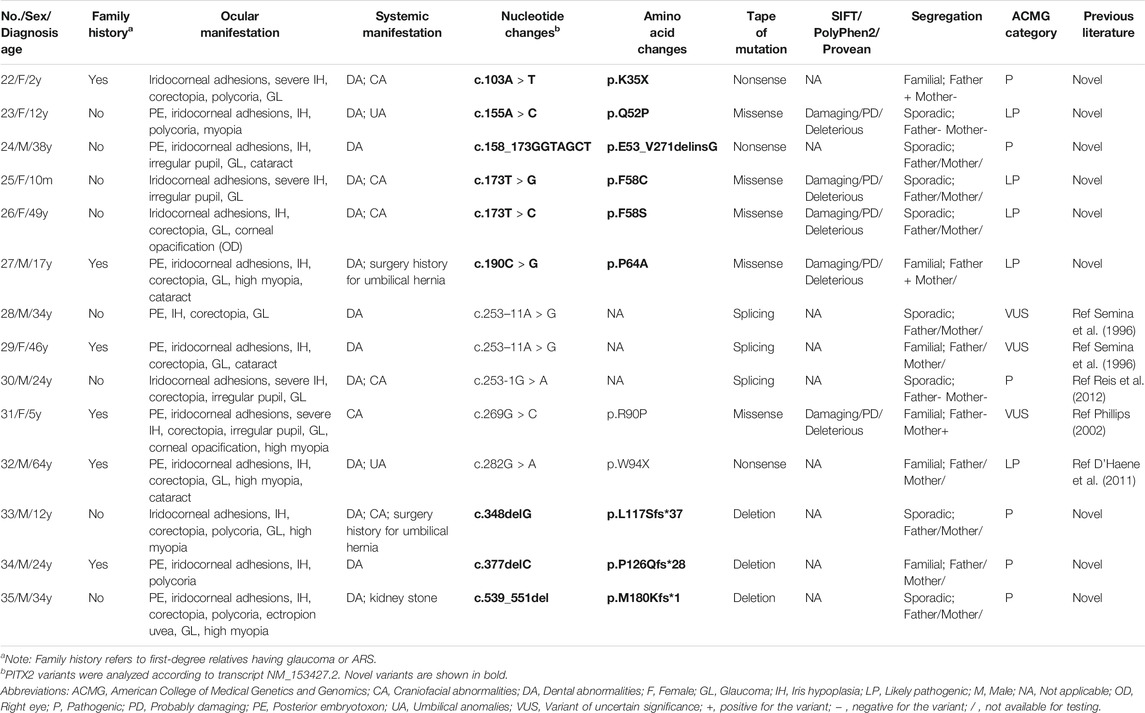
TABLE 2. The genotype and phenotype of ARS patients with PITX2 mutations.
Clinical Manifestations and Genotype-Phenotype Correlations
Except for the two patients carrying gross deletions, other patients were divided into three groups: FOXC1 group (patients carrying FOXC1 variants, n = 19), PITX2 group (patients carrying PITX2 variants, n = 14), and negative group (patients without FOXC1 or PITX2 variants, n = 20). The median age at diagnosis was 5.0 (0.1–40.0) years in the FOXC1 group, 24.0 (0.8–64.0) years in the PITX2 group, and 15.0 (0.3–46.0) years in the negative group. The diagnosis age of the FOXC1 group was significantly lesser than that of the PITX2 group (p = 0.006) and the negative group (p = 0.048). However, there was no significant difference in the diagnosis age between PITX2 group and the negative group (p > 0.99). The ratio of patients that were diagnosed before the age of 1 year was statistically significantly different among the three groups (p = 0.007). It was 47.4% (9/19) in the FOXC1 group, but only 7.1% (1/14) in the PITX2 group and 10.0% (2/20) in the negative group.
The patients presented different degrees of anterior chamber and angle anomalies. Iridocorneal adhesion was the most common ocular feature (Tables 1–3), and this presented as iridocorneal tissue adhesions across the anterior chamber angle, observed by gonioscope or UBM (Figure 3). Posterior embryotoxon, which refers to a prominent and centrally displaced Schwalbe’s line (Lines et al., 2002), was another important characteristic (Figure 3). Iris hypoplasia, corectopia, pupillary anomalies, and polycoria were also frequently seen in the patients. Other ocular manifestations included corneal opacification, cataract, corneal decompensation, congenital lacrimal duct obstruction, high myopia, amblyopia, nystagmus, retinal detachment, and exotropia.
TABLE 3. The genotype and phenotype of ARS patients without FOXC1 or PITX2 mutations.
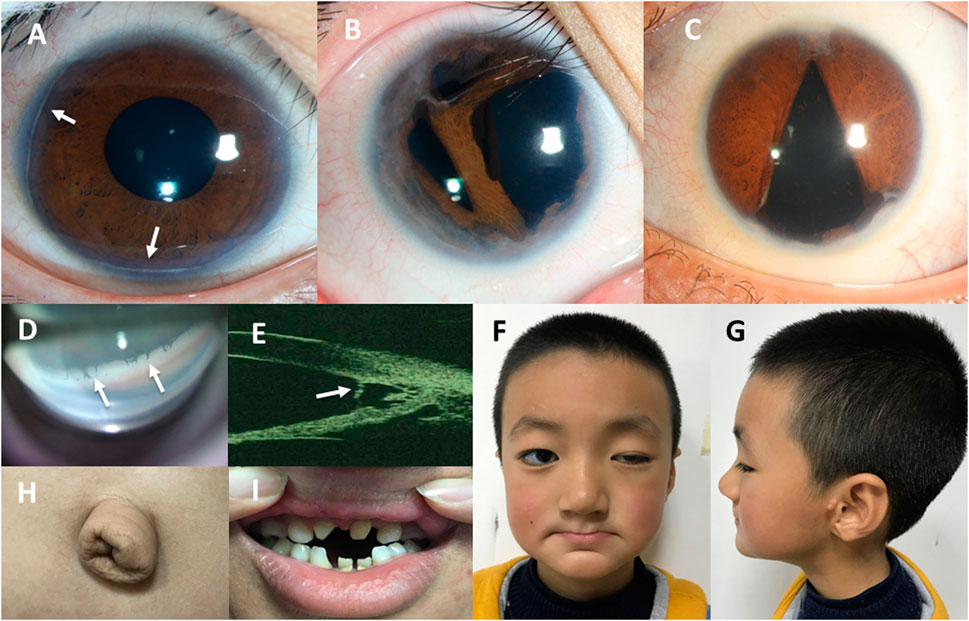
FIGURE 3. Ocular features and systemic manifestations in our patients with ARS. (A) The anterior segment photo of patient No. 13 shows the posterior embryotoxon indicated by white arrowheads. (B) The anterior segment photo of patient No. 23 shows iris stromal hypoplasia and polycoria. (C) The anterior segment photo of patient No. 45 shows irregular pupil and corectopia. (D) The photo of anterior chamber angle under gonioscopy of patient No. 16 shows the iridocorneal adhesions across the anterior chamber angle indicated by white arrowheads. (E) The ultrasound biomicroscopy (UBM) image of the anterior chamber angle of patient No. 44 shows the iris strands bridging the iris to the posterior embryotoxon indicated by a white arrowhead. (F–H). The photographs of patient No. 37 shows craniofacial abnormalities (a broad flat nasal root, maxillary hypoplasia, thin upper lip and everted lower lip) (F,G) and redundant periumbilical skin (H). (I) The photograph of patient No. 41 shows dental anomalies (hypodontia and microdontia).
As the most frequent consequence of ARS, glaucoma was found in 53 patients (96.4%). Apart from one patient (No. 3) with p.Y81_S82insY in FOXC1 gene who presented with unilateral glaucoma, all others presented with bilateral glaucoma. Among the patients with glaucoma, 15.1% (8/53) received medication only, 84.9% (45/53) received both medication and surgical treatments, and 60.4% (32/53) underwent at least two surgeries for glaucoma. Three patients carrying FOXC1 variants (No. 7, No. 8, and No. 10) underwent penetrating keratoplasties because of corneal decompensation.
Furthermore, 36 (65.5%) patients presented with systemic anomalies. The most common systemic manifestations detected in our patients were dental and craniofacial abnormalities. Tooth agenesis presented as hypodontia and microdontia (Figure 3). The craniofacial abnormalities mainly presented as a broad flat nasal root, thin upper lip, and an everted lower lip (Figure 3), which could be seen in almost every ARS patient with facial abnormalities. Other craniofacial features presented as hypertelorism, telecanthus, prominent forehead, and maxillary hypoplasia. Redundant periumbilical skin was also observed in the PITX2 group and negative group. Two patients in the PITX2 group had histories of undergoing surgery for umbilical hernia. Four patients with FOXC1 variants or gross deletions spanning FOXC1 had congenital heart diseases. Other systemic manifestations in the patients included hearing loss, growth disorder, depressive disorder, and kidney stone. The prevalence of systemic manifestations was statistically significantly lower in the FOXC1 group (4/19, 21.1%) than in the PITX2 group (14/14, 100%, p < 0.001) and in the negative group (16/20, 80.0%, p < 0.001); however, there was no significant difference between the PITX2 group and the negative group (p = 0.13).
Discussion
In this study, we investigated 55 Chinese patients with ARS from independent families, and analyzed their genotypes, phenotypes, and genotype-phenotype correlations.
ARS is a rare disease with autosomal dominant inheritance. All of the patients in our study who carried variants are heterozygous. FOXC1 and PITX2 were the two major causative genes of ARS, which has genetic heterogeneity. To date, the variant rates of FOXC1 and PITX2 in individuals with ARS have been reported in a limited number of studies. In an ASD cohort mainly from Belgium or Netherlands, FOXC1 and PITX2 disruptions were recorded in 24% (19/80) and 16% (13/80) of patients, respectively (D’Haene et al., 2011). Whereas, combined FOXC1 and PITX2 variants were found to account for 63% of ARS probands (24/38) in a multi-racial study, with 8% (3/38) harboring FOXC1 variants and 55% (21/38) harboring PITX2 variants (Reis et al., 2012). In another study of 20 patients with ARS from Southeast China, PITX2 variants were detected in 55% (11/20) of patients, while no FOXC1 variants were detected (Wang et al., 2018). The variant rates of FOXC1 and PITX2 varied widely among these studies, and this could be due to variations in inclusion criteria and study populations, and limited sample size. In our study, FOXC1 variants, gross deletions spanning FOXC1, and PITX2 variants in total were detected in 63.6% (35/55) of patients with ARS; FOXC1 variants were detected in 34.5% (19/55) of probands, gross deletions of 6p25 were detected in 3.6% (2/55) of probands and PITX2 variants were detected in 25.5% (14/55) of probands. There were no pathogenic variants found in the other 20 patients. The pathogenic genes of these patients still require exploration. By increasing the number of studied patients, we have greatly expanded the genetic spectrum of Chinese patients with ARS.
Eighteen FOXC1 variants were detected in 19 patients. Most of our detected variants were located in the forkhead domain, which is a conserved 110 amino acid sequence (Figure 1). The forkhead domain is shared by all FOX family proteins and is highly conserved in evolution (Benayoun et al., 2011). The transcription factor encoded by FOXC1 binds with DNA through the forkhead domain; therefore, the forkhead domain is vital to the localization of FOXC1 to the nucleus, and to the interaction between FOXC1 protein and DNA (Benayoun et al., 2011; Seifi et al., 2017). Forkhead domain variants will impair the translocation, DNA-binding capacity and specificity, and transactivation of FOXC1 (Lehmann et al., 2003; Berry et al., 2006), and ultimately lead to loss of function of FOXC1. FOXC1 is widely expressed in the mesenchyme, and it is important for the regulation of embryogenesis, cell migration, and differentiation (Lehmann et al., 2003; Aldinger et al., 2009; Benayoun et al., 2011; Seifi et al., 2017). Thus, FOXC1 disruptions could lead to abnormal development disorders of the ocular anterior segment and other non-ocular tissues (Lehmann et al., 2003; Chrystal et al., 2021). Deletions are a common form of FOXC1 variants. To date, 25 small deletions and 35 gross deletions have been detected according to the Human Gene Mutation Database. Similarly, four small deletions and two gross deletions spanning the entire FOXC1 gene were detected in our patients. Two patients who carried gross deletions had an early age of onset and various multisystemic phenotypes. Patient No. 20 carried a 1.2 Mb deletion, and presented with ocular malformations and craniofacial abnormalities, while patient No. 21 carried a 1.95 Mb deletion, and presented with hydrocephalus, congenital heart disease, and growth disorder, in addition to the ocular malformations. Gross deletions in chromosome 6p25 cause variable clinical features due to the genes involved and the size of the deletions (Fan et al., 2020). Common features include ocular malformations, hydrocephalus, and hearing loss, as well as craniofacial, cardiac, skeletal, and renal malformations (Gould et al., 2004), which were also observed in our patients.
Thirteen PITX2 variants were found in 14 patients with ARS in our study, including nine novel variants. All missense mutations and two nonsense mutations were located in the 60-amino-acid homeodomain, which is a highly conservative sequence (Figure 1). The homeodomain is responsible for DNA binding and is essential for the activity of the PITX2 protein as a transcription regulator (Priston et al., 2001; Tumer and Bach-Holm 2009). As previously reported, most PITX2 missense mutations are located in this homeodomain (Tumer and Bach-Holm 2009; Lewis et al., 2017), which could affect all known PITX2 isoforms and lead to loss of function of the PITX2 protein (Tumer and Bach-Holm 2009; Hendee et al., 2018). Studies using experimental mouse or zebrafish models have demonstrated that PITX2 plays a key role in embryonic development. Mice with a homozygous null mutation of Pitx2 were found to have a severe embryonic lethal phenotype with abnormal eye development, defective body-wall closure, abnormal craniofacial development, arrested tooth development, and other numerous abnormalities (Gage et al., 1999; Kitamura et al., 1999; Lin et al., 1999; Lu et al., 1999). Both mice heterozygous for a Pitx2 null and zebrafish homozygous for a Pitx2 mutant were found to have ARS-related ocular malformations (Chen and Gage 2016; Hendee et al., 2018). The PITX2 variants in our patients could disrupt the structure and the transcriptional activity of the PITX2 protein, and ultimately lead to ocular anterior segment and developmental disorders of non-ocular structures.
Our patients displayed a wide range of variability and severity in ocular and systemic manifestations. The spectrum of ARS ocular manifestations mainly included posterior embryotoxon, iris hypoplasia, iridocorneal adhesions, corectopia, and polycoria, consistent with previous studies (Strungaru et al., 2007; Tumer and Bach-Holm 2009; Reis et al., 2012; Souzeau et al., 2017). Due to anterior segment dysgenesis, glaucoma is the most common and serious consequence of ARS. As previously reported, 50–85% of patients with ARS develop glaucoma (Alward 2000; Souzeau et al., 2017; Wang et al., 2018). In our group, 96.4% of patients had glaucoma, and most of them were diagnosed with ARS and glaucoma simultaneously, which was probably because all patients were recruited at glaucoma clinics. The main systemic features in our patients were dental and craniofacial abnormalities. Notably, two patients (No. 27 and No. 33) in the PITX2 group had histories of undergoing surgery for umbilical hernias. It has been reported that redundant periumbilical skin is frequently mistaken for an umbilical hernia (Alward 2000); thus, there was a possibility that these surgeries were unnecessary.
Furthermore, the genotype-phenotype analysis in our study revealed that the FOXC1 variants were associated with an earlier age of diagnosis and a lower prevalence of systemic features. Patients in the FOXC1 group presented with a significantly earlier age of diagnosis compared to the PITX2 group and the negative group. Furthermore, the proportion of patients aged younger than 1 year was significantly different among the three groups: 47.4% in the FOXC1 group, 7.1% in the PITX2 group, and 10.0% in the negative group (p = 0.007). Similarly, Souzeau et al. reported that the age of glaucoma diagnosis was significantly lower in FOXC1 carriers than in the PITX2 carriers (Souzeau et al., 2017). Almost half of our patients with FOXC1 variants presented with ocular manifestation before the age of 1 year, and may present systemic manifestations later in life according to our findings and the literature (Siggs et al., 2019). Thus, long term follow-up and comprehensive physical examination are important in these patients. Interestingly, the prevalence of systemic features in the FOXC1 group was significantly lower than that in the PITX2 group (100%, p < 0.001) and in the negative group (80.0%, p < 0.001). Furthermore, FOXC1 was more likely to be associated with congenital heart disease, while PITX2 was always associated with dental anomalies and/or umbilical anomalies, and this is supported by previous studies (Strungaru et al., 2007; Reis et al., 2012). Our study also illustrates that patients without FOXC1 or PITX2 variants had a similar age of diagnosis, and similar prevalence and spectrum of systemic features compared to those harboring PITX2 variants. This suggests that the undiscovered pathogenic genes of ARS might have a similar function to the PITX2 gene.
Conclusion
To our best knowledge, this study enrolled the largest number of Chinese patients with ARS to date. In total, 55 Chinese patients with ARS from independent families were studied both clinically and genetically. We found that 63.6% of patients carried 33 FOXC1 variants, PITX2 variants or gross deletions spanning FOXC1, out of which 23 were novel. The genotype and phenotype spectrums of Chinese patients with ARS have been greatly expanded by our study. Because of the early age of onset, high risk for glaucoma, and combined systematic disorders, genetic testing is recommended for patients with ARS, in order to make an early and precise diagnosis.
Data Availability Statement
The data presented in the study are deposited in the LOVD (www.lovd.nl), which is a large Open-Source DNA variation database system. Here is the link: https://databases.lovd.nl/shared/variants/FOXC1?search_var_status=%3D%22Marked%22%7C%3D%22Public%22#object_id=VariantOnTranscript%2CVariantOnGenome&id=FOXC1&search_transcriptid=00008069&search_owned_by_=youjia%20Zhang&page_size=100&page=1; https://databases.lovd.nl/shared/variants/PITX2?search_var_status=%3D%22Marked%22%7C%3D%22Public%22#object_id=VariantOnTranscript%2CVariantOnGenome&id=PITX2&order=VariantOnTranscript%2FDNA%2CASC&search_transcriptid=00024018&search_owned_by_=youjia%20Zhang&page_size=100&page=1.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of the Eye and ENT Hospital of Fudan University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
Concept and design, YC and XS; Literature search, YZ and YC; Clinical studies, YZ, YC, XC, LW, and XS; Experimental studies, YZ and YC; Data acquisition, YZ, YC, XC, LW, and XS; Data analysis, YZ and YC; Statistical analysis, YZ and YC; Manuscript preparation, YZ; Manuscript editing and manuscript review, YZ and YC.
Funding
This work was supported by the funds from the Shanghai Committee of Science and Technology, China (Grant no. 20S31905800), and the National Natural Science Foundation of China (Grant no. 81870692), and the Clinical Research Plan of SHDC (Grant no. SHDC2020CR6029). The authors were supported by grants from the National Key Research and Development Program of China (Grant no. 2020YFA0112700), from the State Key Program of National Natural Science Foundation of China (Grant no. 82030027), and from the subject of major projects of National Natural Science Foundation of China (Grant no. 81790641). The sponsor or funding organization had no role in the design or conduct of this research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are grateful to the biobank of the Eye and ENT Hospital of Fudan University. The authors would like to thank Haijun Zhu, Minjie Xu, Xilu Wang, Hao Yuan, and Yanxin Shi from Amplicongene (Shanghai, China) for the helpful bioinformatics analysis. The authors would also like to thank all of the patients and their families.
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 7, 248–249. doi:10.1038/nmeth0410-248
Aldinger, K. A., Lehmann, O. J., Hudgins, L., Chizhikov, V. V., Bassuk, A. G., Ades, L. C., et al. (2009). FOXC1 Is Required for normal Cerebellar Development and Is a Major Contributor to Chromosome 6p25.3 Dandy-Walker Malformation. Nat. Genet. 41, 1037–1042. doi:10.1038/ng.422
Alward, W. L. M. (2000). Axenfeld-Rieger Syndrome in the Age of Molecular Genetics. Am. J. Ophthalmol. 130, 107–115. doi:10.1016/s0002-9394(00)00525-0
Benayoun, B. A., Caburet, S., and Veitia, R. A. (2011). Forkhead Transcription Factors: Key Players in Health and Disease. Trends Genet. 27, 224–232. doi:10.1016/j.tig.2011.03.003
Berry, F. B., Lines, M. A., Oas, J. M., Footz, T., Underhill, D. A., Gage, P. J., et al. (2006). Functional Interactions between FOXC1 and PITX2 Underlie the Sensitivity to FOXC1 Gene Dose in Axenfeld-Rieger Syndrome and Anterior Segment Dysgenesis. Hum. Mol. Genet. 15, 905–919. doi:10.1093/hmg/ddl008
Cella, W., Cabral de Vasconcellos, J. P., Barbosa de Melo, M. n., Kneipp, B., Costa, F. F., Longui, C. A., et al. (2006). Structural Assessment of PITX2, FOXC1, CYP1B1, and GJA1 Genes in Patients with Axenfeld-Rieger Syndrome with Developmental Glaucoma. Invest. Ophthalmol. Vis. Sci. 47, 1803–1809. doi:10.1167/iovs.05-0979
Chang, T. C., Summers, C. G., Schimmenti, L. A., and Grajewski, A. L. (2012). Axenfeld-Rieger Syndrome: New Perspectives: Figure 1. Br. J. Ophthalmol. 96, 318–322. doi:10.1136/bjophthalmol-2011-300801
Chen, L., and Gage, P. J. (2016). Heterozygous Pitx2 Null Mice Accurately Recapitulate the Ocular Features of Axenfeld-Rieger Syndrome and Congenital Glaucoma. Invest. Ophthalmol. Vis. Sci. 57, 5023–5030. doi:10.1167/iovs.16-19700
Choi, Y., and Chan, A. P. (2015). PROVEAN Web Server: a Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 31, 2745–2747. doi:10.1093/bioinformatics/btv195
Crystal, P. W., French, C. R., Jean, F., Havrylov, S., van Baarle, S., Peturson, A. M., et al. (2021). The Axenfeld-Rieger Syndrome Gene FOXC1 Contributes to Left-Right Patterning. Genes (Basel) 12 (2), 170. doi:10.3390/genes12020170
Cox, M. P., Peterson, D. A., and Biggs, P. J. (2010). SolexaQA: At-A-Glance Quality Assessment of Illumina Second-Generation Sequencing Data. BMC Bioinf. 11, 485. doi:10.1186/1471-2105-11-485
D'Haene, B., Meire, F., Claerhout, I., Kroes, H. Y., Plomp, A., Arens, Y. H., et al. (2011). Expanding the Spectrum of FOXC1 and PITX2 Mutations and Copy Number Changes in Patients with Anterior Segment Malformations. Invest. Ophthalmol. Vis. Sci. 52, 324–333. doi:10.1167/iovs.10-5309
Fan, S. P., Lee, N. C., and Lin, C. H. (2020). Novel Phenotype of 6p25 Deletion Syndrome Presenting Juvenile Parkinsonism and Brain Calcification. Mov. Disord. 35, 1457–1462. doi:10.1002/mds.28079
Gage, P. J., Suh, H., and Camper, S. A. (1999). Dosage Requirement of Pitx2 for Development of Multiple Organs. Development 126, 4643–4651. doi:10.1242/dev.126.20.4643
Gould, D. B., Jaafar, M. S., Addison, M. K., Munier, F., Ritch, R., MacDonald, I. M., et al. (2004). Phenotypic and Molecular Assessment of Seven Patients with 6p25 Deletion Syndrome: Relevance to Ocular Dysgenesis and Hearing Impairment. BMC Med. Genet. 5, 17. doi:10.1186/1471-2350-5-17
Hendee, K. E., Sorokina, E. A., Muheisen, S. S., Reis, L. M., Tyler, R. C., Markovic, V., et al. (2018). PITX2 Deficiency and Associated Human Disease: Insights from the Zebrafish Model. Hum. Mol. Genet. 27, 1675–1695. doi:10.1093/hmg/ddy074
Kawase, C., Kawase, K., Taniguchi, T., Sugiyama, K., Yamamoto, T., Kitazawa, Y., et al. (2001). Screening for Mutations of Axenfeld-Rieger Syndrome Caused by FOXC1 Gene in Japanese Patients. J. Glaucoma 10, 477–482. doi:10.1097/00061198-200112000-00007
Kitamura, K., Miura, H., Miyagawa-Tomita, S., Yanazawa, M., Katoh-Fukui, Y., Suzuki, R., et al. (1999). Mouse Pitx2 Deficiency Leads to Anomalies of the Ventral Body wall, Heart, Extra- and Periocular Mesoderm and Right Pulmonary Isomerism. Development 126, 5749–5758. doi:10.1242/dev.126.24.5749
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the Effects of Coding Non-synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 4, 1073–1081. doi:10.1038/nprot.2009.86
Lehmann, O. J., Sowden, J. C., Carlsson, P., Jordan, T., and Bhattacharya, S. S. (2003). Fox's in Development and Disease. Trends Genet. 19, 339–344. doi:10.1016/s0168-9525(03)00111-2
Lewis, C. J., Hedberg-Buenz, A., DeLuca, A. P., Stone, E. M., Alward, W. L. M., and Fingert, J. H. (2017). Primary Congenital and Developmental Glaucomas. Hum. Mol. Genet. 26, R28–R36. doi:10.1093/hmg/ddx205
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. doi:10.1093/bioinformatics/btp324
Lin, C. R., Kioussi, C., O'Connell, S., Briata, P., Szeto, D., Liu, F., et al. (1999). Pitx2 Regulates Lung Asymmetry, Cardiac Positioning and Pituitary and Tooth Morphogenesis. Nature 401, 279–282. doi:10.1038/45803
Lines, M. A., Kozlowski, K., and Walter, M. A. (2002). Molecular Genetics of Axenfeld-Rieger Malformations. Hum. Mol. Genet. 11, 1177–1187. doi:10.1093/hmg/11.10.1177
Lu, M.-F., Pressman, C., Dyer, R., Johnson, R. L., and Martin, J. F. (1999). Function of Rieger Syndrome Gene in Left-Right Asymmetry and Craniofacial Development. Nature 401, 276–278. doi:10.1038/45797
Nishimura, D. Y., Swiderski, R. E., Alward, W. L. M., Searby, C. C., Patil, S. R., Bennet, S. R., et al. (1998). The Forkhead Transcription Factor Gene FKHL7 Is Responsible for Glaucoma Phenotypes Which Map to 6p25. Nat. Genet. 19, 140–147. doi:10.1038/493
Nishimura, D. Y., Searby, C. C., Alward, W. L., Walton, D., Craig, J. E., Mackey, D. A., et al. (2001). A Spectrum of FOXC1 Mutations Suggests Gene Dosage as a Mechanism for Developmental Defects of the Anterior Chamber of the Eye. Am. J. Hum. Genet. 68, 364–372. doi:10.1086/318183
Panicker, S. G., Sampath, S., Mandal, A. K., Reddy, A. B., Ahmed, N., and Hasnain, S. E. (2002). Novel Mutation in FOXC1 wing Region Causing Axenfeld-Rieger Anomaly. Invest. Ophthalmol. Vis. Sci. 43, 3613–3616.
Phillips, J. C. (2002). Four Novel Mutations in the PITX2 Gene in Patients with Axenfeld-Rieger Syndrome. Ophthalmic Res. 34, 324–326. doi:10.1159/000065602
Plagnol, V., Curtis, J., Epstein, M., Mok, K. Y., Stebbings, E., Grigoriadou, S., et al. (2012). A Robust Model for Read Count Data in Exome Sequencing Experiments and Implications for Copy Number Variant Calling. Bioinformatics 28, 2747–2754. doi:10.1093/bioinformatics/bts526
Priston, M., Kozlowski, K., Gill, D., Letwin, K., Buys, Y., Levin, A. V., et al. (2001). Functional Analyses of Two Newly Identified PITX2 Mutants Reveal a Novel Molecular Mechanism for Axenfeld-Rieger Syndrome. Hum. Mol. Genet. 10, 1631–1638. doi:10.1093/hmg/10.16.1631
Reis, L. M., Tyler, R. C., Volkmann Kloss, B. A., Schilter, K. F., Levin, A. V., Lowry, R. B., et al. (2012). PITX2 and FOXC1 Spectrum of Mutations in Ocular Syndromes. Eur. J. Hum. Genet. 20, 1224–1233. doi:10.1038/ejhg.2012.80
Richards, S., Aziz, N., Aziz, N., Bale, S., Bick, D., Das, S., et al. Acmg Laboratory Quality Assurance Committee (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423. doi:10.1038/gim.2015.30
Seifi, M., and Walter, M. A. (2018). Axenfeld-Rieger Syndrome. Clin. Genet. 93, 1123–1130. doi:10.1111/cge.13148
Seifi, M., Footz, T., Taylor, S. A. M., and Walter, M. A. (2017). Comparison of Bioinformatics Prediction, Molecular Modeling, and Functional Analyses of FOXC1 Mutations in Patients with Axenfeld-Rieger Syndrome. Hum. Mutat. 38, 169–179. doi:10.1002/humu.23141
Semina, E. V., Reiter, R., Leysens, N. J., Alward, W. L. M., Small, K. W., Datson, N. A., et al. (1996). Cloning and Characterization of a Novel Bicoid-Related Homeobox Transcription Factor Gene, RIEG, Involved in Rieger Syndrome. Nat. Genet. 14, 392–399. doi:10.1038/ng1296-392
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, Scalable Generation of High‐quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 7, 539. doi:10.1038/msb.2011.75
Siggs, O. M., Souzeau, E., Pasutto, F., Dubowsky, A., Smith, J. E. H., Taranath, D., et al. (2019). Prevalence of FOXC1 Variants in Individuals with a Suspected Diagnosis of Primary Congenital Glaucoma. JAMA Ophthalmol. 137 (4), 348–355. doi:10.1001/jamaophthalmol.2018.5646
Souzeau, E., Siggs, O. M., Zhou, T., Galanopoulos, A., Hodson, T., Taranath, D., et al. (2017). Glaucoma Spectrum and Age-Related Prevalence of Individuals with FOXC1 and PITX2 Variants. Eur. J. Hum. Genet. 25, 839–847. doi:10.1038/ejhg.2017.59
Strungaru, M. H., Dinu, I., and Walter, M. A. (2007). Genotype-Phenotype Correlations in Axenfeld-Rieger Malformation and Glaucoma Patients with FOXC1 and PITX2 Mutations. Invest. Ophthalmol. Vis. Sci. 48, 228–237. doi:10.1167/iovs.06-0472
Tümer, Z., and Bach-Holm, D. (2009). Axenfeld-Rieger Syndrome and Spectrum of PITX2 and FOXC1 Mutations. Eur. J. Hum. Genet. 17, 1527–1539. doi:10.1038/ejhg.2009.93
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 38, e164. doi:10.1093/nar/gkq603
Keywords: glaucoma, next generation sequence, genotype-phenotype correlation, FOXC1 gene, PITX2 gene, Axenfeld Rieger syndrome
Citation: Zhang Y, Chen X, Wang L, Sun X and Chen Y (2021) Heterogeneity of Axenfeld–Rieger Syndrome: Molecular and Clinical Findings in Chinese Patients. Front. Genet. 12:732170. doi: 10.3389/fgene.2021.732170
Received: 28 June 2021; Accepted: 24 September 2021;
Published: 20 October 2021.
Edited by:
Alfredo Brusco, University of Turin, ItalyReviewed by:
Fabio Sirchia, Neurological Institute Foundation Casimiro Mondino (IRCCS), ItalyCecilia Mancini, Bambino Gesù Children Hospital (IRCCS), Italy
Copyright © 2021 Zhang, Chen, Wang, Sun and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinghuai Sun, eGhzdW5Ac2htdS5lZHUuY24=; Yuhong Chen, eXVob25nY2hlbkBmdWRhbi5lZHUuY24=