 Pierre Caron†
Pierre Caron† Enrico Pobega
Enrico Pobega Sophie E. Polo
Sophie E. Polo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 01 September 2021
Sec. Human and Medical Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.730696
This article is part of the Research TopicProtecting the Code: DNA Double-Strand Break Repair Pathway ChoiceView all 19 articles
In response to DNA double-strand breaks (DSBs), chromatin modifications orchestrate DNA repair pathways thus safeguarding genome integrity. Recent studies have uncovered a key role for heterochromatin marks and associated factors in shaping DSB repair within the nucleus. In this review, we present our current knowledge of the interplay between heterochromatin marks and DSB repair. We discuss the impact of heterochromatin features, either pre-existing in heterochromatin domains or de novo established in euchromatin, on DSB repair pathway choice. We emphasize how heterochromatin decompaction and mobility further support DSB repair, focusing on recent mechanistic insights into these processes. Finally, we speculate about potential molecular players involved in the maintenance or the erasure of heterochromatin marks following DSB repair, and their implications for restoring epigenome function and integrity.
The organization of the genome into chromatin in the nuclear space serves to precisely orchestrate cellular functions by controlling gene expression. While the euchromatin compartment is generally accessible and associated with active gene transcription, heterochromatin is more condensed and mostly transcriptionally silent (Allshire and Madhani, 2018). Beyond this general definition, heterochromatin domains are actually quite diverse, in localization, regulation, and function. Constitutive heterochromatin is highly conserved between different cell types, rich in repeated sequences, and plays critical roles in the maintenance of chromosomal architecture and stability (Janssen et al., 2018; Penagos-Puig and Furlan-Magaril, 2020). The bulk of constitutive heterochromatin forms at pericentromeric regions, which are involved in the control of chromosomal segregation (Saksouk et al., 2015). Likewise, telomeres adopt a specific constitutive heterochromatin structure that serves to shield chromosomal ends from aberrant DNA repair, thus protecting chromosome integrity (de Lange, 2002; Allshire and Madhani, 2018; Lim and Cech, 2021). Facultative heterochromatin, in contrast, is developmentally regulated and varies across cell types. Its main function is to silence gene regions that should not be expressed in a specific developmental or somatic context (Trojer and Reinberg, 2007). The inactive X chromosome (Xi) is a typical example of facultative heterochromatin, which is established early during female mammalian development for the dosage compensation of X-linked genes (Galupa and Heard, 2018). Heterochromatin also forms at the nuclear periphery through interactions with the nuclear lamina leading to lamina-associated domains (LADs), which play an important role in chromosome organization and gene repression (van Steensel and Belmont, 2017).
The establishment and maintenance of the silent state in heterochromatin domains involve DNA methylation and repressive histone post-translational modifications (PTMs). While constitutive and facultative heterochromatin are both enriched in DNA methylation, they show specific histone PTMs. Constitutive heterochromatin is enriched in H3K9me2/3, which is bound by heterochromatin protein 1 (HP1), a factor that plays a crucial role in heterochromatin assembly (Saksouk et al., 2015). Facultative heterochromatin instead shows an enrichment of H3K27me3 and H2AK119ub (Galupa and Heard, 2018), and the facultative heterochromatin mark H3K27me3 is also enriched in LADs (van Steensel and Belmont, 2017). In addition to DNA and histone modifications, heterochromatin domains incorporate specific histone variants (Martire and Banaszynski, 2020), such as centromere protein A (CENP-A) at centromeres and macroH2A1 in the Xi, and associate with architectural factors that determine the three-dimensional chromatin structure. In this review, DNA and histone modifications, histone readers, histone variants, and architectural factors enriched in heterochromatin domains are collectively referred to as heterochromatin marks or features. Remodeling factors (Clapier et al., 2017) provide another layer of regulation of heterochromatin accessibility by affecting nucleosome positioning. All these factors come into play to shape heterochromatin domains and mediate their function (Allshire and Madhani, 2018).
One of the major functions of heterochromatin is to ensure a tight control of transcriptional states, which is key for maintaining genome integrity and cell fate (Janssen et al., 2018). Nevertheless, heterochromatin domains also represent challenging environments for DNA metabolic activities, including DNA replication and DNA damage repair (Fortuny and Polo, 2018). Indeed, these domains are late replicating, highly compact, and often encompass repetitive sequences, which contributes to replication stress and fuels genome instability. Heterochromatin repeats are also prone to instability through ectopic recombination leading to deletions or translocations. Moreover, the highly compacted state and the low transcriptional activity in heterochromatin domains impede several repair pathways (Fortuny and Polo, 2018). These obstacles can be circumvented by alterations of the heterochromatin structure during the repair process.
Among the many types of DNA lesions, highly cytotoxic DNA double-strand breaks (DSBs) are repaired by multiple pathways with different levels of fidelity. Non-homologous end joining (NHEJ) is predominant and proceeds by direct ligation of DNA ends, while homologous recombination (HR) requires an initial resection of the DNA ends followed by recombination with a homologous template, usually the sister chromatid, which restricts HR to the S and G2 phases of the cell cycle (Chen et al., 2018; Zhao et al., 2020). Single-strand annealing (SSA) is based on homology on the same DNA strand and repairs DSBs between repeated sequences, leading to large deletions (Bhargava et al., 2016). DNA double-strand break repair by microhomology-mediated end joining (MMEJ) is also highly mutagenic, as it relies on short microhomology sequences that are exposed after end resection, and always generates small indels (Sallmyr and Tomkinson, 2018). The choice between several DSB repair pathways with different degrees of mutagenicity is thus decisive for the maintenance of genomic stability and is subject to complex regulatory mechanisms (Scully et al., 2019), including at the chromatin level. Recent studies have uncovered the key role of heterochromatin marks in dictating DSB repair pathway choice. DNA double-strand break repair, in turn, involves alterations in heterochromatin organization and heterochromatin marks, which need to be reverted to preserve epigenome integrity.
Here, by focusing on recent discoveries in the field, we provide an overview of our current knowledge of the interplay between heterochromatin marks and DSB repair and discuss potential mechanisms that preserve the integrity of heterochromatin domains.
Heterochromatin marks and associated factors not only play critical roles in transcriptional silencing but also contribute to regulate DSB repair pathway choice, in part by controlling the recruitment of DSB repair factors. This regulation has been observed both in heterochromatin domains where heterochromatin marks are present before damage infliction and in euchromatin domains where DSBs trigger the deposition of specific heterochromatin marks. In this section, we discuss recent studies that provided new mechanistic insights into the regulation exerted by heterochromatin marks on DSB repair.
Histone PTMs constitute an important layer of epigenomic information with a broad impact on chromatin organization and function; some of these marks define heterochromatin domains and have been shown to regulate DSB repair responses.
For instance, a well-known PTM enriched in constitutive heterochromatin is H3K9me3. As previously described, an increase of this mark was observed at break sites in mammalian cells, both in heterochromatin and in euchromatin regions (Ayrapetov et al., 2014; Tsouroula et al., 2016; Natale et al., 2017), and several players in the H3K9me3 pathway – writers (SUV39H1/2, SETDB1) and readers (HP1, TIP60) – were shown to promote DSB repair by HR (Sun et al., 2009; Baldeyron et al., 2011; Soria and Almouzni, 2013; Tang et al., 2013; Alagoz et al., 2015; Jacquet et al., 2016). In line with these studies, the H3K9 methyltransferase SET domain bifurcated histone lysine methyltransferase 1 (SETDB1) was also shown to regulate alternative lengthening of telomeres (ALT) in mouse cells by creating an H3K9me3-rich heterochromatin environment that facilitates recombination (Gauchier et al., 2019).
Recent studies have elucidated the mechanism underlying the deposition of H3K9me3 around DSBs and described new interactions between this histone modification and the regulation of DSB repair. It was shown that H3K9me3 actually depends on another damage-induced PTM on histone H4. The DSB sensor complex MRE11-RAD50-NBS1 indeed recruits UFM1-specific ligase 1, leading to the conjugation of a ubiquitin-like protein to histone H4 lysine 31, a process known as ufmylation. This histone PTM is bound by the serine/threonine-protein kinase 38 (STK38), which in turn recruits the H3K9 methyltransferase suppressor of variegation 3–9 homolog 1 (SUV39H1) leading to the trimethylation of H3K9 around DSBs (Qin et al., 2019, 2020). The local increase of H3K9me3 at DSB sites seems to be crucial for HR as shown in human cancers with elevated levels of oncometabolites that inhibit the lysine demethylase KDM4B. This causes an aberrant constitutive hypermethylation of H3K9 instead of a local increase at break sites, which diverts TIP60 away from the DSBs, thereby impairing HR activation (Sulkowski et al., 2020).
The local increase of H3K9me3 points toward a heterochromatinization phenomenon that may be necessary for HR repair. However, one also has to consider how the DSB repair machinery handles breaks in heterochromatin regions that are already decorated with this mark. Indeed, HR could lead to mutagenic recombination in heterochromatin compartments due to their highly repetitive nature. Several strategies actually serve to prevent HR in H3K9me3-containing heterochromatin domains. In mouse cells for instance, resection of the DNA ends leads to their relocalization to the periphery of pericentromeric heterochromatin, where recombination takes place (Tsouroula et al., 2016). In Drosophila melanogaster in contrast, DSBs in pericentromeric heterochromatin trigger the recruitment of Drosophila lysine demethylase 4a, which demethylates H3K9me3 and H3K56me3, another conserved pericentromeric heterochromatin mark (Jack et al., 2013). This demethylase channels repair to NHEJ by inhibiting the recruitment of early HR factors to heterochromatic DSBs (Janssen et al., 2019).
In addition to H3K9me3, H3K27me3 also decorates heterochromatin regions, such as those associated with the lamina, and DSBs in these H3K27me3-enriched regions show increased repair by MMEJ (Lemaître et al., 2014; Schep et al., 2021). Interestingly, chemical inhibition of H3K27 and not of H3K9 methyltransferases shifted the MMEJ/NHEJ balance toward NHEJ (Schep et al., 2021), arguing that the H3K27me3 heterochromatin mark either stimulates MMEJ or inhibits NHEJ. However, the underlying molecular mechanisms still need to be elucidated and the impact of H3K27me3 on HR is unknown. Similar to H3K9me3, H3K27me3 was also found increased at DSBs in some studies but with conflicting results in other studies (reviewed in Ferrand et al., 2020), so further work is needed to clarify the status of this mark at DSBs in and outside heterochromatin.
Besides H3K9me3 and H3K27me3, other histone modifications play a role in DSB repair regulation in heterochromatin domains. For instance, several H3K36me2-specific histone methyltransferases, including multiple myeloma SET domain-containing protein (MMSET), promote NHEJ at deprotected telomeres in mouse cells. Interestingly, the involvement of H3K36me2 seems to occur downstream of DSB recognition and repair pathway choice (de Krijger et al., 2020).
While the mechanisms through which H3K9me3 impacts DSB repair are now quite well characterized, how other histone marks, like H3K27me3 and H3K36me2, influence this process is still unknown. Further studies are necessary to determine whether those marks modulate the recruitment of specific DSB repair factors to chromatin. In addition, it will be interesting to investigate whether histone PTMs also underlie the differential regulation of HR between centromeric and pericentromeric heterochromatin observed in mouse cells (Tsouroula et al., 2016). Homologous recombination of centromeric DSBs is indeed licensed in G1, in addition to S/G2, which could rely on histone marks decorating centromeres, such as H3K4me2, H3K36me2, and H3 acetylation (Chan and Wong, 2012).
Besides histone modifications, another layer of chromatin regulation builds upon the incorporation of histone variants (Martire and Banaszynski, 2020), some of which are enriched in heterochromatin and regulate DSB repair. Such regulation occurs in already histone variant-enriched heterochromatin domains and also upon the accumulation of these variants at euchromatic DSBs.
The histone variant MacroH2A1, for instance, is enriched in facultative heterochromatin domains on autosomes and on the inactive X chromosome (Costanzi and Pehrson, 1998; Changolkar and Pehrson, 2006; Gamble et al., 2010). Remarkably, the macroH2A1 gene expresses two splicing isoforms: macroH2A1.1 and macroH2A1.2, which exhibit antagonistic properties in the regulation of DSB repair pathway choice in mammalian cells. MacroH2A1.2 accumulates at DSBs in an ataxia telangiectasia mutated (ATM)-dependent manner and stimulates DSB repair by HR by promoting the recruitment of breast cancer type 1 susceptibility protein (BRCA1; Khurana et al., 2014). Similarly, macroH2A1.2 deposition at sites of replication stress by the histone chaperone facilitates chromatin transcription (FACT) forms a chromatin environment amenable for BRCA1 recruitment (Kim et al., 2017). In human cells lacking the chromatin remodeler Alpha thalassemia/mental retardation syndrome X-linked (ATRX), macroH2A1.2 is also highly enriched at telomeres and contributes to ALT, a HR-mediated process (Kim et al., 2019). Mechanistically, macroH2A1.2 collaborates with the histone demethylase KDM5A to promote both DSB repair by HR and transcriptional silencing at breaks (Kumbhar et al., 2021). MacroH2A1.1 in contrast supports MMEJ, a mutagenic DSB repair pathway (Sebastian et al., 2020). The preferential interaction of macroH2A1.1 with MMEJ repair factors, including poly(ADP-ribose) polymerase 1 (PARP1), is likely linked to the ability of this isoform to bind ADP-ribose, a property that is not shared with macroH2A1.2 (Sebastian et al., 2020). MacroH2A1.2-deficient cells display X-chromosome instability due to defective HR and enhanced MMEJ. Interestingly, loss of macroH2A1.1 rescues the X-chromosome instability observed in macroH2A1.2-deficient cells (Sebastian et al., 2020). This nicely illustrates how histone variants exert antagonistic control on DSB repair pathway choice and genome integrity in facultative heterochromatin.
Another histone variant that may regulate DSB repair is CENP-A, which defines centromeric heterochromatin. There is conflicting evidence regarding CENP-A accumulation post DSBs (Zeitlin et al., 2009; Ambartsumyan et al., 2010; Helfricht et al., 2013), and the link between CENP-A and DSB repair has not yet been explored. It will be interesting to investigate whether CENP-A can contribute to licensing HR in G1 at centromeric DSBs (Tsouroula et al., 2016). Furthermore, the CENP-A chaperone HJURP may have functional connections to DSB repair (Kato et al., 2007).
In addition to histone variants and modifications, several heterochromatin-associated factors play a central role in DSB repair, including histone readers, architectural factors, and chromatin remodelers.
Chromatin remodelers were shown to regulate chromatin relaxation at heterochromatic DSBs in mammalian cells. In this respect, imitation switch (ISWI)-class and chromodomain-helicase-DNA binding (CHD)-class chromatin remodelers play antagonistic roles. CHD3 promotes heterochromatin compaction, but is released from chromatin following ATM activation, while ACF1 and SNF2H (ISWI class) are recruited to the damage site and lead to heterochromatin decompaction, which allows Artemis-dependent NHEJ (Klement et al., 2014). A recent study put forward the involvement of another chromatin remodeler in stimulating DSB repair by HR in heterochromatin. The human chromatin remodeler lymphoid-specific helicase (HELLS), through its ATPase activity, indeed promotes HR of heterochromatic DSBs in G2 cells exposed to ionizing radiation by facilitating end resection through CTBP-interacting protein (CtIP) recruitment (Kollárovič et al., 2020). Whether the function of HELLS in HR repair of heterochromatic breaks is linked to its ability to promote macroH2A1.2 deposition (Ni et al., 2020; Xu et al., 2021) is an intriguing possibility that deserves further investigation. The involvement of multiple remodelers, some of which having opposing activities, likely allows a fine-tuning of heterochromatin compaction during DSB repair, with dynamic changes over time after DSB induction.
Among readers of heterochromatin-specific modifications, HP1 is recruited to DSBs arising in euchromatin and heterochromatin domains, and a major regulator of DSB repair, with HP1 isoforms having different effects on DSB repair pathways: HP1α and β stimulate HR at the resection step, while HP1γ inhibits this pathway (Baldeyron et al., 2011; Soria and Almouzni, 2013). In line with these findings, HP1γ depletion, but not that of HP1α and β, negatively impacts Ku80 recruitment to heterochromatic DSBs in mouse cells (Tsouroula et al., 2016), suggesting that the HP1γ isoform may play a role in NHEJ. A possible mechanism through which some HP1 isoforms channel DSB repair toward HR might rely on the direct binding of HP1 to BRCA1-associated RING domain 1 (BARD1) in response to DSBs, which promotes retention of the BARD1-BRCA1 complex stimulating CtIP-dependent resection (Wu et al., 2015).
Finally, the heterochromatin-enriched architectural factor structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1) contributes both to transcriptional silencing and to DSB repair. It is still unknown if both functions of SMCHD1 are mechanistically connected. This protein indeed plays a crucial role in mammalian X chromosome inactivation (Blewitt et al., 2008) and is involved in the silencing of specific autosomal genes (Gendrel et al., 2013; Mould et al., 2013). In addition, SMCHD1 is recruited to DNA damage foci (Coker and Brockdorff, 2014; Tang et al., 2014) and is highly enriched on deprotected telomeres in human cells (Vancevska et al., 2020), pointing to a role in the DSB response. SMCHD1 actually contributes to DSB repair pathway choice by promoting NHEJ while inhibiting HR, as shown using cell reporter systems (Tang et al., 2014). Consistent with this, SMCHD1 stimulates 53BP1 foci formation and impairs BRCA1 foci formation following cell treatment with the radiomimetic drug zeocin (Tang et al., 2014). SMCHD1 also promotes the fusion of unprotected telomeres, which relies on NHEJ; however, the function of SMCHD1 seems to be upstream of DSB repair at telomeres through the stimulation of ATM-dependent damage signaling (Vancevska et al., 2020).
Together, these studies illustrate that several heterochromatin marks, including histone trimethylation, histone variants, and non-histone proteins, regulate DSB repair pathway choice (Figure 1). Interestingly, some marks with opposing activities on DSB repair are enriched in the same heterochromatin domain, as observed for macroH2A1.1, 1.2, SMCHD1, and H3K27me3 on the Xi. This might suggest an interplay between heterochromatin marks, which could allow a fine regulation of DSB repair pathways. Notably, in addition to HR, NHEJ, and MMEJ, DSB repair by SSA also operates in heterochromatin, in particular when HR is compromised (Janssen et al., 2016; Tsouroula et al., 2016), but whether heterochromatin marks stimulate SSA is still unknown. Further studies are necessary to clarify the mechanisms through which heterochromatin marks modulate DSB repair and to assess the combinatorial effects of these marks.
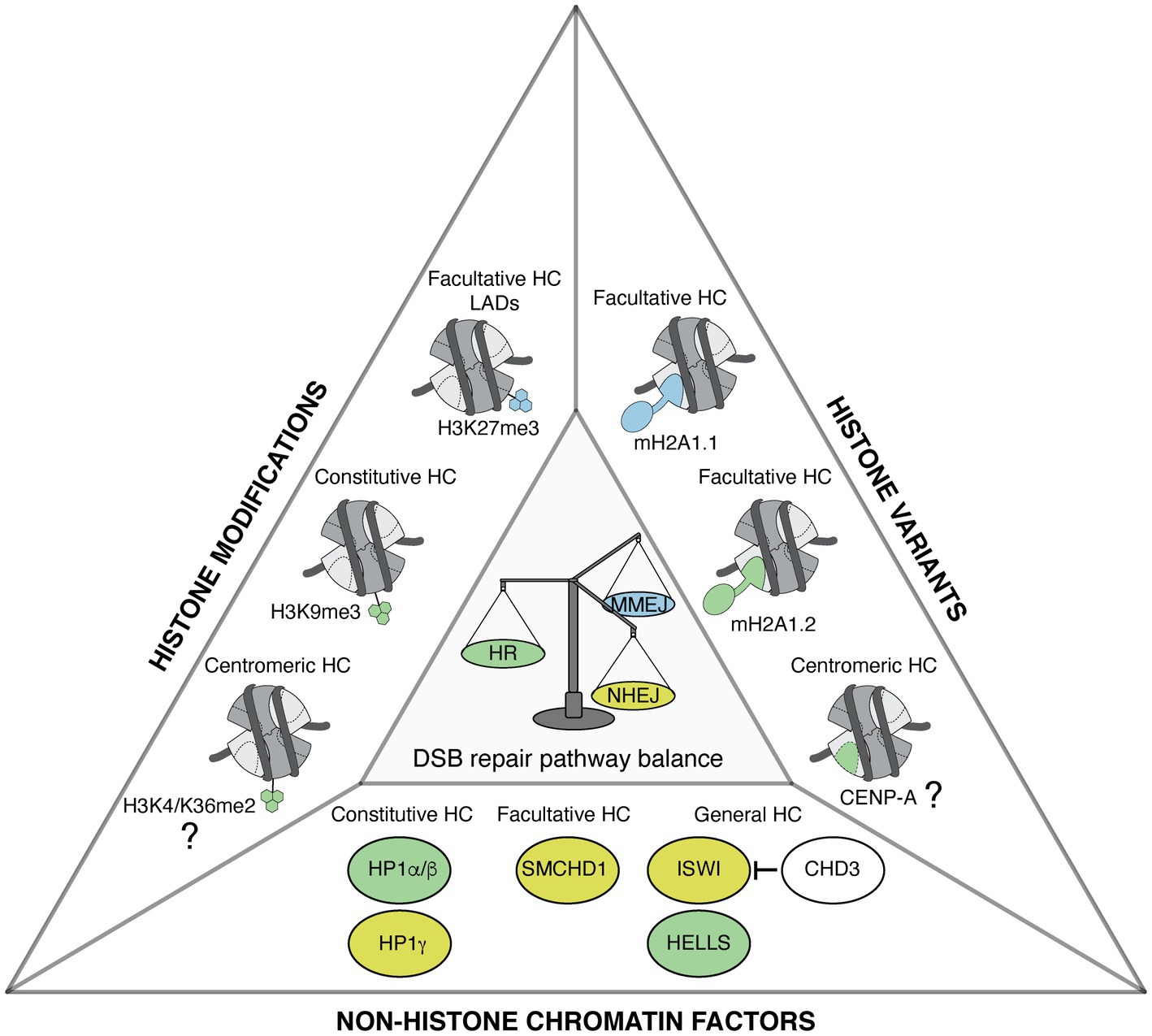
Figure 1. Heterochromatin features govern DNA double-strand break (DSB) repair pathway choice. Representation of heterochromatic (HC) histone modifications, histone variants, and non-histone chromatin factors that modulate DSB repair pathway choice in mammalian cells. Features that favor homologous recombination (HR), non-homologous end joining (NHEJ), and microhomology-mediated end joining (MMEJ) are shown in green, yellow, and blue, respectively. The types of heterochromatin enriched in these features are indicated when known. The contribution of centromeric histone variant and modifications to promoting HR in G1 is still to be determined, as indicated by the question marks.
While the choice of repair pathway is influenced by the chromatin context, DSB repair itself leads to changes in heterochromatin organization, which can have a profound impact on its function and thus on the maintenance of genomic integrity.
Heterochromatin is a highly compacted nucleoprotein structure that can be seen as an obstacle for the detection of DNA lesions and their repair. However, in mammals and D. melanogaster, it was observed that DSB repair kinetics were comparable between heterochromatin and euchromatin (Goodarzi et al., 2008; Janssen et al., 2016). This is achieved, at least in part, through the decompaction of heterochromatin, which facilitates DSB signaling and repair. Mechanistically, heterochromatin decompaction is regulated by chromatin remodelers, as discussed above, and by the ATM kinase, which phosphorylates KRAB-associated protein 1 (KAP-1) thus triggering its eviction from chromatin (Ziv et al., 2006; Goodarzi et al., 2008). Heterochromatin decompaction has been observed in several heterochromatin compartments in response to DSBs induced by ionizing radiation or by site-specific nucleases, including the Xi compartment in female mammalian cells (Müller et al., 2013) and pericentromeric heterochromatin domains in Drosophila and mouse cells (Chiolo et al., 2011; Tsouroula et al., 2016). Of note, the expansion of pericentromeric regions is not specific to the DSB response as it is also observed upon UV damage detection by DNA damage-binding protein 2 (DDB2), which triggers the eviction of linker histone H1 from chromatin (Fortuny et al., 2021). H1 eviction is also reported in response to DSBs (Strickfaden et al., 2016; Clouaire et al., 2018; Li et al., 2018), but whether linker histone eviction participates to heterochromatin decompaction post DSBs is not yet known.
Mechanistically, chromatin decompaction is accompanied by DSB repositioning outside of pericentromeric heterochromatin domains, favoring repair completion through HR, as shown in both Drosophila and mouse cells (Chiolo et al., 2011; Tsouroula et al., 2016; Amaral et al., 2017). In both systems, the resection of the broken ends occurs within pericentromeric heterochromatin and triggers their migration to the periphery of these domains in mouse cells, and to the nuclear periphery in Drosophila cells, where RAD51-mediated recombination takes place (Chiolo et al., 2011; Tsouroula et al., 2016). A similar process may occur in response to DSBs in the Xi. Indeed, while 53BP1 is found both within and outside of the Xi, phosphorylated RPA localizes at the Xi periphery, indicating a possible relocalization of the breaks undergoing resection, to be repaired by HR (Müller et al., 2013). Functionally, such relocalization of breaks outside heterochromatin domains is thought to prevent aberrant ectopic recombination between repeated sequences.
A role for the nucleoskeleton and molecular motors in the relocalization of heterochromatic breaks was put forward in several species (Figure 2). In Drosophila for instance, nuclear actin filaments and myosins promote the relocalization of pericentromeric DSBs to the nuclear periphery (Caridi et al., 2018; Dialynas et al., 2019), with a contribution of sumoylation and of structural maintenance of chromosomes 5/6 proteins (SMC5/6) in anchoring the breaks to the nuclear periphery (Ryu et al., 2015). Mechanisms appear to be distinct in mammalian cells where SMC5/6 proteins are dispensable, and anchoring of breaks to the nuclear periphery does not occur (Tsouroula et al., 2016). Nuclear actin drives the migration of a subset of breaks undergoing HR also in mammalian cells (Schrank et al., 2018), but whether those correspond to heterochromatic breaks is not known. Similar to DSBs in pericentromeric heterochromatic repeats, DSBs in nucleolar repeats trigger chromatin mobility. Indeed, in human cells, nucleolar DSBs relocalize to the periphery of nucleoli where they are repaired by HR (van Sluis and McStay, 2015). A recent study provided the first clue to molecular players controlling the relocalization of nucleolar breaks, with a role for myosin chaperones and actin-related proteins (Marnef et al., 2019). The linker of the nucleoskeleton and cytoskeleton (LINC) complex, embedded in the nuclear envelope, also contributes to nucleolar DSB mobility, which involves nuclear envelope invaginations that connect nucleoli (Marnef et al., 2019). Similarly, DSB repair by NHEJ in heterochromatin domains invokes microtubule-mediated chromatin mobility, as reported for the fusion of uncapped telomeres in mouse cells, which is promoted by 53BP1-dependent chromatin mobility through the LINC complex (Dimitrova et al., 2008; Lottersberger et al., 2015). This microtubule-mediated heterochromatin mobility stimulates NHEJ of dysfunctional telomeres (Lottersberger et al., 2015). Together, these studies put forward the role of the nucleoskeleton in regulating damaged heterochromatin mobility to support DSB repair in several contexts (Figure 2). Considering the emerging role of phase separation in regulating DNA damage responses (Spegg and Altmeyer, 2021), we can envision molecular condensates as part of an alternative or cooperative mechanism to control DSB dynamics.
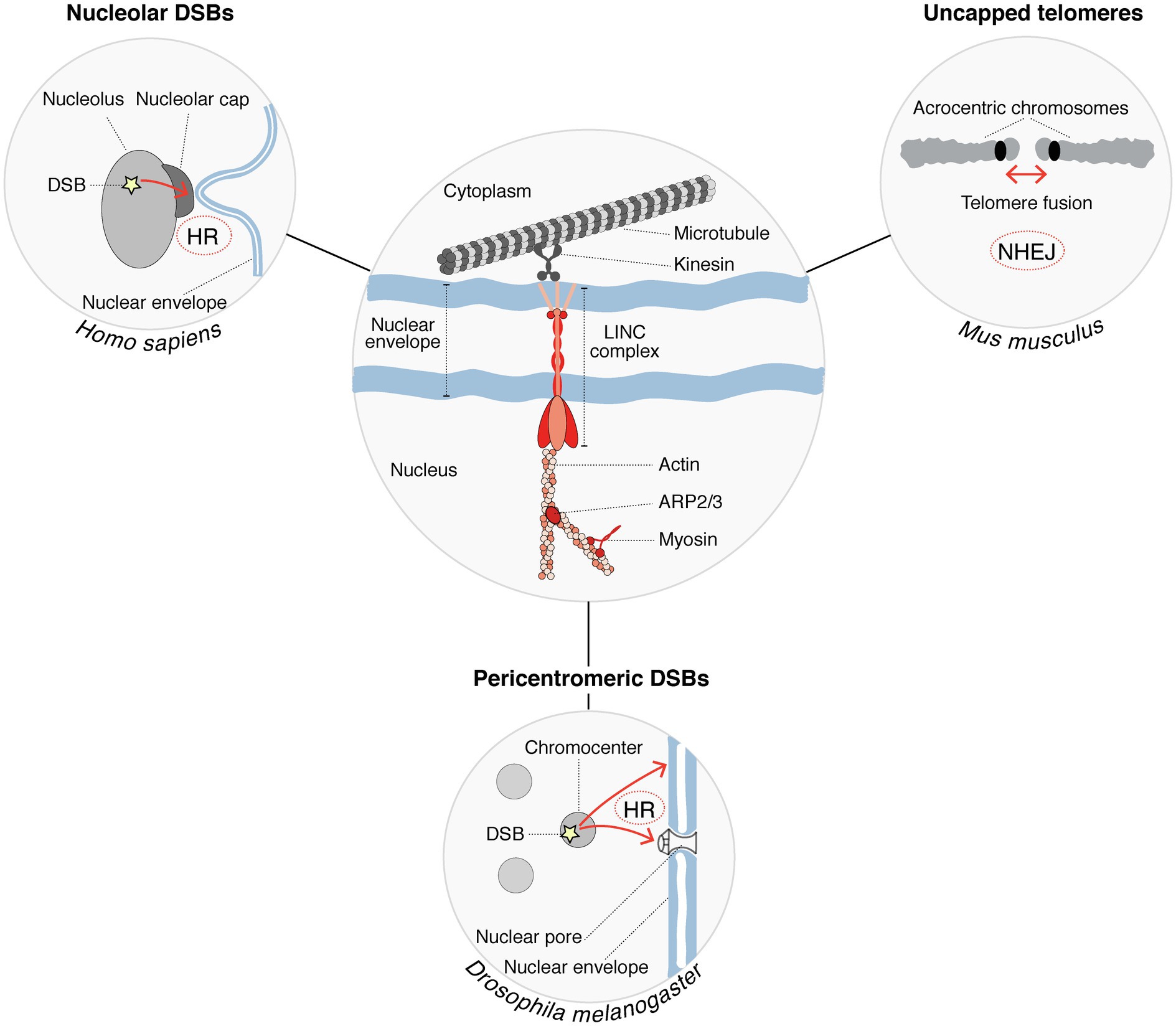
Figure 2. Cytoskeleton factors regulate the mobility of damaged heterochromatin to support DSB repair. Nucleoskeleton factors and molecular motors (shown in red in the center circle) are involved in the mobility of heterochromatic DSBs (DSB mobility is represented by red arrows in the peripheral circles). Chromatin mobility is further supported by microtubules, which provide mechanical forces. This process is conserved in several species and promotes the repair of DSBs occurring within different heterochromatin compartments. DSB repair pathways operating in each case are indicated.
Heterochromatin regions play a crucial role in silencing transposable elements and in regulating the segregation and stability of chromosomes (Allshire and Madhani, 2018). Thus, the maintenance of heterochromatin features is essential to preserve genome integrity and cell identity. However, very little is known regarding whether and how heterochromatin organization is faithfully re-established after DSB repair. One would assume that the heterochromatin compaction state would be restored and that repaired loci would retrieve their original positions inside heterochromatin domains. To address these questions, long-term kinetic analyses should be carried out post DSB induction in order to follow changes in heterochromatin organization during the course of the repair process and even beyond DSB repair completion. A refined analysis of heterochromatin folding during DSB repair would also be needed. Several recent studies have shed light on the impact of DSBs on chromatin folding in the nuclear space and on the importance of 3D chromatin organization in shaping DSB responses by exploiting chromatin conformation capture and super-resolution microscopy (Natale et al., 2017; Ochs et al., 2019; Sanders et al., 2020; Arnould et al., 2021). Similar approaches would help to determine whether heterochromatin compartments retrieve their original topology after DSB repair and to dissect the underlying molecular mechanisms.
Despite dramatic changes in heterochromatin organization following DNA breaks, some heterochromatin marks are maintained during the DSB repair process, as shown for H3K9me3 in mouse pericentromeric heterochromatin (Tsouroula et al., 2016; Natale et al., 2017) but not in Drosophila, where H3K9me3 levels decrease post DSB (Janssen et al., 2019). The mechanisms supporting the maintenance or the restoration of H3K9me3 within these heterochromatic domains are not yet elucidated; however, it is tempting to envision a similar response to what is observed in UV-damaged pericentromeric heterochromatin domains in mouse cells, where the histone methyltransferase SETDB1 is recruited and coordinates the maintenance of H3K9me3 with new H3 deposition during UV damage repair (Fortuny et al., 2021).
DNA double-strand breaks within transcribed genes trigger the incorporation of heterochromatin-specific histone variants and histone post-translational modifications, leading to a transient heterochromatinization of the damaged locus, which contributes to transcriptional silencing (Figure 3). Among the many regulators of transcriptional silencing at DSBs (Caron et al., 2019), heterochromatin marks play a pivotal role. For instance, mono-ubiquitylation of H2A on Lys 119 is induced in the vicinity of DSBs and governs transcriptional silencing (Shanbhag et al., 2010). Interestingly, the removal of H2AK119ub involves the deubiquitinase ubiquitin-specific peptidase 16 (USP16) and is crucial for transcription restart after DSB repair (Shanbhag et al., 2010). Silencing at DSBs is also contributed to by the deposition of H3K27me3 through the PARP1-EZH2 axis (Abu-Zhayia et al., 2018). However, how this histone mark is removed once the break is repaired and whether it is required to license transcription restart is still unknown. The histone demethylase ubiquitously-transcribed tetratricopeptide repeat X (UTX) was shown to diminish H3K27me3 levels in response to ionizing radiation (Rath et al., 2018). It is thus tempting to speculate that the removal of H3K27me3 upon DSB repair may be mediated by UTX, contributing to transcription restart. The transient heterochromatinization at euchromatic breaks also involves H3K9me2/3 deposition (Ayrapetov et al., 2014; Khurana et al., 2014). H3K9 dimethylation is deposited by PR/SET domain 2 (PRDM2), recruited to DSBs in a manner dependent on the histone variant macroH2A1.2 (Khurana et al., 2014). This histone variant also inhibits transcription at DSBs by stimulating H3K4me3 demethylation by KDM5A (Kumbhar et al., 2021). Further studies will be needed to investigate reversal mechanisms of these heterochromatin marks after DSB repair, including the removal of macroH2A1.2, H3K9me2/3, and their importance for transcription recovery.
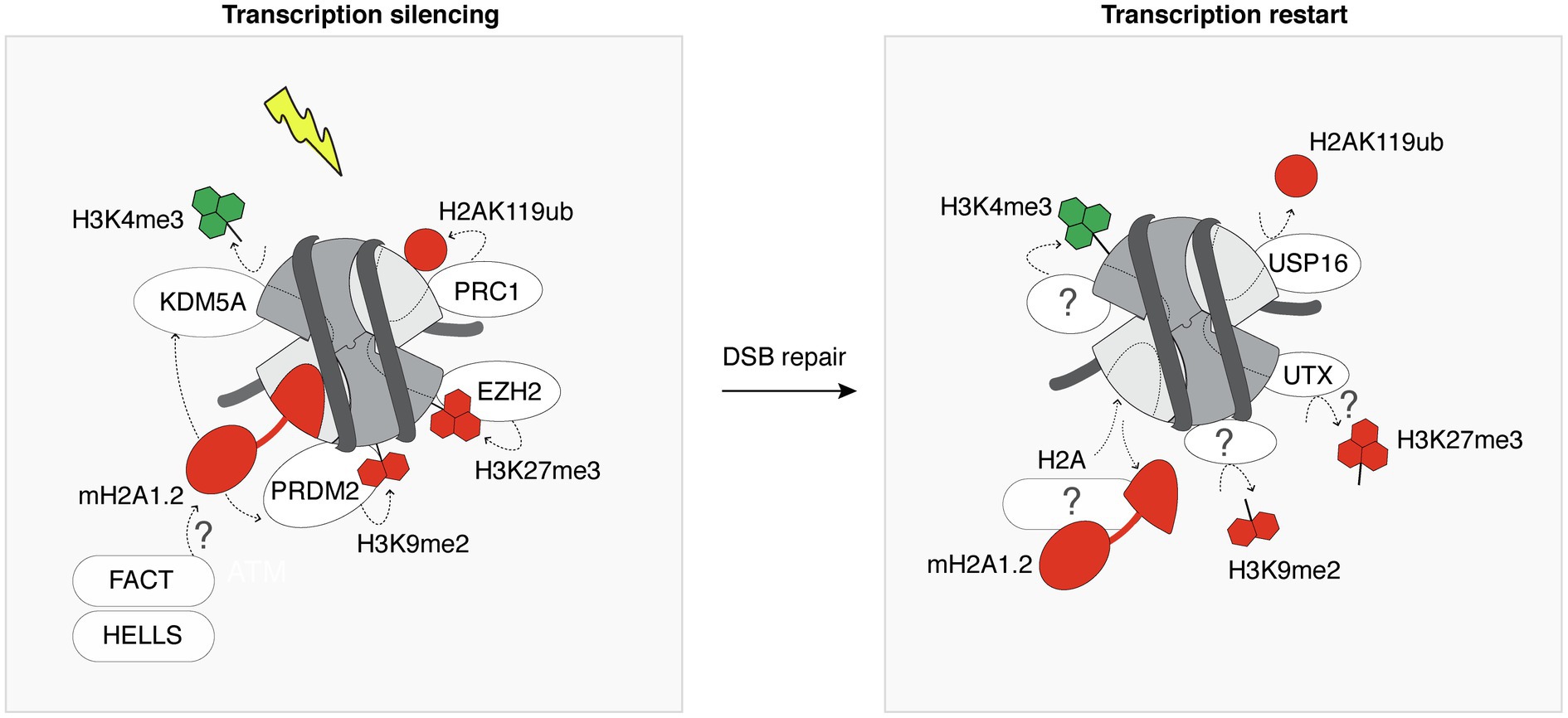
Figure 3. Heterochromatin features are transiently established following DSBs. Following DSB induction, the chromatin surrounding the lesion is modified by the deposition of heterochromatin-specific histone variants and post-translational modifications (red) at the expense of active marks (green). This contributes to silence transcription in the vicinity of the DSB. For simplicity, all chromatin marks are represented on a single nucleosome flanking a DSB. Most of the players that promote the recovery of the pre-existing marks after completion of DSB repair are still unknown, as indicated by the question marks. White ovals and rectangles represent histone modifying enzymes and histone chaperone/remodeler, respectively.
During the last decade, a growing number of studies have highlighted the key contribution of histone post-translational modifications and factors implicated in heterochromatin formation in the response to DSBs. Heterochromatin marks, either pre-existing in heterochromatin domains or de novo established in euchromatin, indeed play a central role in regulating DSB repair pathway choice. Thus, heterochromatin features should not be considered as barriers to DSB repair but as fine-tuners of the DSB response. While our knowledge of the DSB repair pathways that operate in different heterochromatin domains is increasing, the players involved in restoring heterochromatin organization and in erasing heterochromatin marks from euchromatin regions after DSB repair are still unknown. Beyond histone variants and modifications, another crucial epigenetic mark enriched in heterochromatin domains is DNA methylation. Interestingly, DSB repair alters DNA methylation patterns (Sriraman et al., 2020), but little is known about the mechanisms allowing DNA methylation restoration. Furthermore, the DNA methyltransferase DNMT1 can read heterochromatin histone marks thus protecting cells against ionizing radiation (Ren et al., 2020, 2021). These findings suggest a potential role for DNA methylation in controlling DSB repair responses, which is still to be elucidated. Future work will shed light on these mechanisms and on the interplay between different heterochromatin marks in regulating DSB responses. This will help move toward a better characterization of genome and epigenome maintenance processes whose defects underlie pathological disorders.
All authors contributed to write this review and approved the submitted version.
Research in the SP Lab is supported by the European Research Council (ERC-2018-CoG-818625), the French National Research Agency (ANR-18-CE12-0017-01), and the Labex “Who am I?” (ANR-11-LABX-0071; ANR-18-IDEX-0001). PC is recipient of a post-doctoral grant from the Fondation pour la Recherche Médicale (ARF201909009206). EP is a recipient of a PhD fellowship from the H2020 ITN aDDRess (812829). SP is an EMBO Young Investigator.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Alexandre Plessier and Beatrice Rondinelli for critical reading of the manuscript.
Abu-Zhayia, E. R., Awwad, S. W., Ben-Oz, B. M., Khoury-Haddad, H., and Ayoub, N. (2018). CDYL1 fosters double-strand break-induced transcription silencing and promotes homology-directed repair. J. Mol. Cell Biol. 10, 341–357. doi: 10.1093/jmcb/mjx050
Alagoz, M., Katsuki, Y., Ogiwara, H., Ogi, T., Shibata, A., Kakarougkas, A., et al. (2015). SETDB1, HP1 and SUV39 promote repositioning of 53BP1 to extend resection during homologous recombination in G2 cells. Nucleic Acids Res. 43, 7931–7944. doi: 10.1093/nar/gkv722
Allshire, R. C., and Madhani, H. D. (2018). Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 19, 229–244. doi: 10.1038/nrm.2017.119
Amaral, N., Ryu, T., Li, X., and Chiolo, I. (2017). Nuclear dynamics of heterochromatin repair. Trends Genet. 33, 86–100. doi: 10.1016/j.tig.2016.12.004
Ambartsumyan, G., Gill, R. K., Perez, S. D., Conway, D., Vincent, J., Dalal, Y., et al. (2010). Centromere protein A dynamics in human pluripotent stem cell self-renewal, differentiation and DNA damage. Hum. Mol. Genet. 19, 3970–3982. doi: 10.1093/hmg/ddq312
Arnould, C., Rocher, V., Finoux, A.-L., Clouaire, T., Li, K., Zhou, F., et al. (2021). Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature 590, 660–665. doi: 10.1038/s41586-021-03193-z
Ayrapetov, M. K., Gursoy-Yuzugullu, O., Xu, C., Xu, Y., and Price, B. D. (2014). DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. U. S. A. 111, 9169–9174. doi: 10.1073/pnas.1403565111
Baldeyron, C., Soria, G., Roche, D., Cook, A. J. L., and Almouzni, G. (2011). HP1alpha recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J. Cell Biol. 193, 81–95. doi: 10.1083/jcb.201101030
Bhargava, R., Onyango, D. O., and Stark, J. M. (2016). Regulation of single-Strand annealing and its role in genome maintenance. Trends Genet. 32, 566–575. doi: 10.1016/j.tig.2016.06.007
Blewitt, M. E., Gendrel, A.-V., Pang, Z., Sparrow, D. B., Whitelaw, N., Craig, J. M., et al. (2008). SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat. Genet. 40, 663–669. doi: 10.1038/ng.142
Caridi, C. P., D'Agostino, C., Ryu, T., Zapotoczny, G., Delabaere, L., Li, X., et al. (2018). Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 559, 54–60. doi: 10.1038/s41586-018-0242-8
Caron, P., van der Linden, J., and van Attikum, H. (2019). Bon voyage: a transcriptional journey around DNA breaks. DNA Repair (Amst) 82:102686. doi: 10.1016/j.dnarep.2019.102686
Chan, F. L., and Wong, L. H. (2012). Transcription in the maintenance of centromere chromatin identity. Nucleic Acids Res. 40, 11178–11188. doi: 10.1093/nar/gks921
Changolkar, L. N., and Pehrson, J. R. (2006). macroH2A1 histone variants are depleted on active genes but concentrated on the inactive X chromosome. Mol. Cell. Biol. 26, 4410–4420. doi: 10.1128/MCB.02258-05
Chen, C.-C., Feng, W., Lim, P. X., Kass, E. M., and Jasin, M. (2018). Homology-directed repair and the role of BRCA1, BRCA2, and related proteins in genome integrity and cancer. Annu Rev Cancer Biol 2, 313–336. doi: 10.1146/annurev-cancerbio-030617-050502
Chiolo, I., Minoda, A., Colmenares, S. U., Polyzos, A., Costes, S. V., and Karpen, G. H. (2011). Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144, 732–744. doi: 10.1016/j.cell.2011.02.012
Clapier, C. R., Iwasa, J., Cairns, B. R., and Peterson, C. L. (2017). Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 18, 407–422. doi: 10.1038/nrm.2017.26
Clouaire, T., Rocher, V., Lashgari, A., Arnould, C., Aguirrebengoa, M., Biernacka, A., et al. (2018). Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 72, 250–262.e6. doi: 10.1016/j.molcel.2018.08.020
Coker, H., and Brockdorff, N. (2014). SMCHD1 accumulates at DNA damage sites and facilitates the repair of DNA double-strand breaks. J. Cell Sci. 127, 1869–1874. doi: 10.1242/jcs.140020
Costanzi, C., and Pehrson, J. R. (1998). Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 393, 599–601. doi: 10.1038/31275
de Krijger, I., van der Torre, J., Peuscher, M. H., Eder, M., and Jacobs, J. J. L. (2020). H3K36 dimethylation by MMSET promotes classical non-homologous end-joining at unprotected telomeres. Oncogene 39, 4814–4827. doi: 10.1038/s41388-020-1334-0
de Lange, T. (2002). Protection of mammalian telomeres. Oncogene 21, 532–540. doi: 10.1038/sj.onc.1205080
Dialynas, G., Delabaere, L., and Chiolo, I. (2019). Arp2/3 and Unc45 maintain heterochromatin stability in Drosophila polytene chromosomes. Exp. Biol. Med. (Maywood) 244, 1362–1371. doi: 10.1177/1535370219862282
Dimitrova, N., Chen, Y.-C., Spector, D., and de Lange, T. (2008). 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456, 524–528. doi: 10.1038/nature07433
Ferrand, J., Plessier, A., and Polo, S. E. (2020). Control of the chromatin response to DNA damage: histone proteins pull the strings. Semin. Cell Dev. Biol. 113, 75–87. doi: 10.1016/j.semcdb.2020.07.002
Fortuny, A., Chansard, A., Caron, P., Chevallier, O., Leroy, O., Renaud, O., et al. (2021). Imaging the response to DNA damage in heterochromatin domains reveals core principles of heterochromatin maintenance. Nat. Commun. 12:2428. doi: 10.1038/s41467-021-22575-5
Fortuny, A., and Polo, S. E. (2018). The response to DNA damage in heterochromatin domains. Chromosoma 127, 291–300. doi: 10.1007/s00412-018-0669-6
Galupa, R., and Heard, E. (2018). X-chromosome inactivation: a crossroads Between chromosome architecture and gene regulation. Annu. Rev. Genet. 52, 535–566. doi: 10.1146/annurev-genet-120116-024611
Gamble, M. J., Frizzell, K. M., Yang, C., Krishnakumar, R., and Kraus, W. L. (2010). The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev. 24, 21–32. doi: 10.1101/gad.1876110
Gauchier, M., Kan, S., Barral, A., Sauzet, S., Agirre, E., Bonnell, E., et al. (2019). SETDB1-dependent heterochromatin stimulates alternative lengthening of telomeres. Sci. Adv. 5:eaav3673. doi: 10.1126/sciadv.aav3673
Gendrel, A.-V., Tang, Y. A., Suzuki, M., Godwin, J., Nesterova, T. B., Greally, J. M., et al. (2013). Epigenetic functions of smchd1 repress gene clusters on the inactive X chromosome and on autosomes. Mol. Cell. Biol. 33, 3150–3165. doi: 10.1128/MCB.00145-13
Goodarzi, A. A., Noon, A. T., Deckbar, D., Ziv, Y., Shiloh, Y., Löbrich, M., et al. (2008). ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 31, 167–177. doi: 10.1016/j.molcel.2008.05.017
Helfricht, A., Wiegant, W. W., Thijssen, P. E., Vertegaal, A. C., Luijsterburg, M. S., and van Attikum, H. (2013). Remodeling and spacing factor 1 (RSF1) deposits centromere proteins at DNA double-strand breaks to promote non-homologous end-joining. Cell Cycle 12, 3070–3082. doi: 10.4161/cc.26033
Jack, A. P. M., Bussemer, S., Hahn, M., Pünzeler, S., Snyder, M., Wells, M., et al. (2013). H3K56me3 is a novel, conserved heterochromatic mark that largely but not completely overlaps with H3K9me3 in both regulation and localization. PLoS One 8:e51765. doi: 10.1371/journal.pone.0051765
Jacquet, K., Fradet-Turcotte, A., Avvakumov, N., Lambert, J.-P., Roques, C., Pandita, R. K., et al. (2016). The TIP60 complex regulates bivalent chromatin recognition by 53BP1 through direct H4K20me binding and H2AK15 acetylation. Mol. Cell 62, 409–421. doi: 10.1016/j.molcel.2016.03.031
Janssen, A., Breuer, G. A., Brinkman, E. K., van der Meulen, A. I., Borden, S. V., van Steensel, B., et al. (2016). A single double-strand break system reveals repair dynamics and mechanisms in heterochromatin and euchromatin. Genes Dev. 30, 1645–1657. doi: 10.1101/gad.283028.116
Janssen, A., Colmenares, S. U., and Karpen, G. H. (2018). Heterochromatin: Guardian of the genome. Annu. Rev. Cell Dev. Biol. 34, 265–288. doi: 10.1146/annurev-cellbio-100617-062653
Janssen, A., Colmenares, S. U., Lee, T., and Karpen, G. H. (2019). Timely double-strand break repair and pathway choice in pericentromeric heterochromatin depend on the histone demethylase dKDM4A. Genes Dev. 33, 103–115. doi: 10.1101/gad.317537.118
Kato, T., Sato, N., Hayama, S., Yamabuki, T., Ito, T., Miyamoto, M., et al. (2007). Activation of Holliday junction recognizing protein involved in the chromosomal stability and immortality of cancer cells. Cancer Res. 67, 8544–8553. doi: 10.1158/0008-5472.CAN-07-1307
Khurana, S., Kruhlak, M. J., Kim, J., Tran, A. D., Liu, J., Nyswaner, K., et al. (2014). A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 8, 1049–1062. doi: 10.1016/j.celrep.2014.07.024
Kim, J., Sturgill, D., Sebastian, R., Khurana, S., Tran, A. D., Edwards, G. B., et al. (2017). Replication stress shapes a protective chromatin environment across fragile genomic regions. Mol. Cell 69, 36–57. doi: 10.1016/j.molcel.2017.11.021
Kim, J., Sun, C., Tran, A. D., Chin, P.-J., Ruiz, P. D., Wang, K., et al. (2019). The macroH2A1.2 histone variant links ATRX loss to alternative telomere lengthening. Nat. Struct. Mol. Biol. 26, 213–219. doi: 10.1038/s41594-019-0192-3
Klement, K., Luijsterburg, M. S., Pinder, J. B., Cena, C. S., Del Nero, V., Wintersinger, C. M., et al. (2014). Opposing ISWI- and CHD-class chromatin remodeling activities orchestrate heterochromatic DNA repair. J. Cell Biol. 207, 717–733. doi: 10.1083/jcb.201405077
Kollárovič, G., Topping, C. E., Shaw, E. P., and Chambers, A. L. (2020). The human HELLS chromatin remodelling protein promotes end resection to facilitate homologous recombination and contributes to DSB repair within heterochromatin. Nucleic Acids Res. 48, 1872–1885. doi: 10.1093/nar/gkz1146
Kumbhar, R., Sanchez, A., Perren, J., Gong, F., Corujo, D., Medina, F., et al. (2021). Poly(ADP-ribose) binding and macroH2A mediate recruitment and functions of KDM5A at DNA lesions. J. Cell Biol. 220:e202006149. doi: 10.1083/jcb.202006149
Lemaître, C., Grabarz, A., Tsouroula, K., Andronov, L., Furst, A., Pankotai, T., et al. (2014). Nuclear position dictates DNA repair pathway choice. Genes Dev. 28, 2450–2463. doi: 10.1101/gad.248369.114
Li, Z., Li, Y., Tang, M., Peng, B., Lu, X., Yang, Q., et al. (2018). Destabilization of linker histone H1.2 is essential for ATM activation and DNA damage repair. Cell Res. 28, 756–770. doi: 10.1038/s41422-018-0048-0
Lim, C. J., and Cech, T. R. (2021). Shaping human telomeres: from shelterin and CST complexes to telomeric chromatin organization. Nat. Rev. Mol. Cell Biol. 22, 283–298. doi: 10.1038/s41580-021-00328-y
Lottersberger, F., Karssemeijer, R. A., Dimitrova, N., and de Lange, T. (2015). 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell 163, 880–893. doi: 10.1016/j.cell.2015.09.057
Marnef, A., Finoux, A.-L., Arnould, C., Guillou, E., Daburon, V., Rocher, V., et al. (2019). A cohesin/HUSH- and LINC-dependent pathway controls ribosomal DNA double-strand break repair. Genes Dev. 33, 1175–1190. doi: 10.1101/gad.324012.119
Martire, S., and Banaszynski, L. A. (2020). The roles of histone variants in fine-tuning chromatin organization and function. Nat. Rev. Mol. Cell Biol. 21, 522–541. doi: 10.1038/s41580-020-0262-8
Mould, A. W., Pang, Z., Pakusch, M., Tonks, I. D., Stark, M., Carrie, D., et al. (2013). Smchd1 regulates a subset of autosomal genes subject to monoallelic expression in addition to being critical for X inactivation. Epigenetics Chromatin 6:19. doi: 10.1186/1756-8935-6-19
Müller, I., Merk, B., Voss, K.-O., Averbeck, N., Jakob, B., Durante, M., et al. (2013). Species conserved DNA damage response at the inactive human X chromosome. Mutat. Res. 756, 30–36. doi: 10.1016/j.mrgentox.2013.04.006
Natale, F., Rapp, A., Yu, W., Maiser, A., Harz, H., Scholl, A., et al. (2017). Identification of the elementary structural units of the DNA damage response. Nat. Commun. 8:15760. doi: 10.1038/ncomms15760
Ni, K., Ren, J., Xu, X., He, Y., Finney, R., Braun, S. M. G., et al. (2020). LSH mediates gene repression through macroH2A deposition. Nat. Commun. 11:5647. doi: 10.1038/s41467-020-19159-0
Ochs, F., Karemore, G., Miron, E., Brown, J., Sedlackova, H., Rask, M.-B., et al. (2019). Stabilization of chromatin topology safeguards genome integrity. Nature 574, 571–574. doi: 10.1038/s41586-019-1659-4
Penagos-Puig, A., and Furlan-Magaril, M. (2020). Heterochromatin as an important driver of genome organization. Front. Cell Dev. Biol. 8:579137. doi: 10.3389/fcell.2020.579137
Qin, B., Yu, J., Nowsheen, S., Wang, M., Tu, X., Liu, T., et al. (2019). UFL1 promotes histone H4 ufmylation and ATM activation. Nat. Commun. 10:1242. doi: 10.1038/s41467-019-09175-0
Qin, B., Yu, J., Nowsheen, S., Zhao, F., Wang, L., and Lou, Z. (2020). STK38 promotes ATM activation by acting as a reader of histone H4 ufmylation. Sci. Adv. 6:eaax8214. doi: 10.1126/sciadv.aax8214
Rath, B. H., Waung, I., Camphausen, K., and Tofilon, P. J. (2018). Inhibition of the histone H3K27 demethylase UTX enhances tumor cell Radiosensitivity. Mol. Cancer Ther. 17, 1070–1078. doi: 10.1158/1535-7163.MCT-17-1053
Ren, W., Fan, H., Grimm, S. A., Guo, Y., Kim, J. J., Yin, J., et al. (2020). Direct readout of heterochromatic H3K9me3 regulates DNMT1-mediated maintenance DNA methylation. Proc. Natl. Acad. Sci. U. S. A. 117, 18439–18447. doi: 10.1073/pnas.2009316117
Ren, W., Fan, H., Grimm, S. A., Kim, J. J., Li, L., Guo, Y., et al. (2021). DNMT1 reads heterochromatic H4K20me3 to reinforce LINE-1 DNA methylation. Nat. Commun. 12:2490. doi: 10.1038/s41467-021-22665-4
Ryu, T., Spatola, B., Delabaere, L., Bowlin, K., Hopp, H., Kunitake, R., et al. (2015). Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat. Cell Biol. 17, 1401–1411. doi: 10.1038/ncb3258
Saksouk, N., Simboeck, E., and Déjardin, J. (2015). Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin 8:3. doi: 10.1186/1756-8935-8-3
Sallmyr, A., and Tomkinson, A. E. (2018). Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 293, 10536–10546. doi: 10.1074/jbc.TM117.000375
Sanders, J. T., Freeman, T. F., Xu, Y., Golloshi, R., Stallard, M. A., Hill, A. M., et al. (2020). Radiation-induced DNA damage and repair effects on 3D genome organization. Nat. Commun. 11:6178. doi: 10.1038/s41467-020-20047-w
Schep, R., Brinkman, E. K., Leemans, C., Vergara, X., van der Weide, R. H., Morris, B., et al. (2021). Impact of chromatin context on Cas9-induced DNA double-strand break repair pathway balance. Mol. Cell 81, 2216–2230.e10. doi: 10.1016/j.molcel.2021.03.032
Schrank, B. R., Aparicio, T., Li, Y., Chang, W., Chait, B. T., Gundersen, G. G., et al. (2018). Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature 559, 61–66. doi: 10.1038/s41586-018-0237-5
Scully, R., Panday, A., Elango, R., and Willis, N. A. (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 20, 698–714. doi: 10.1038/s41580-019-0152-0
Sebastian, R., Hosogane, E. K., Sun, E. G., Tran, A. D., Reinhold, W. C., Burkett, S., et al. (2020). Epigenetic regulation of DNA repair pathway choice by MacroH2A1 splice variants ensures genome stability. Mol. Cell 79, 836–845.e7. doi: 10.1016/j.molcel.2020.06.028
Shanbhag, N. M., Rafalska-Metcalf, I. U., Balane-Bolivar, C., Janicki, S. M., and Greenberg, R. A. (2010). ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981. doi: 10.1016/j.cell.2010.04.038
Soria, G., and Almouzni, G. (2013). Differential contribution of HP1 proteins to DNA end resection and homology-directed repair. Cell Cycle 12, 422–429. doi: 10.4161/cc.23215
Spegg, V., and Altmeyer, M. (2021). Biomolecular condensates at sites of DNA damage: more than just a phase. DNA Repair (Amst) 106:103179. doi: 10.1016/j.dnarep.2021.103179
Sriraman, A., Debnath, T. K., Xhemalce, B., and Miller, K. M. (2020). Making it or breaking it: DNA methylation and genome integrity. Essays Biochem. 64, 687–703. doi: 10.1042/EBC20200009
Strickfaden, H., McDonald, D., Kruhlak, M. J., Haince, J.-F., Th’ng, J. P. H., Rouleau, M., et al. (2016). Poly(ADP-ribosyl)ation-dependent Transient Chromatin Decondensation and Histone Displacement following Laser Microirradiation. J. Biol. Chem. 291, 1789–1802. doi: 10.1074/jbc.M115.694992
Sulkowski, P. L., Oeck, S., Dow, J., Economos, N. G., Mirfakhraie, L., Liu, Y., et al. (2020). Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 582, 586–591. doi: 10.1038/s41586-020-2363-0
Sun, Y., Jiang, X., Xu, Y., Ayrapetov, M. K., Moreau, L. A., Whetstine, J. R., et al. (2009). Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 11, 1376–1382. doi: 10.1038/ncb1982
Tang, J., Cho, N. W., Cui, G., Manion, E. M., Shanbhag, N. M., Botuyan, M. V., et al. (2013). Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 20, 317–325. doi: 10.1038/nsmb.2499
Tang, M., Li, Y., Zhang, X., Deng, T., Zhou, Z., Ma, W., et al. (2014). Structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1) promotes non-homologous end joining and inhibits homologous recombination repair upon DNA damage. J. Biol. Chem. 289, 34024–34032. doi: 10.1074/jbc.M114.601179
Trojer, P., and Reinberg, D. (2007). Facultative heterochromatin: is there a distinctive molecular signature? Mol. Cell 28, 1–13. doi: 10.1016/j.molcel.2007.09.011
Tsouroula, K., Furst, A., Rogier, M., Heyer, V., Maglott-Roth, A., Ferrand, A., et al. (2016). Temporal and spatial uncoupling of DNA double Strand break repair pathways within mammalian heterochromatin. Mol. Cell 63, 293–305. doi: 10.1016/j.molcel.2016.06.002
van Sluis, M., and McStay, B. (2015). A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 29, 1151–1163. doi: 10.1101/gad.260703.115
van Steensel, B., and Belmont, A. S. (2017). Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791. doi: 10.1016/j.cell.2017.04.022
Vancevska, A., Ahmed, W., Pfeiffer, V., Feretzaki, M., Boulton, S. J., and Lingner, J. (2020). SMCHD1 promotes ATM-dependent DNA damage signaling and repair of uncapped telomeres. EMBO J. 39:e102668. doi: 10.15252/embj.2019102668
Wu, W., Nishikawa, H., Fukuda, T., Vittal, V., Asano, M., Miyoshi, Y., et al. (2015). Interaction of BARD1 and HP1 is required for BRCA1 retention at sites of DNA damage. Cancer Res. 75, 1311–1321. doi: 10.1158/0008-5472.CAN-14-2796
Xu, X., Ni, K., He, Y., Ren, J., Sun, C., Liu, Y., et al. (2021). The epigenetic regulator LSH maintains fork protection and genomic stability via MacroH2A deposition and RAD51 filament formation. Nat. Commun. 12:3520. doi: 10.1038/s41467-021-23809-2
Zeitlin, S. G., Baker, N. M., Chapados, B. R., Soutoglou, E., Wang, J. Y. J., Berns, M. W., et al. (2009). Double-strand DNA breaks recruit the centromeric histone CENP-A. Proc. Natl. Acad. Sci. U. S. A. 106, 15762–15767. doi: 10.1073/pnas.0908233106
Zhao, B., Rothenberg, E., Ramsden, D. A., and Lieber, M. R. (2020). The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 21, 765–781. doi: 10.1038/s41580-020-00297-8
Keywords: chromatin mobility, chromatin remodeling factors, DNA double-strand break repair pathway choice, heterochromatin, histone variants, histone modifications
Citation: Caron P, Pobega E and Polo SE (2021) DNA Double-Strand Break Repair: All Roads Lead to HeterochROMAtin Marks. Front. Genet. 12:730696. doi: 10.3389/fgene.2021.730696
Edited by:
Matthias Altmeyer, University of Zurich, SwitzerlandReviewed by:
Kyle Miller, The University of Texas at Austin, United StatesCopyright © 2021 Caron, Pobega and Polo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie E. Polo, c29waGllLnBvbG9AdS1wYXJpcy5mcg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.