 Xiaojing Xu1†
Xiaojing Xu1† Juan Zhao1†
Juan Zhao1† Chao Wang1Xiaoxuan Qu1Menglong Ran2,3
Chao Wang1Xiaoxuan Qu1Menglong Ran2,3 Fang Ye1Ming Shen1
Fang Ye1Ming Shen1 Kundi Wang1
Kundi Wang1 Qi Zhang1*
Qi Zhang1*- 1Department of Pediatrics, China-Japan Friendship Hospital, Beijing, China
- 2Department of Dermatology and Venereology, Peking University First Hospital, Beijing, China
- 3Beijing Key Laboratory of Molecular Diagnosis on Dermatoses, Beijing, China
Objectives: The aim of this study was to determine the molecular etiology and clinical manifestations of a pair of Chinese twins affected with epidermolysis bullosa simplex. Pediatricians should pay attention to the early genetic diagnosis of this disease.
Methods: Histopathological examination of HE-stained skin, electron microscopy of biopsied normal skin, and whole-exome sequencing was performed to assess pathogenicity and conservation of detected mutations. Two years later, the cutaneous and extracutaneous manifestations of the twins were comprehensively evaluated.
Results: A de novo pathogenic variant c.2T>C (p.M1T) in KLHL24 (NM_017,644) was identified in both twins. The characteristics of extensive skin defects on the extremities at birth and the tendency to lesson with increasing age were confirmed. No positive sensitive markers, such as B-type natriuretic peptide, cardiac troponin I, for cardiac dysfunction were detected.
Conclusions: The de novo pathogenic variants c.2T>C (p.M1T) in KLHL24 (NM_017,644) contributes to the development of epidermolysis bullosa. Genetic diagnosis at birth or early infancy can better predict the disease prognosis and guide the treatment.
Introduction
Inherited epidermolysis bullosa (EB) is a genetically heterogeneous disorder, characterized by skin fragility annexed with the formation of blisters and skin erosion in response to minor mechanical trauma.1 Currently, over twenty genes that encode structural proteins within keratin intermediate filaments, focal adhesion desmosome cell junctions, and hemidesmosome attachment complexes have been reported in the pathogenesis of EB. (Has and Fischer, 2018; Vahidnezhad et al., 2018). Clinically, EB is classified into four major groups based on the plane of cleavage within the skin, viz. epidermolysis bullosa simplex (EBS), junctional epidermolysis bullosa (JEB), dystrophic epidermolysis bullosa (DEB), and Kindler epidermolysis bullosa (KEB) (Bardhan et al., 2020). The diagnosis and classification of EB are based on the morphological analysis of a skin sample using immunohistologic methods and on the analysis of the pathogenic variants of the candidate genes. (Has and He, 2016; Has and Fischer, 2018). As the most common type of EB, EBS is mainly caused by monoallelic pathogenic variants in KRT5 (MIM: 148,040) and KRT14 (MIM: 148,066), which encode keratin 5 and keratin 14, respectively. In addition, some cases of EBS were reported to associate with other pathogenic variants in PLEC (plectin), EXPH5 (exophilin-5), DST (dystonin, 230-kDa bullous pemphigoid antigen), and CD151 (member of the tetraspanin superfamily). (Karamatic Crew et al., 2004; Groves et al., 2010; McGrath et al., 2012; Bolling et al., 2014; Bardhan et al., 2020). Recently, pathogenic variants in KLHL24 (MIM: 611,295) encoding the Kelch-like protein 24 have been identified in cases with skin fragility and progressive thickening of the nails by whole-exome sequencing. To date, about 40 individuals with monoallelic pathogenic variants of KLHL24 have been reported. (He et al., 2016; Lin et al., 2016; Lee et al., 2017; Alkhalifah et al., 2018; Yenamandra et al., 2018; Grilletta, 2019; Hachem et al., 2019; Schwieger-Briel et al., 2019). KLHL24 is part of the family of more than 40 genes with a Kelch-like motif, and it partially forms the ubiquitin-ligase complex. (Dhanoa et al., 2013). These pathogenic variants caused the loss of the first 28 amino acids of the encoded protein. The mutant protein promotes excessive ubiquitination and degradation of KRT14. The above observations have invoked a new mechanism that is germane to inherited skin blistering, namely, dysregulation of autoubiquitination. (He et al., 2016). Most KLHL24 positive patients carry a heterozygous pathogenic variant in the first codon that affects translation initiation. (He et al., 2016; Lin et al., 2016; Lee et al., 2017; Alkhalifah et al., 2018; Yenamandra et al., 2018).
The clinical diagnosis for EB can be difficult at birth or in the early infancy, even for experienced dermatologists, particularly without an established family history of the disease. An important part of EB research lies in the diagnosis and classification of the disease at the early stage. The optimal treatment regime for disease complications has to be assessed for a long time. Here, we reported a case of twin boys with de novo KLHL24 pathogenic variants. This is the first study to describe the pathogenic variant in KLHL24 affecting Chinese twins. The twin brothers were diagnosed, screened, and treated effectively at the early stage by pediatricians. Meanwhile, cutaneous and extracutaneous manifestations were evaluated at the age of two. This case report will help pediatricians, not confined to dermatologists, to pay enough attention to the early diagnosis and long-term management of EB.
Case Report
The dichorionic twin boys in this report were born at the 32nd week of gestation as a result of a large intracranial hemorrhage in their mother, who had a history of multiple spontaneous abortions under diverse complications, including antiphospholipid antibody syndrome and subclinical hypothyroidism during pregnancy, and she took multiple medications during pregnancy, including methylprednisolone, hydroxychloroquine, and aspirin. The older brother’s body weight was 1,380 g, less than third percentile of typical boys of the same gestational age. At birth, he presented with extensive areas of denuded skin involving the limbs, knees, wrist joints, and ankle joints (Figure 1A). The younger brother’s body weight was 1,650 g, which ranges between the 25th and 50th percentiles for boys of the same gestational age. His skin had the same appearance as his brother’s. New skin defects occurred on the twins faces after positive-pressure ventilation. At birth, both the white blood cell count and neutrophil count of the two brothers were low. The white blood cell count of the elder brother was 1.94*109/L and the neutrophil count was 0.2*109/L, and that of the younger brother was 1.58*109/L and 0.29*109/L respectively. The course was complicated by Serratia marcescens sepsis as a result of preterm labor. The twins homocysteine levels at birth were 4.42 and 4.61 μmol/L (normal ≤15 μmol/L) respectively. The results of echocardiography indicated congenital heart disease and atrial septal defect (secondary foramen type), and the degree of interruption was 6 and 5 mm, respectively. The remaining systemic examination was normal. Histopathological examination of hematoxylin-eosin (HE)–stained skin and electron microscopy (EM) of normal skin biopsy were performed in a reference center for EB. In the older brother, pathology showed no epidermis or intradermal vascular hyperplasia (Figure 1B). EM revealed cleavage within the epidermal basal layer, some epidermal cells with a large amount of melanin deposition, reduction in the local density of the superficial dermis, and partial basal cell degeneration with vacuolar changes (Figure 1C). In his younger brother, histopathological examination showed no epidermis and dermis with only a lipid membrane structure. EM also revealed cleavage within the epidermal basal layer and the substrate, incomplete basal cells within the dermis, and reduction in the local density of the superficial dermis (Figure 1D). These results collectively suggest EBS. Soon thereafter, a whole-exome sequencing analysis was performed of peripheral blood DNA for this family. We identified the de novo variants c.2T>C (p.M1T) in KLHL24 (NM_017,644) from the two boys, which were previously reported to be pathogenic. (He et al., 2016). We have applied ACMG, PolyPhen-2 and PROVEN criteria to prove the pathogenicity of this mutation c.2T>C. Moreover, variant c.2T>C was absent in cohorts of healthy control subjects in dbSNP, 1,000 Genomes, the Exome Variant Server, and the ExAC Browser in previous report (He et al., 2016). Sanger sequencing confirmed these de novo pathogenic variants (Figure 1E). Treatment consisted largely of supportive care, including wound care, as well as prevention and treatment of complications. Mupirocin ointment and recombinant bovine basic fibroblast growth factor were mixed at a 1:1 ratio, then above medicine was applied on the oil gauze, and finally the oil gauze was covered the skins wound. The treatment was carried out every other day on the twin boys. After about 1 month, the stability of the skins was enhanced gradually, above skin treatments were carried on when necessary.
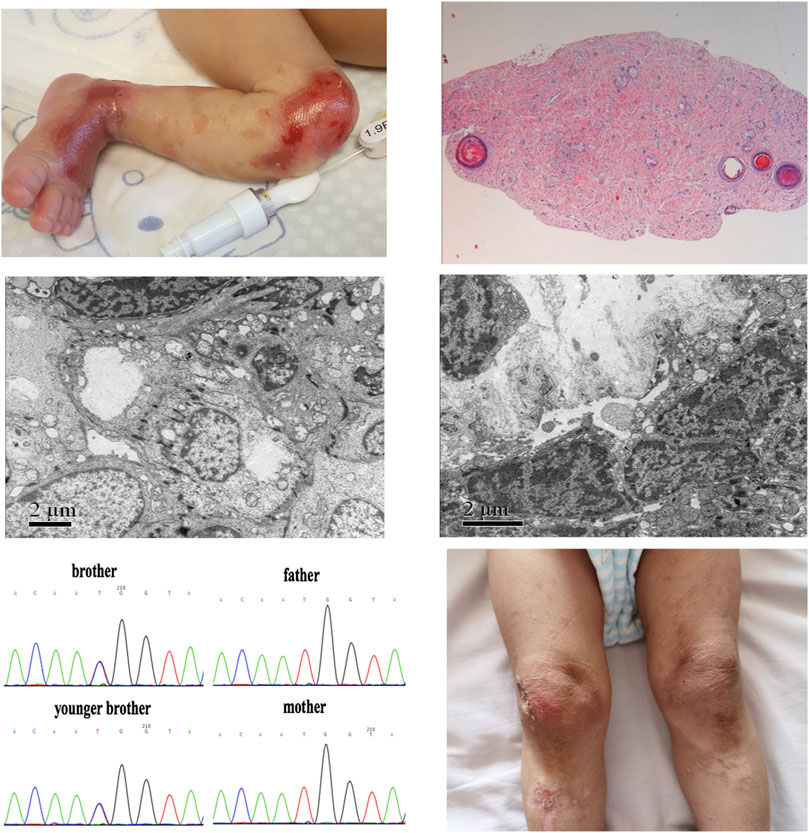
FIGURE 1. Skin image, pathology, and de novo pathogenic variants of KLHL24 in the twin brothers. (A)Picture of the older brother’s left leg at birth demonstrated congenital skin defects. (B)Histopathological examination of HE-stained skin in the older brother (C) Skin performance under EM in the older brother (D) Skin performance under EM in the younger brother (E) De novo pathogenic variants of c.2T>C (p.M1T) in KLHL24 of this ancestry (F) Picture of younger brother’s legs at the age of 2 years demonstrated skin damage, pigmentation and scar.
At the age of 2 years, we followed up with the two brothers. There were old scars, pigmentation, nail thickening, and yellowing, no joint contracture and functional damage, and few new blisters (Figure 1F). Their head circumferences were below the third percentile of boys after adjusted age, and their heights and body weights were between the third and 10th percentiles. Considering the existence of extrauterine growth retardation, it may be related to the lack of functional training, regular follow-up, and the late addition of complementary food. Both brothers were assessed for Gesell Developmental Observation, and they showed mild retardation in adaptability, fine movement and personal social interaction, moderate retardation in language, and normal serum myocardial enzymes (except the MB isoenzyme of creatine kinase is higher than normal in elder brother). The elder brother’s brain MRI showed myelination delay, and echocardiography showed atrial septal defect (5 mm) and a small amount of tricuspid regurgitation, yet the results of the younger brother’s brain MRI and echocardiography were normal.
Discussion
KLHL24 was first reported by He et al., (He et al., 2016), who discovered heterozygous pathogenic variants of this gene in 5 unrelated individuals with EBS. (Lin et al., 2016). KLHL24 is expressed in multiple tissues, including heart, brain, liver, skeleton muscle, kidney, pancreas, placenta, lung, and peripheral blood, as well as in the main skin cell types: keratinocytes, fibroblasts, and melanocytes. (He et al., 2016; Lin et al., 2016). It was of interest to note that all 26 previously reported patients harbored monoallelic pathogenic variants in the KLHL24 translation start codon, c.1A > G, c.1A > T, c.2T > C, c.3G > T, c.3G > A, with a high rate of de novo and recurrent pathogenic variants. (Has and Fischer, 2018), (Has, 2017) In our case, it is the first to describe c.2T>C pathogenic variant in KLHL24 affecting twins in China and it was correlated with EB simplex. He et al. (He et al., 2016) also found truncated KLHL24 resulting from the start codon mutations and the use of a downstream methionine initiation codon. Abnormal intermediate filaments in keratinocytes and fibroblasts, with evidence for irregular and fragmented KRT14, and data to support an altered balance in the stability and degradation of this keratin (He et al., 2016).
The clinical manifestations of our cases included extensive skin defects on the extremities at birth, leaving hypopigmentation and atrophy with a whirled pattern, in accordance with the characteristic of KLHL24. In addition, early blistering occurred often in the trunk and upper limbs, especially after the compression or friction. These lesions were typically healed quickly with subtle atrophic scarring. Based on the results of other studies, blistering persists throughout childhood but tends to lesson with increasing age. (Hachem et al., 2019). Nail defects and oral ulceration are common, whereas transient milia also occur. Dyspigmentation is not a prominent feature. Other reported features include cutaneous findings, such as loss of dermatoglyphics, hypohidrosis, and congenital malrotation of the great toenails, besides mental problems. (Yenamandra et al., 2018). Whether these symptoms are related to KLHL24 requires further investigations. After 2 years of follow-up in our study, we found that the skin defects became milder, nails became thicken and yellowing, the oral ulceration was not obvious.
Indeed, the wide tissue distributions of KLHL24 suggest that pathogenic variants could affect organs other than the skin. Schwieger et al. (Schwieger-Briel et al., 2019) found evidence of dilated cardiomyopathy in 8 of 20 EBS-KLHL24 patients (40%), with the youngest being 25 years. He et al. (He et al., 2016) also noticed dilated cardiomyopathy in a 43 year-old patient, although the age of onset in this patient was unclear. In our report, the cardiac ultrasound examinations of the boys indicated congenital heart disease (atrial septal defect). Other sensitive markers, such as B-type natriuretic peptide, cardiac troponin I, for cardiac dysfunction were proved negative until hospital discharge (except for the MB isoenzyme of creatine kinase is higher than normal in the elder brother). Because of the skin damage and other problems caused by the disease, the children need special care in daily life which produced heavy financial burden for the family. At 2 years of birth, they were comprehensively evaluated by pediatricians, dermatologists, and neurologists. The dichorionic twin boys in this report were born at the 32nd week of gestation. At the 2-year follow-up, except for old scars, we only found the elder brother’s brain MRI showed delayed myelination, and echocardiography showed atrial septal defect (5 mm) and a small amount of tricuspid regurgitation. Their phenotypes were compared with previous cases reported with c.2T>C variant in KLHL24 (Table 1). As myelination is a progression phenotype, later examinations will be required to confirm their brain developmental status. Congenital heart disease is very common in premature infants. Clinicians also need further follow-ups to determine the future treatment plan. Future cardiac complications may emerge with age, and this should be a focus of the treating physician. Considerably different phenotypes of pathogenic variants have been reported within EB subtypes. There were correlations between phenotypes and genotypes in EB. KLHL24 pathogenic variants were associated with the mild phenotype, such as EB simplex.
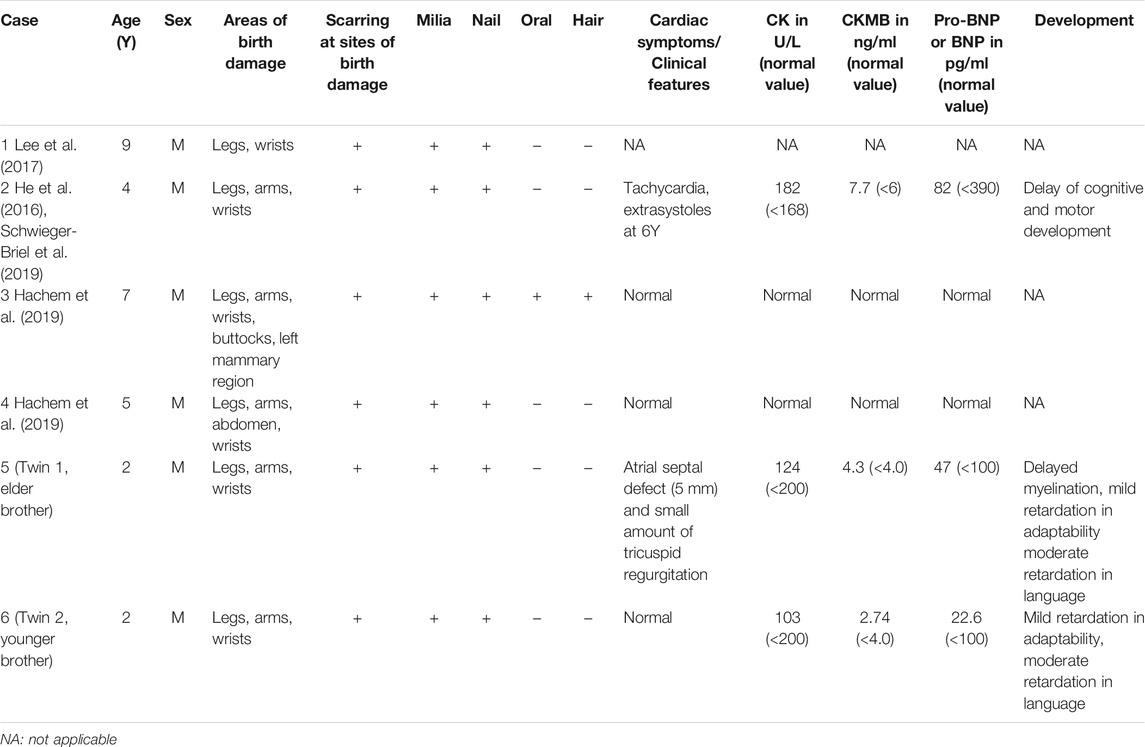
TABLE 1. Summary of clinical features of individuals with c.2T>C pathogenic variant in KLHL24.
Although there is currently no effective treatment for EB, genetic diagnosis at an early age can better predict the prognosis and guide the treatment. We here recommend both genetic and prenatal diagnoses to reduce the incidence of this disease and improve the quality of life.
This study was supported by the Medical and health science and technology innovation project of the Chinese Academy of Medical Sciences (2018-12M-1–003), and approved by the Ethical Committee, China-Japan Friendship Hospital. All study protocols and the report of the clinical research findings are in accordance with federal laws and institutional regulations in China and approved by the institutional review board. Written informed consent was obtained from the father (legal guardian) of the twins to agree to the participation and reporting of the clinical records, as well as the publication of this case report and all information and any accompanying images from the family.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by China-Japan Friendship Hospital. Written informed consent to participate in this study was provided by the participants legal guardian/next of kin.
Author Contributions
XX, JZ: Responsible for the diagnosis and treatment of patients, put forward research ideas, collect datas, write papers MR: Responsible for the extraction, examination and interpretation the results of the cutaneous pathological CW, XQ, FY: Responsible for the concrete treatment of patients) MS, KW, QZ: Responsible for guiding and proposing appropriate treatment plans).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors acknowledge the work of Yali Ren, associate chief physician, from the Electron Microscope Lab of Peking University First Hospital, and thank for the nursing service of pediatric nurses in China-Japan Friendship Hospital.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.729628/full#supplementary-material
References
Alkhalifah, A., Chiaverini, C., Charlesworth, A., Has, C., and Lacour, J.-P. (2018). Burnlike Scars: A Sign Suggestive of KLHL24-Related Epidermolysis Bullosa Simplex. Pediatr. Dermatol. 35, e193–e195. doi:10.1111/pde.13443
Bardhan, A., Bruckner-Tuderman, L., Chapple, I. L. C., Fine, J.-D., Harper, N., Has, C., et al. (2020). Epidermolysis Bullosa. Nat. Rev. Dis. Primers 6, 78. doi:10.1038/s41572-020-0210-0
Bolling, M. C., Jongbloed, J. D. H., Boven, L. G., Diercks, G. F. H., Smith, F. J. D., Irwin McLean, W. H., et al. (2014). Plectin Mutations Underlie Epidermolysis Bullosa Simplex in 8% of Patients. J. Invest. Dermatol. 134, 273–276. doi:10.1038/jid.2013.277
Dhanoa, B. S., Cogliati, T., Satish, A. G., Bruford, E. A., and Friedman, J. S. (2013). Update on the Kelch-like (KLHL) Gene Family. Hum. Genomics 7, 13. doi:10.1186/1479-7364-7-13
Grilletta, E. A. (2019). Cardiac Transplant for Epidermolysis Bullosa Simplex with KLHL24 Mutation-Associated Cardiomyopathy. JAAD Case Rep. 5, 912–914. doi:10.1016/j.jdcr.2019.08.009
Groves, R. W., Liu, L., Dopping-Hepenstal, P. J., Markus, H. S., Lovell, P. A., Ozoemena, L., et al. (2010). A Homozygous Nonsense Mutation within the Dystonin Gene Coding for the Coiled-Coil Domain of the Epithelial Isoform of BPAG1 Underlies a New Subtype of Autosomal Recessive Epidermolysis Bullosa Simplex. J. Invest. Dermatol. 130, 1551–1557. doi:10.1038/jid.2010.19
Hachem, M., Barresi, S., Diociaiuti, A., Boldrini, R., Condorelli, A., Capoluongo, E., et al. (2019). Phenotypic Features of Epidermolysis Bullosa Simplex Due to KLHL24 Mutations in 3 Italian Cases. Acta Derm Venerol 99, 238–239. doi:10.2340/00015555-3046
Has, C., and Fischer, J. (2018). Inherited Epidermolysis Bullosa: New Diagnostics and New Clinical Phenotypes. Exp. Dermatol.. doi:10.1111/exd.13668
Has, C., and He, Y. (2016). Research Techniques Made Simple: Immunofluorescence Antigen Mapping in Epidermolysis Bullosa. J. Invest. Dermatol. 136, e65–e71. doi:10.1016/j.jid.2016.05.093
Has, C. (2017). The "Kelch" Surprise: KLHL24, a New Player in the Pathogenesis of Skin Fragility. J. Invest. Dermatol. 137, 1211–1212. doi:10.1016/j.jid.2017.02.011
He, Y., Maier, K., Leppert, J., Hausser, I., Schwieger-Briel, A., Weibel, L., et al. (2016). Monoallelic Mutations in the Translation Initiation Codon of KLHL24 Cause Skin Fragility. Am. J. Hum. Genet. 99, 1395–1404. doi:10.1016/j.ajhg.2016.11.005
Karamatic Crew, V., Burton, N., Kagan, A., Green, C. A., Levene, C., Flinter, F., et al. (2004). CD151, the First Member of the Tetraspanin (TM4) Superfamily Detected on Erythrocytes, Is Essential for the Correct Assembly of Human Basement Membranes in Kidney and Skin. Blood 104, 2217–2223. doi:10.1182/blood-2004-04-1512
Lee, J. Y. W., Liu, L., Hsu, C.-K., Aristodemou, S., Ozoemena, L., Ogboli, M., et al. (2017). Mutations in KLHL24 Add to the Molecular Heterogeneity of Epidermolysis Bullosa Simplex. J. Invest. Dermatol. 137, 1378–1380. doi:10.1016/j.jid.2017.01.004
Lin, Z., Li, S., Feng, C., Yang, S., Wang, H., Ma, D., et al. (2016). Stabilizing Mutations of KLHL24 Ubiquitin Ligase Cause Loss of Keratin 14 and Human Skin Fragility. Nat. Genet. 48, 1508–1516. doi:10.1038/ng.3701
McGrath, J. A., Stone, K. L., Begum, R., Simpson, M. A., Dopping-Hepenstal, P. J., Liu, L., et al. (2012). Germline Mutation in EXPH5 Implicates the Rab27B Effector Protein Slac2-B in Inherited Skin Fragility. Am. J. Hum. Genet. 91, 1115–1121. doi:10.1016/j.ajhg.2012.10.012
Schwieger-Briel, A., Fuentes, I., Castiglia, D., Barbato, A., Greutmann, M., Leppert, J., et al. (2019). Epidermolysis Bullosa Simplex with KLHL24 Mutations Is Associated with Dilated Cardiomyopathy. J. Invest. Dermatol. 139, 244–249. doi:10.1016/j.jid.2018.07.022
Vahidnezhad, H., Youssefian, L., Saeidian, A. H., Mahmoudi, H., Touati, A., Abiri, M., et al. (2018). Recessive Mutation in Tetraspanin CD151 Causes Kindler Syndrome-like Epidermolysis Bullosa with Multi-Systemic Manifestations Including Nephropathy. Matrix Biol. 66, 22–33. doi:10.1016/j.matbio.2017.11.003
Keywords: KLHL24, de novo pathogenic variants, epidermolysis bullosa, skin defect, follow-up
Citation: Xu X, Zhao J, Wang C, Qu X, Ran M, Ye F, Shen M, Wang K and Zhang Q (2021) Case Report: De novo KLHL24 Gene Pathogenic Variants in Chinese Twin Boys With Epidermolysis Bullosa Simplex. Front. Genet. 12:729628. doi: 10.3389/fgene.2021.729628
Received: 23 June 2021; Accepted: 15 October 2021;
Published: 05 November 2021.
Edited by:
Ming Li, Shanghai Jiaotong University, ChinaReviewed by:
Leslie Matalonga, Center for Genomic Regulation (CRG), SpainKunju Zhu, Jinan University, China
Copyright © 2021 Xu, Zhao, Wang, Qu, Ran, Ye, Shen, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qi Zhang, emhhbmdxaWtleWFuQDE2My5jb20=
†Xiaojing Xu and Juan Zhao have contributed equally to this work and share first authorship