 Robin Vroman
Robin Vroman Anne-Marie Malfait2
Anne-Marie Malfait2 Fransiska Malfait
Fransiska Malfait Delfien Syx
Delfien Syx
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 12 October 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.726474
This article is part of the Research TopicMolecular Mechanisms of Heritable Connective Tissue DisordersView all 9 articles
The Ehlers–Danlos syndromes (EDS) are a group of heritable connective tissues disorders mainly characterized by skin hyperextensibility, joint hypermobility and generalized tissue fragility. Currently, 14 EDS subtypes each with particular phenotypic features are recognized and are caused by genetic defects in 20 different genes. All of these genes are involved in the biosynthesis and/or fibrillogenesis of collagens at some level. Although great progress has been made in elucidating the molecular basis of different EDS subtypes, the pathogenic mechanisms underlying the observed phenotypes remain poorly understood, and consequentially, adequate treatment and management options for these conditions remain scarce. To date, several animal models, mainly mice and zebrafish, have been described with defects in 14 of the 20 hitherto known EDS-associated genes. These models have been instrumental in discerning the functions and roles of the corresponding proteins during development, maturation and repair and in portraying their roles during collagen biosynthesis and/or fibrillogenesis, for some even before their contribution to an EDS phenotype was elucidated. Additionally, extensive phenotypical characterization of these models has shown that they largely phenocopy their human counterparts, with recapitulation of several clinical hallmarks of the corresponding EDS subtype, including dermatological, cardiovascular, musculoskeletal and ocular features, as well as biomechanical and ultrastructural similarities in tissues. In this narrative review, we provide a comprehensive overview of animal models manifesting phenotypes that mimic EDS with a focus on engineered mouse and zebrafish models, and their relevance in past and future EDS research. Additionally, we briefly discuss domestic animals with naturally occurring EDS phenotypes. Collectively, these animal models have only started to reveal glimpses into the pathophysiological aspects associated with EDS and will undoubtably continue to play critical roles in EDS research due to their tremendous potential for pinpointing (common) signaling pathways, unveiling possible therapeutic targets and providing opportunities for preclinical therapeutic interventions.
The Ehlers–Danlos syndromes (EDS) comprise a group of rare heritable connective tissue disorders, clinically hallmarked by skin hyperextensibility and fragility, generalized joint hypermobility, abnormal wound healing, easy bruising, and widespread connective tissue friability (Malfait et al., 2017). Additional clinical features differ among the EDS subtypes and include potentially life-threatening cardiovascular complications, fragility of hollow organs, involvement of the musculoskeletal and/or ocular system as well as chronic, widespread pain, which can all contribute to severe disability, compromising patients’ quality of life, and/or early mortality (Malfait et al., 2020).
Originally suspected to be a disorder affecting the collagen “wickerwork” based on electron microscopy studies (Jansen, 1955), early biochemical and genetic studies indeed identified defects in the primary structure of fibrillar collagens (types I, III, and V) or their modifying enzymes in human patients displaying different subtypes of EDS (Beighton et al., 1998). More recently, defects affecting other extracellular matrix (ECM) molecules (e.g., tenascin-X, enzymes involved in glycosaminoglycan biosynthesis of proteoglycans, etc.) further expanded the molecular and clinical heterogeneity of this syndrome. These findings laid the basis for the revised International Classification of the Ehlers–Danlos Syndromes, published in 2017, which provides a regrouping of EDS based on the underlying genetic defect and affected pathways (Malfait et al., 2017). To date, 14 distinct EDS subtypes are recognized, of which 13 have been molecularly elucidated and are caused by defects in 20 different genes (Malfait et al., 2017, 2020). All of these EDS-related defects compromise proper collagen biosynthesis, fibrillogenesis and/or supramolecular organization of collagen fibrils at some level as demonstrated by altered collagen fibril ultrastructure on transmission electron microscopy (TEM) in patients’ dermis (Hausser and Anton-Lamprecht, 1994; Proske et al., 2006). Although for many EDS types the scope of both genetic and allelic heterogeneity is well appreciated, the pathways linking the genetic changes to the phenotype remain largely elusive. An overview of EDS-related genes and proteins and their corresponding clinical presentation is provided in Table 1.
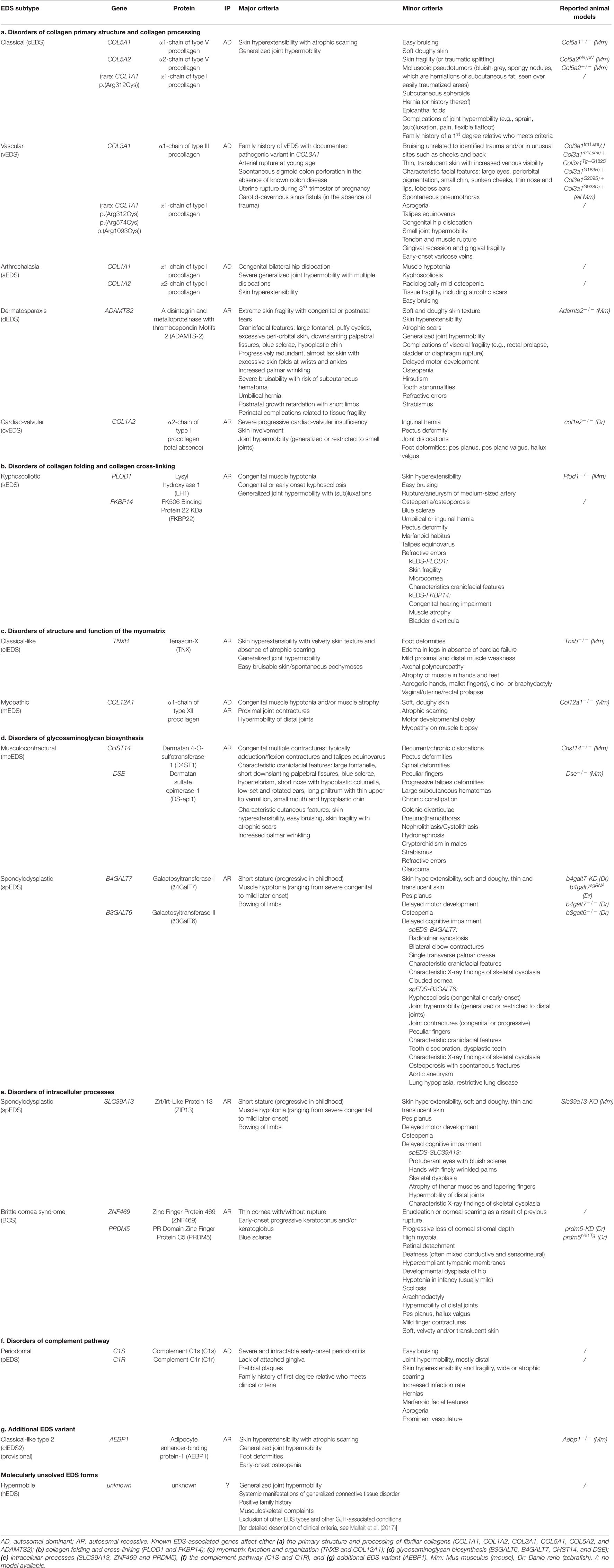
Table 1. Overview of the Ehlers-Danlos syndrome (EDS) types, causal gene and protein, inheritance pattern (IP) and the major and minor clinical criteria as defined by the 2017 International EDS Classification with the reported animal models indicated.
Fibrillar collagens are trimeric proteins consisting of either three identical (homotrimer) or genetically distinct (heterotrimer) polypeptide chains, referred to as pro-α-chains, which contain repeating Gly-Xaa-Yaa triplets, comprising glycine and two other amino acids, and which form a long uninterrupted triple helical domain, flanked by globular carboxy- (C)- and amino- (N-) terminal propeptides.
Biosynthesis and fibrillogenesis have been extensively studied for the prototypic and most abundant type I collagen and are complex processes requiring the orchestrated action of several modifying enzymes, chaperones and ECM molecules (Figure 1A; Myllyharju and Kivirikko, 2004; Canty and Kadler, 2005). In brief, nascent pro-α-chains are heavily post-translationally modified, followed by association of the C-terminal propeptides of three pro-α-chains which assemble into a trimeric procollagen molecule propagating to the N-terminus in a zipperlike fashion. These procollagens are subsequently transported to the extracellular environment and N- and C-propeptides are proteolytically removed, resulting in the formation of a collagen molecule that can then assemble into highly ordered cross-striated fibrils and fibers (Birk and Brückner, 2010). Collagen fibrils are usually composed of different fibrillar collagen types and collagen fibril assembly occurs in a tissue-specific way, requiring the concerted action of several assisting proteins, proposed to be categorized into three general classes: organizers (e.g., fibronectin, integrins), nucleators (e.g., type V collagen) and regulators [e.g., small leucine-rich proteoglycans (SLRPs), fibril-associated collagens with interrupted triple helices (FACIT), and glycoproteins] (Kadler et al., 2008). As fibrillogenesis proceeds, fibril growth occurs through linear and lateral fusion of intermediate collagen fibrils which are subsequently stabilized by the formation of intra- and inter-molecular cross-links (Birk, 2001; Canty and Kadler, 2005; Greenspan, 2005).
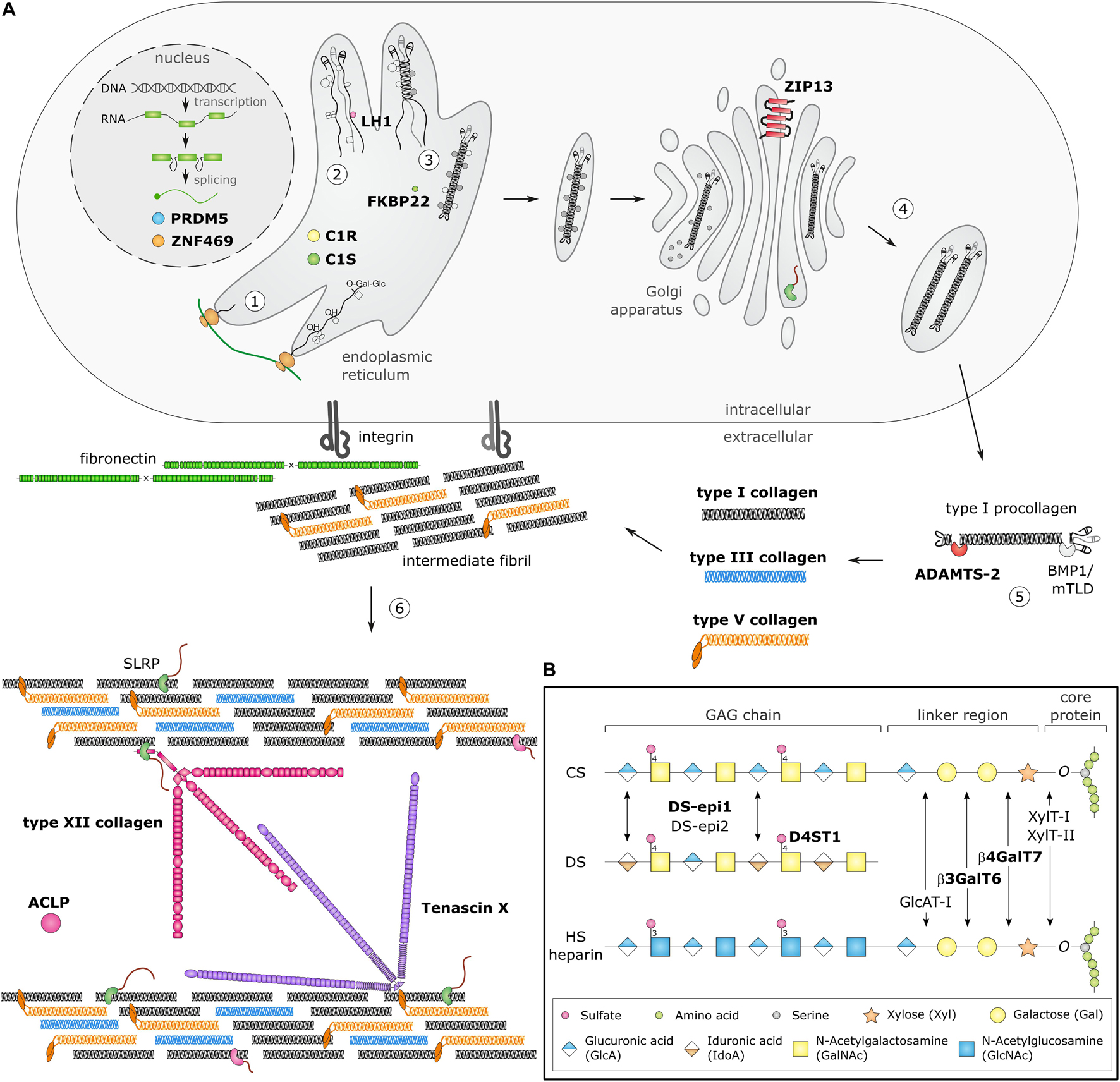
Figure 1. Schematic overview of collagen and glycosaminoglycan (GAG) biosynthesis and collagen fibrillogenesis. Molecules defective in Ehlers-Danlos syndromes (EDS) are highlighted in bold. (A) Fibrillar collagen biosynthesis starts with transcription and translation of pro-α-chains (step 1). Nascent pro-α-chains are heavily post-translationally modified by several proline and lysine hydroxylases and galactosyltransferases (step 2). The association of the C-terminal propeptides of three pro-α-chains, initiates triple helix formation which propagates to the N-terminus in a zipperlike fashion and is assisted by several molecular chaperones (step 3). The trimeric procollagen molecules aggregate laterally, are transported in secretory vesicles and are eventually directed to the extracellular environment (step 4). Removal of the N- and C-propeptides, by ADAMTS-2 and BMP-1/mTLD, respectively, results in the formation of a collagen molecule (step 5) that can then assemble into highly ordered striated fibrils. The tissue-specific assembly of collagen fibrils requires the concerted action of several assisting proteins, categorized as organizers, nucleators and regulators (step 6). At the plasma membrane, fibronectin and integrins serve as organizers of fibril assembly. Some collagens, such as type V collagen, function as nucleators, which initiate immature fibril assembly at the cell surface. Type V collagen co-assembles with type I collagen into heterotypic fibrils with the entire triple helical domain of type V collagen embedded within the fibril, whereas its partially processed N-propeptide domain protrudes to the fibril surface and controls fibrillogenesis by sterically hindering the addition of collagen monomers. The intermediate fibrils are then deposited into the extracellular matrix (ECM). Stabilization of these fibrils is provided by interactions with regulators such as the small leucine-rich proteoglycan (SLRP) decorin, tenascin-X and type XII collagen, which influence the rate of assembly, size and structure of the collagen fibrils. As fibrillogenesis proceeds, fibril growth occurs through linear and lateral fusion of intermediate collagen fibrils which are subsequently stabilized by the formation of covalent intra- and inter-molecular cross-links. (B) GAG biosynthesis starts with the synthesis of a proteoglycan core protein which is subsequently modified by several Golgi-resident enzymes. Initially, a common linker region containing four monosaccharides is formed. Biosynthesis of this tetrasaccharide linker region starts with the stepwise addition of a xylose (Xyl) residue to a specific serine residue of the core protein, catalyzed by xylosyltransferase-I and II (XylT-I/-II). Subsequently, two galactose (Gal) residues are added by galactosyltransferase-I (GalT-I or β4GalT7) and galactosyltransferase-II (GalT-II or β3GalT6). Finally, the addition of a glucuronic acid (GlcA), catalyzed by glucuronosyltransferase-I (GlcAT-I) completes the formation of the linker region. The alternating addition of either N-acetyl-glucosamine (GlcNAc) or N-galactosyl-glucosamine (GalNAc) and GlcA defines the composition of the GAG-chain and subdivides proteoglycans into heparan sulfate (HS) proteoglycans and chondroitin sulfate (CS)/dermatan sulfate (DS) proteoglycans. The GAG-chains are then further modified by epimerization and sulfation. DS synthesis necessitates the epimerization of GlcA towards iduronic acid (IdoA), which is catalyzed by DS epimerases–1 and -2 (DS-epi1 and DS-epi2). Subsequently, dermatan 4-O-sulfotransferase-1 (D4ST1) is able to catalyze 4-O-sulfation of GalNAc, thereby preventing back-epimerization of the adjacent IdoA.
Insights into the function of some of the proteins involved in these processes have been irrevocably intertwined with the identification of genetic defects in human disorders, including EDS, but also with the study of animal models.
Animals have been extensively used as experimental models for human diseases in biomedical research and have successfully provided tools for studying the function of a particular protein, understanding pathogenic cellular and molecular mechanisms in relevant tissues as well as the preclinical development and testing of therapeutic options for a wide variety of human diseases (Okechukwu, 2018). Traditionally, mainly rodents [mice (Mus musculus) and rats (Rattus norvegicus)] were used as experimental animals due to their anatomical, physiological and genetic similarity to humans. More recently, zebrafish (Danio rerio) have gained popularity in biomedical research and have emerged as a promising animal model with several advantages, such as small size, easy genetic manipulation, high fecundity, external fertilization, large offspring, transparency of rapidly developing embryos, and low maintenance cost (Kimmel, 1989; Lieschke and Currie, 2007).
In nature, several spontaneously occurring animals presenting EDS features exist. Additionally, over the past two decades, engineered animal models of EDS have been developed (Figure 2), which have increased our knowledge about the phenotype and our understanding of the mechanisms underlying these conditions.
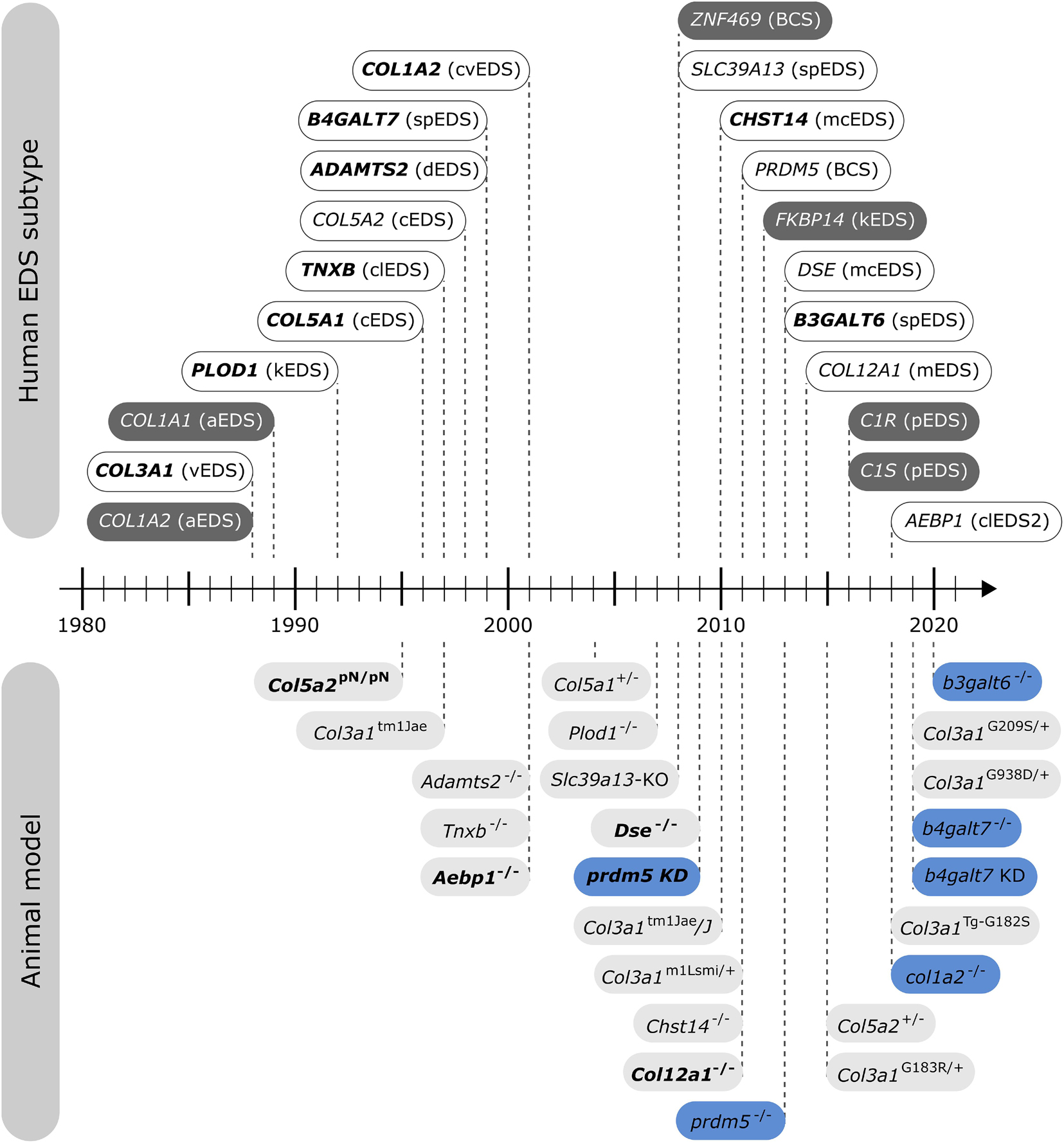
Figure 2. Timeline illustrating the first identification of molecular defects in human EDS (above timeline) and the first description of engineered animal models targeting an EDS-associated gene (below timeline). Human genes associated with EDS without a corresponding animal model are indicated in dark gray. Note that for some human EDS subtypes, biochemical and/or ultrastructural findings preceded the identification of the molecular defect. Mouse models are depicted in light gray and zebrafish models in blue. Bold indicates what was first, either the identification of the human disease gene or the generation of the engineered animal model.
The aim of this narrative review is to provide an overview of available animal models with defects in EDS-related genes, describe how these animals recapitulate the human phenotype, and how these models have assisted the research community in broadening our knowledge on these conditions and respective protein defects. Therefore, we searched PubMed for a combination of the individual EDS-related genes, combined with the following terms: “Ehlers–Danlos syndrome,” “EDS,” “mouse,” “animal,” and “model.”
Below, we provide an overview of published mouse and zebrafish models with defects in EDS-associated genes, organized according to the underlying genetic and pathogenetic mechanisms as defined in the 2017 EDS classification (Malfait et al., 2017). A detailed summary of the available models is presented in Figures 3, 4 and Supplementary Table 1 for mouse models and Figures 5, 6 and Supplementary Table 2 for zebrafish models. Although other (often conditional) mouse models affecting an EDS-related gene have been reported, an extensive description of these models is beyond the scope of this review (for a brief summary, see Supplementary Information).
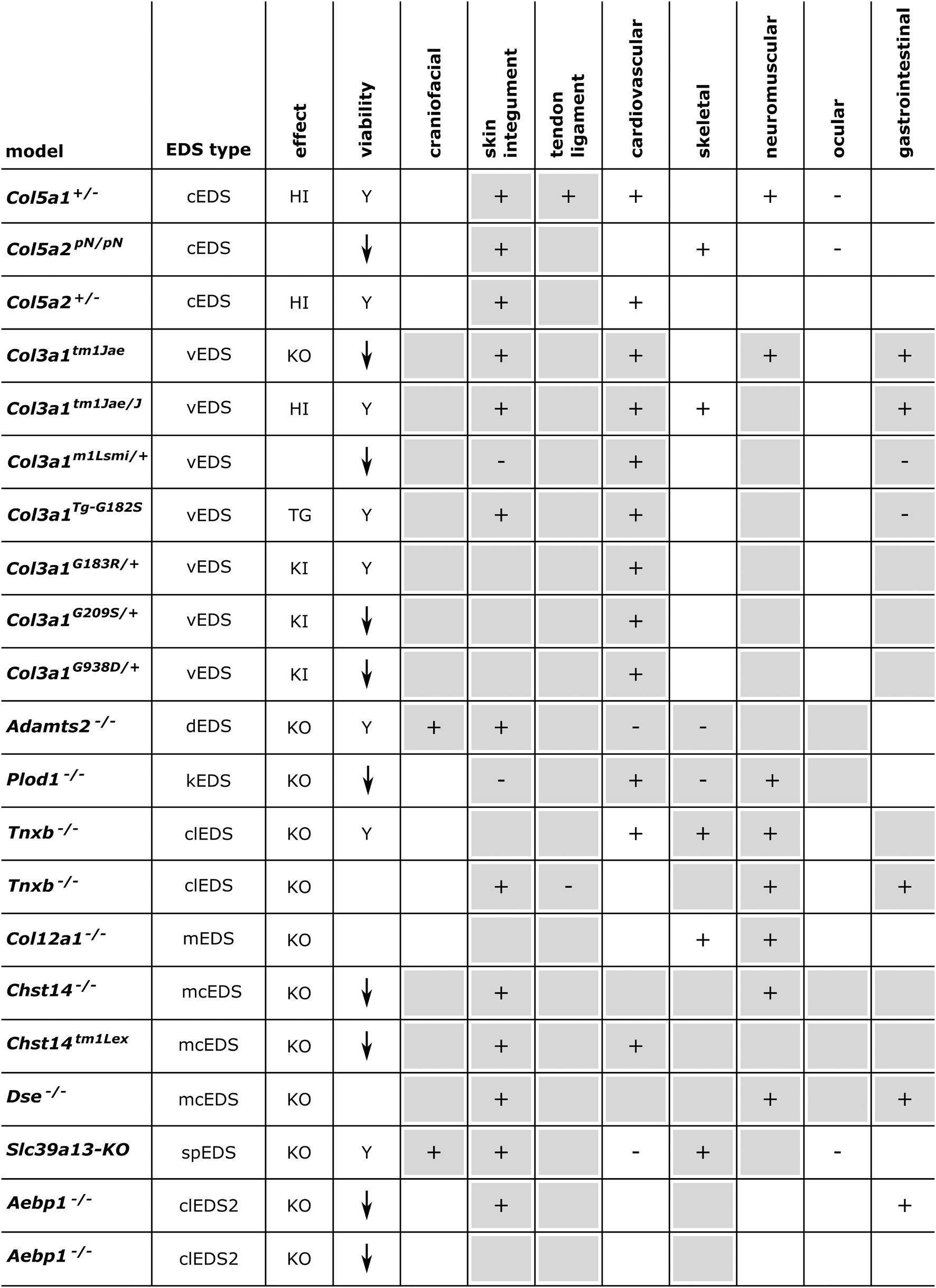
Figure 3. Phenotypic characteristics of mouse models with defects in EDS-associated genes. The presence or absence of a phenotype in mice is indicated with “+” or “-”, respectively, when investigated. A detailed description of the murine phenotypes can be found in Supplementary Table 1. The major and minor clinical characteristics in human EDS patients as defined in the International EDS Classification, published in 2017, are indicated with a gray background (Malfait et al., 2017). HI, haploinsufficiency; KO, (homozygous) knockout; TG, transgenic; KI, knock-in; Y, viable; ↓, decreased survival rate.
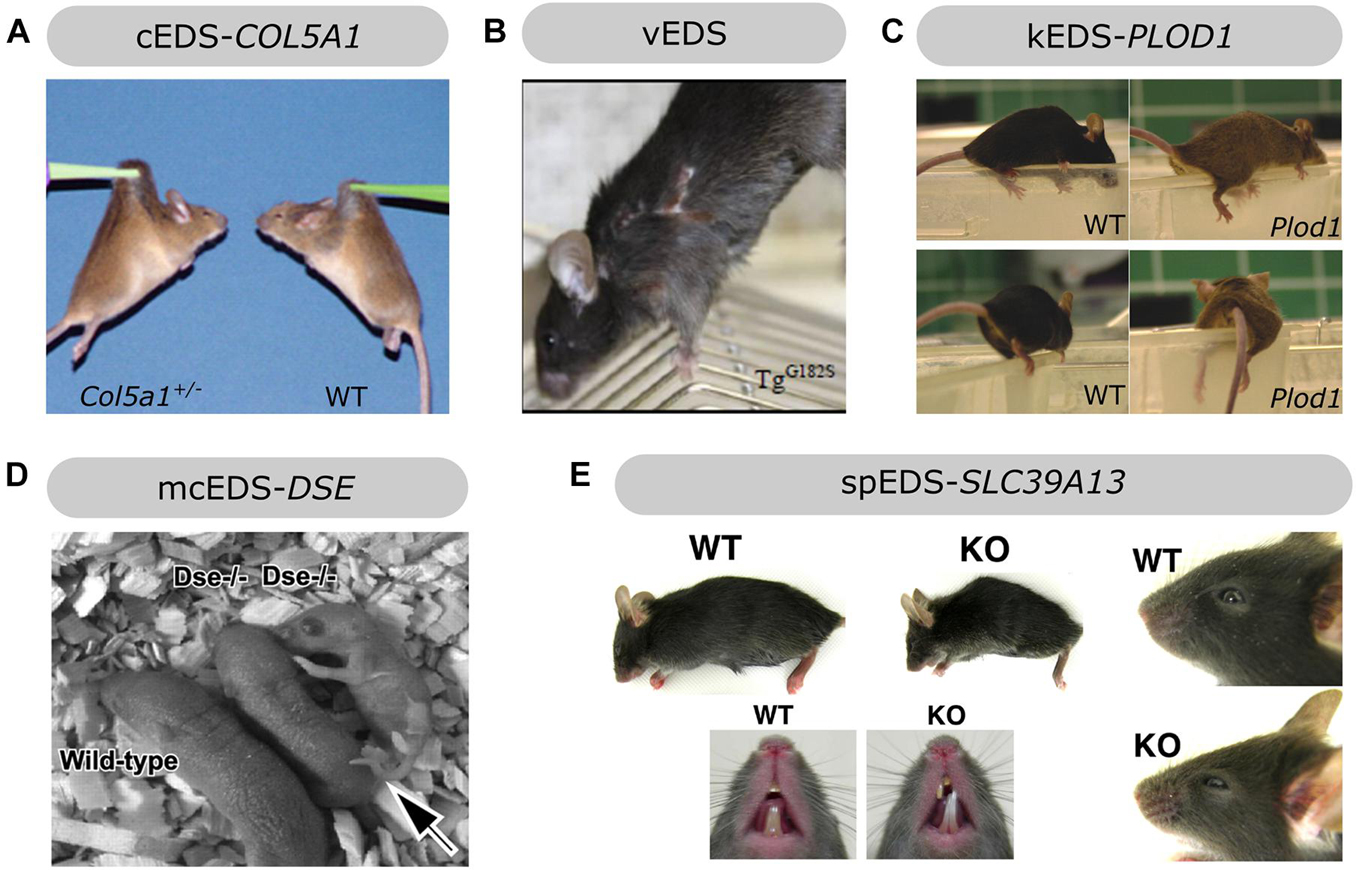
Figure 4. Overview of the phenotypic findings in some mouse models of EDS. (A) Col5a1+/– mouse model of cEDS. Images adapted from Wenstrup et al. (2006). (B) Col3a1Tg–G182S mouse model of vEDS. Image adapted from D’hondt et al. (2018). (C) Plod1–/– mouse model of kEDS-PLOD1. Images adapted from Takaluoma et al. (2007). (D) Dse–/– mouse model of mcEDS-DSE. Image adapted from Maccarana et al. (2009). (E) Slc39A13-KO mouse model of spEDS-SLC39A13. Images adapted from Fukada et al. (2008). Images depicted in (A,C,D,E) were used under the Creative Commons License and image B with license number 5090710991420.
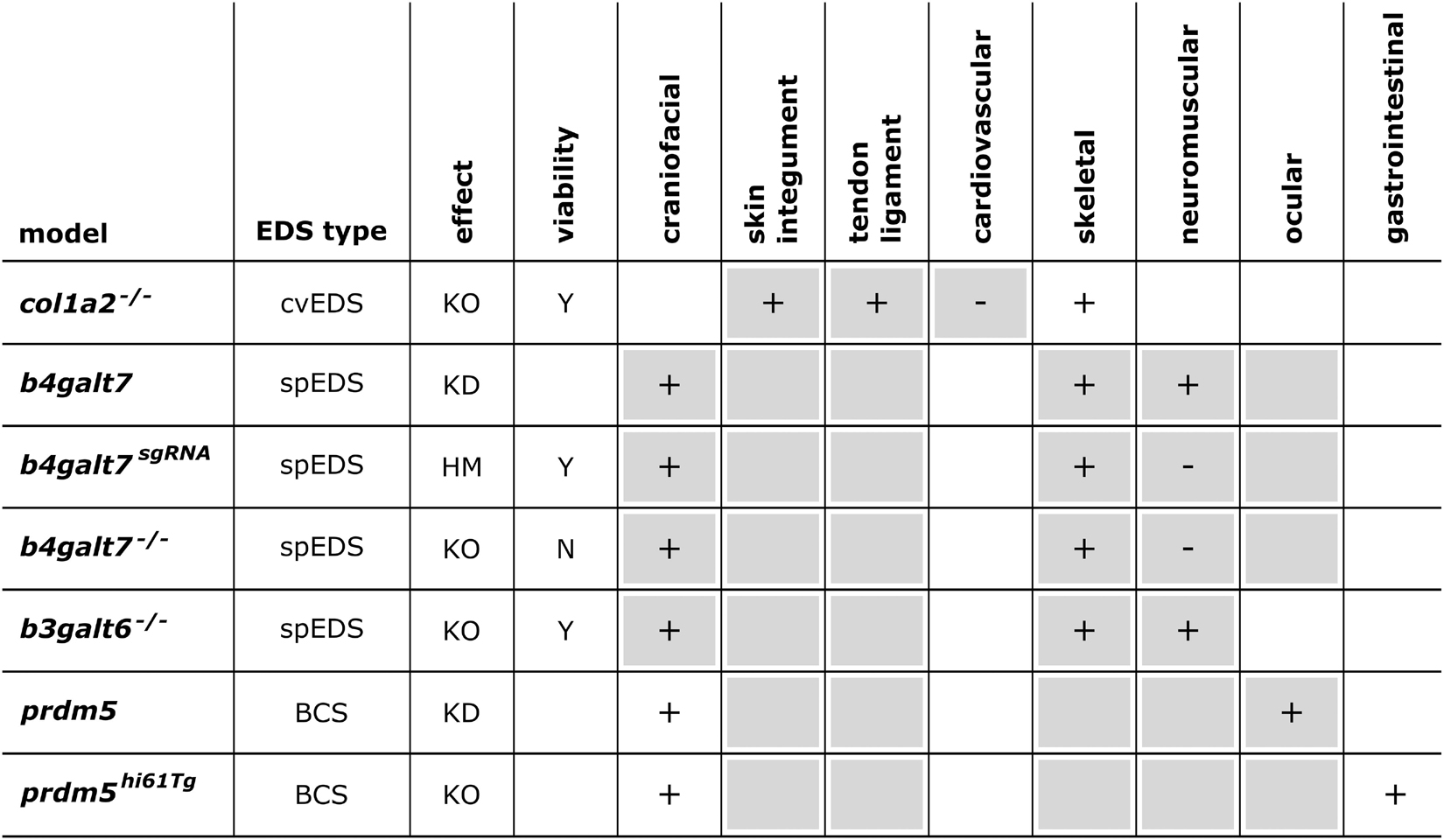
Figure 5. Phenotypic characteristics of zebrafish models with defects in EDS-associated genes. The presence or absence of a phenotype in the zebrafish model is indicated with “+” or “-”, respectively, when investigated. A detailed description of the zebrafish phenotypes can be found in Supplementary Table 2. The major and minor clinical characteristics in humans EDS patients as defined in the International EDS Classification, published in 2017, are indicated with a gray background (Malfait et al., 2017). BCS, brittle cornea syndrome; KO, knockout; KD, (morpholino-based) knockdown; HM, hypomorphic; Y, viable; N, not viable.

Figure 6. Overview of the phenotypic findings in zebrafish models of EDS. (A) Adult col1a2–/– zebrafish model of cvEDS. Images adapted from Gistelinck et al. (2018). (B) Larval b4galt7 morphant, crispant and knockout models of spEDS-B4GALT7. Scale bars: 1 mm. Images adapted from Delbaere et al. (2019a). (C) Adult b3galt6–/– zebrafish model of spEDS-B3GALT6. Images adapted from Delbaere et al. (2020). (D) Morpholino (MO)-based knockdown of prdm5 in zebrafish larvae. Images adapted from Meani et al. (2009). Images depicted in (A,C,D) were used under the Creative Commons License and image B with license number 5078981221357.
Classical EDS (cEDS) is the most common, molecularly defined EDS subtype. It is characterized by generalized joint hypermobility, skin hyperextensibility and skin fragility, manifesting as easy splitting of the skin upon minor trauma, which, in combination with delayed wound healing, results in the formation of widened atrophic scarring (Bowen et al., 2017). It is inherited in an autosomal dominant fashion and about 90% of cEDS patients harbor a heterozygous mutation in either the COL5A1 or COL5A2 gene, encoding the pro-α1- and pro-α2-chain of type V collagen, respectively. The majority of these mutations result in a non-functional COL5A1 allele and give rise to COL5A1 haploinsufficiency with reduced type V collagen protein levels, while mutations in COL5A1 or COL5A2 that lead to a structural defect (e.g., glycine substitution or in-frame exon skip) have a dominant negative effect (Symoens et al., 2012; Colman et al., 2021). The most common isoform of type V collagen is the [α1(V)]2α2(V) heterotrimer found in skin, tendon, ligaments, cornea, and bone, where it forms heterotypic fibrils with type I collagen. Other isoforms, including the embryonic [α1(V)]3 homotrimer and the α1(V)α2(V)α3(V) heterotrimer mainly found in placenta, are less abundant. Although type V collagen is quantitatively of low abundance (about 2–5% of total collagen in bone, tendon, and dermis), it plays a key role during collagen fibrillogenesis by initiating fibril assembly and regulating heterotypic type I/V collagen fibril diameter through its partially retained α1(V)-N-propeptide that protrudes beyond the fibril surface (Birk, 2001; Wenstrup et al., 2004).
Several murine models have been generated to study the functional and regulatory roles of type V collagen during collagen fibrillogenesis, and as such these models also provide insights into cEDS pathogenesis.
Complete absence of the pro-α1(V)-chains in homozygous Col5a1 knockout (Col5a1–/–) mice was shown to result in lethality around embryonic day 10 due to cardiovascular failure. Ultrastructural evaluation of Col5a1–/– embryos using TEM revealed the virtual absence of predermal mesenchymal fibril formation despite the presence of normal amounts of type I collagen. These findings revealed a critical regulatory role of type V collagen in the initiation of early fibril formation and nucleation of type I collagen fibril assembly during early murine embryogenesis (Wenstrup et al., 2004). Complete loss of the pro-α1(V)-chains has not been reported in humans, most probably because it is lethal.
Heterozygous Col5a1 knockout (Col5a1+/–) mice are viable and mimic the most common molecular defect associated with cEDS, i.e., COL5A1 haploinsufficiency, leading to approximately 50% reduction in type V collagen content (Wenstrup et al., 2006). Similar to cEDS patients, the skin of Col5a1+/– mice was hyperextensible with decreased tensile strength (Figure 4A). Ultrastructural analysis showed decreased fibril density in the subscapular dermis, correlating with the decreased collagen content, and consistent with dysfunctional regulation of collagen fibril nucleation when type V collagen is limited. Additionally, two different fibril subpopulations were observed: relatively normal symmetrical fibrils with slightly larger diameters, and very large, structurally aberrant fibrils with irregular contours, so called “cauliflower” fibrils, representing an unregulated assembly of type I collagen which virtually lacks type V collagen and disrupted lateral fibril growth (Wenstrup et al., 2004, 2006). These ultrastructural abnormalities are highly reminiscent of the alterations in dermis of cEDS patients (Vogel et al., 1979). Most Col5a1+/– mice older than 6 months developed spontaneous, non-healing wounds indicative of skin fragility (DeNigris et al., 2015). Wound repair studies demonstrated slower in vivo closure of a subscapular skin wound in Col5a1+/– mice. Subsequent in vitro studies on dermal fibroblasts from Col5a1+/– mice showed that this impaired wound healing is likely attributed to an interplay of decreases in proliferation rate, attachment properties to components of the wound ECM (e.g., types I and III collagen and fibronectin) and migration capacity of Col5a1+/– fibroblasts (DeNigris et al., 2015). The latter finding is consistent with in vitro scratch assays showing delayed migration of dermal fibroblasts from cEDS patients with COL5A1 haploinsufficiency (Viglio et al., 2008; DeNigris et al., 2015).
Although no overt joint hypermobility, another major feature of human cEDS, was described in Col5a1+/– mice (Wenstrup et al., 2006), biomechanical analysis of the flexor digitorum longus (FDL) and patellar tendons showed reduced tensile strength, suggesting increased elasticity. Ultrastructural analyses of FDL tendon showed unexpectedly mild abnormalities in Col5a1+/– mice with less regular cross-sectional profiles and smaller diameter of collagen fibrils (Wenstrup et al., 2011). TEM of patellar tendon showed two distinct subpopulations of smaller and larger diameter fibrils with mostly normal circular fibril cross-section profiles but with larger diameter fibrils in Col5a1+/− mice (Johnston et al., 2017). Functional and structural tendon pathology has also been described in the patellar tendon of cEDS patients with low tendon stiffness, and abnormal ultrastructural findings with various amounts of large and irregular collagen fibrils combined with apparently normal fibrils (Nielsen et al., 2014). Tendons from Col5a1+/– mice showed diminished mechanical recovery potential following bilateral patellar tendon injury with smaller fibril sizes 6 weeks post-injury, indicating further failure in the healing response in Col5a1+/– tendon. These findings point to a role for type V collagen in healing and recovery following injury (Johnston et al., 2017).
Col5a1+/– mice also displayed a vascular phenotype with decreased aortic stiffness and breaking strength, however not associated with ruptures or sudden, premature death (Wenstrup et al., 2006). The heart was morphologically normal, but immunohistochemical analysis revealed increased deposition of type I and III collagen in mitral and aortic valves and ventricular myocardium, supposedly as a compensatory mechanism for disturbed fibrillogenesis due to reduced type V collagen content (Lincoln et al., 2006). Although these findings indicate the importance of type V collagen in vascular and cardiac structure, integrity and/or function, cardiovalvular problems are rarely of clinical significance and severe or life-threatening arterial manifestations are only sporadically observed in cEDS patients (Bowen et al., 2017; Malfait, 2018; Angwin et al., 2020; Colman et al., 2021).
While type V collagen is a quantitatively minor collagen in most tissues, it accounts for 10–20% of total corneal collagen (Birk, 2001). Cornea of Col5a1+/– mice were grossly normal without opacity but showed reduced stromal thickness and a reduced collagen content. At the ultrastructural level, stromal collagen fibril density was decreased, with a single population of consistently larger, cylindrically shaped fibril diameters but unaltered fibril packing (Segev et al., 2006). Intriguingly, mild corneal abnormalities, (e.g., thin and steep but transparent corneas) have been described in few cEDS patients (Segev et al., 2006).
Our group recently reported that both male and female Col5a1+/– mice are hypersensitive to mechanical stimuli applied to the hind paws and abdominal area but not to thermal stimuli. Additionally, female Col5a1+/– mice showed altered climbing behavior with decreased grip strength. Visualization of NaV1.8-positive nociceptors in the glabrous skin of the footpad revealed a decreased intraepidermal nerve fiber density, with fewer nerves crossing the dermis-epidermis junction in Col5a1+/– mice (Syx et al., 2020).
Prior to the elucidation of the molecular basis of human cEDS, involvement of type V collagen in its pathogenesis was already suggested by mice harboring a homozygous in-frame deletion of exon 6 in the Col5a2 gene. The resulting so-called pN/pN mice (Col5a2pN/pN) lack the amino-terminal telopeptide of the pro-α2(V)-chain (Andrikopoulos et al., 1995). Although initially suggested to result in a structurally abnormal α2(V)-chain with a dominant-negative effect, subsequent analyses revealed that the Col5a2 deletion resulted in severely reduced pro-α2(V)-chains levels (despite normal Col5a2 expression levels). As a consequence, compensatory upregulated Col5a1 expression was seen with the predominant formation of [α1(V)]3 homotrimers in skin (Chanut-Delalande et al., 2004). Most homozygous Col5a2pN/pN mice died during the first 48h of postnatal life, with a survival rate of only 5% past weaning age, mainly due to respiratory problems triggered by varying degrees of lordosis and kyphosis. Surviving Col5a2pN/pN mice weighed approximately half of the wild-type littermates at 3 weeks of age, attributed to reduced mobility due to the progressive spinal deformities prohibiting nourishment. They displayed severe skin fragility (with multiple scars and bleeding lacerations) and increased stretchability of the skin, reminiscent of EDS. Histological analyses of the skin of Col5a2pN/pN mice revealed reduced dermal and increased hypodermal thickness, with unusually localized hair follicles in the latter, while ultrastructural analyses demonstrated that dermal collagen fibrils were more disorganized, less tightly packed and heterogeneous in size with areas devoid of banded fibrils (Andrikopoulos et al., 1995; Chanut-Delalande et al., 2004). Dermal fibroblasts derived from Col5a2pN/pN mice produced a sparse network of disorganized, thin fibrils in vitro and also showed increased apoptosis (Chanut-Delalande et al., 2004). These observations underscore the importance of type V collagen and the [α1(V)]2α2(V) heterotrimer for proper cell-matrix interactions during skin development. Additionally, pronounced ultrastructural disorganization of collagen fibrils with an overall increased diameter was also seen in the corneal stroma, which seemed thinner in Col5a2pN/pN mice (Andrikopoulos et al., 1995; Chanut-Delalande et al., 2004).
In 2015, a constitutive Col5a2 knockout (Col5a2–/–) mouse model was created with complete absence of pro-α2(V)-chains (Park et al., 2015). In contrast to Col5a2pN/pN mice, homozygous Col5a2–/– mice were embryonic lethal around 12 days post conception (dpc) due to cardiovascular insufficiency. In contrast to the absence of fibrils in Col5a1–/– embryos, mesenchymal collagen fibrils with abnormally large diameters and abnormal configurations were observed in Col5a2–/– embryos, suggesting that [α1(V)]3 homotrimers can at least partially compensate for the loss of [α1(V)]2α2(V) heterotrimers in the initiation of early embryonic collagen fibril formation.
Whereas heterozygous Col5a2pN/+ mice did not display an overt phenotype, heterozygous Col5a2+/– mice representing Col5a2 haploinsufficiency did survive and showed increased skin extensibility with decreased tensile strength, although less pronounced compared to Col5a1+/– mice. Ultrastructural analyses showed only mild collagen fibril abnormalities in the subscapular dermis of Col5a2+/– mice with irregular collagen fibril contours mainly seen on longitudinal sections, but without the pathognomonic “cauliflower”-shaped dermal collagen fibrils found in cross-sections of cEDS patients and Col5a1+/– mice. Reduced tensile strength and stiffness pointing to increased fragility and elasticity, respectively, were also noted in the aorta of Col5a2+/– mice (Park et al., 2015). Additionally, experimentally increasing blood pressure by angiotensin II administration showed an increased incidence, diameter and severity of abdominal aortic aneurysms with more than half of Col5a2+/– mice dying of aortic arch dissection and rupture (Park et al., 2017). To date, no cEDS patients with COL5A2 haploinsufficiency have been reported.
Taken together, Col5a1+/– mice mimic human cEDS the best since they represent the major molecular defect, i.e., COL5A1 haploinsufficiency, and faithfully recapitulate many of the clinical (including pain), biomechanical, morphological and biochemical features seen in cEDS patients (Wenstrup et al., 2006). To date, no animal models harboring structural defects such as glycine substitutions or in-frame exon skips in the triple helical domain of the pro-α1- or pro-α2-chains of type V collagen have been reported.
Vascular EDS (vEDS) is an autosomal dominant condition mainly characterized by life-threatening complications, including arterial aneurysms, dissections and ruptures, but also bowel perforations/ruptures and ruptures of the gravid uterus, resulting in a reduced life span with a median survival age of 51 years (Pepin et al., 2014; Byers et al., 2017). vEDS patients often have thin, translucent skin, excessive bruising and present with a characteristic facial appearance. To date, the only evidence-based treatment strategy for vEDS that decreases the incidence of arterial rupture is celiprolol, a long-acting β1-receptor antagonist with partial β2-receptor agonist properties used for treatment of hypertension and reducing arterial wall stress, without changing hemodynamic parameters (Ong et al., 2010). Nevertheless, caution is warranted due to some study limitations (Backer and Backer, 2019). The vast majority of vEDS patients harbors a heterozygous defect in the COL3A1 gene, encoding the pro-α1-chain of type III collagen. Most genetic defects are missense or splice site mutations, introducing a glycine substitution or creating an in-frame exon skip within the triple helical domain, respectively. Mutations leading to COL3A1 haploinsufficiency were reported in a small proportion (less than 5%) of vEDS patients and are associated with a delayed onset of complications by almost two decades (Pepin et al., 2014). Type III collagen is a homotrimer consisting of three α1(III)-chains, found abundantly in the wall of arteries, gastrointestinal tract, uterus and skin, where it often co-localizes with type I collagen and is thought to regulate fibril diameter (Epstein and Munderloh, 1975).
Several murine models have been generated to study the role of (defective) type III collagen and vEDS pathogenesis.
The first proposed vEDS model was a homozygous Col3a1 knockout (Col3a1tm1Jae) mouse, which had a 5% survival rate at weaning age with most deaths occurring within 48 h after birth of unknown etiology. Surviving homozygous Col3a1tm1Jae mice appeared normal but were 15% smaller than wild-type mice and had a reduced lifespan (6 months), mainly caused by rupture of large blood vessels and occasionally by intestinal rupture. About 60% of surviving homozygous knockout mice displayed spontaneous skin lesions, including large open wounds. TEM analyses of aorta, skin, intestine, liver, heart, and lung showed a reduced number of collagen fibrils in mutant mice with variable fibril diameters of almost twice the size of those in wild-type mice, hinting at a critical role for type III collagen during collagen fibrillogenesis (Liu et al., 1997). The high mortality of this model precludes preclinical studies. Additionally, complete loss of type III collagen in human patients is rare and not associated with a typical vEDS phenotype. Biallelic COL3A1 mutations result in connective tissue abnormalities with some phenotypic resemblance to vEDS combined with structural brain anomalies. (Plancke et al., 2009; Jørgensen et al., 2014; Horn et al., 2017; Vandervore et al., 2017). In accordance with this human phenotype, the brain of homozygous Col3a1tm1Jae mice showed cobblestone-like cortical malformation with pial basement membrane defects and neuronal overmigration, thereby suggesting a role for type III collagen during brain development (Jeong et al., 2012).
Heterozygous Col3a1 knockout (Col3a1tm1Jae/J) mice, which model Col3a1 haploinsufficiency, were initially reported to be phenotypically normal without spontaneous life-threatening vascular or gastrointestinal complications or premature death up to 2 years of age (Liu et al., 1997). However, more detailed analysis revealed some late-onset histological lesions in the aorta with fragmentation of the internal elastic lamina, which aggravated with age (from 9 to 21 months) and were more pronounced in male (88%) versus female (47%) Col3a1tm1Jae/J mice (Cooper et al., 2010). Additionally, the abdominal aorta had decreased wall strength and stiffness likely due to reduced thickness and collagen content with increased matrix metalloproteinase (MMP) activity, particularly MMP-9 (Cooper et al., 2010; Tae et al., 2012; Goudot et al., 2018). In line with the vascular integrity deficits, experimentally increasing blood pressure by angiotensin II administration increased the susceptibility for thoracic aortic dissection in Col3a1tm1Jae/J mice (Faugeroux et al., 2013). Long-term treatment of Col3a1tm1Jae/J mice with doxycycline, a tetracycline antibiotic and nonspecific MMP inhibitor, aimed at strengthening the arterial walls by decreasing ECM degradation by MMPs, attenuated the decrease in aortic collagen content and prevented the development of spontaneous and stress-induced aortic lesions (Briest and Talan, 2011; Briest et al., 2011; Tae et al., 2012). Colons of Col3a1tm1Jae/J mice showed reduced strength and increased compliance, despite normal histology (Cooper et al., 2010). Bladders of Col3a1tm1Jae/J mice also showed increased compliance, less densely packed collagen fibrils with a broader diameter distribution on ultrastructure of the lamina propria and decreased neurotransmitter function (Stevenson et al., 2006). Col3a1tm1Jae/J mice did not present with a clear skin phenotype, but faster experimental wound closure with increased wound contracture was seen in a collagen gel contraction assay with embryonic dermal fibroblasts in vitro and in vivo in adult mice (>1 year), accompanied by increased myofibroblast differentiation and increased scar tissue formation (Volk et al., 2011). Analysis of the skeleton revealed reduced trabecular bone quantity but no craniofacial abnormalities in Col3a1tm1Jae/J mice (Volk et al., 2014) and a bilateral tibial fracture model showed impaired bone formation and altered remodeling during fracture healing in Col3a1tm1Jae/J mice, without alterations in mechanical bone function (Miedel et al., 2015). These findings suggest a role for type III collagen in both cutaneous and skeletal development and repair. Taken together, haploinsufficient Col3a1tm1Jae/J mice model only a small subset of human vEDS patients (<5%) with a milder phenotype and although this model shows subclinical, age-dependent evidence for vascular and gastrointestinal fragility, preclinical studies are largely limited by the lack of overt disease.
Another mouse model for vEDS was identified serendipitously during a gene-targeting study. Although this model was initially reported to harbor a 185 kb deletion affecting the promotor region and exons 1–39 of Col3a1 thereby introducing a premature termination codon (Smith et al., 2011), subsequent molecular characterization revealed an in-frame deletion of exons 33–39 (Dubacher et al., 2019). Whereas homozygous Col3a1m1Lsmi/m1Lsmi mice were embryonic lethal, heterozygous Col3a1m1Lsmi/+ mice displayed sudden death due to acute aortic dissection, mostly between 4 and 10 weeks of age, with incomplete penetrance (28%) and two times more frequent in males versus females. Prior to aortic dissection, heterozygous Col3a1m1Lsmi/+ mice were indistinguishable from wild-type mice, without hypertension, aneurysms or dilatation of the ascending aorta in vivo and little evidence of cardiovascular dysfunction. However, collagen content in the media of the thoracic aorta was reduced in Col3a1m1Lsmi/+ mice and ultrastructural examination of the thoracic aorta revealed inconsistencies in the elastic laminae structure, tearing of the smooth muscle layer, and a reduced number of collagen fibrils with more variable and larger diameters in the adventitia, similar to findings in the skin (Smith et al., 2011; Dubacher et al., 2019). The thoracic aorta of Col3a1m1Lsmi/+ mice had a lower maximal tensile strength compared to wild-type mice. This biomechanical integrity test of the thoracic aorta was used as a readout for therapeutic intervention and was ameliorated following treatment with the β-blocker celiprolol and MMP inhibitor doxycycline, but not with the angiotensin II receptor type 1 antagonist losartan or β-blocker bisoprolol (Dubacher et al., 2019; Gorosabel et al., 2019). Overall, this model appears to mimic only the vascular features of vEDS, which are more severe compared to Col3a1tm1Jae/J haploinsufficient mice, without overt evidence of skin fragility, gastrointestinal or uterus rupture (Smith et al., 2011).
Since glycine substitutions in the pro-α1(III) triple helical domain are the main molecular defects found in the vast majority of vEDS patients (Pepin et al., 2014), our group developed a transgenic Col3a1 (Col3a1Tg–G182S) mouse model overexpressing a typical glycine substitution, p.(Gly182Ser) (D’hondt et al., 2018), corresponding to the most frequently reported human missense mutation [p.(Gly183Ser)] in type III collagen (Dalgleish, 1998). The skin of Col3a1Tg–G182S mice was thin and easily torn when handled. At 13–14 weeks of age all male (but not female) Col3a1Tg–G182S mice developed spontaneous transdermal wounds, requiring euthanasia before any major vascular manifestations occurred (Figure 4B; D’hondt et al., 2018). These wounds were reminiscent of the dermal phenotype seen in surviving homozygous Col3a1–/– mice (Liu et al., 1997). Total collagen content and tensile strength of 12-week-old Col3a1Tg–G182S mice were severely reduced in the abdominal skin and the thoraco-abdominal aorta with reduced thickness of the adventitia of the aorta, indicative of cutaneous and vascular fragility as seen in vEDS patients. Ultrastructural analysis revealed a reduced amount of collagen fibrils in the dermis and highly variable fibril diameters with a tendency to thicker fibrils in the dermis and the adventitia of the thoracic aorta of Col3a1Tg–G182S mice. The latter also showed abnormal distribution and morphology of smooth muscle cells and reduced contact with the elastic lamina. These clinical and ultrastructural abnormalities were not detected in a control line overexpressing wild-type type III collagen (Col3a1Tg–WT). Overall, these findings confirmed a key role for type III collagen during collagen fibrillogenesis in the dermis and vasculature (D’hondt et al., 2018).
Two groups independently created heterozygous Col3a1 knock-in mouse models harboring different glycine substitutions. The Col3a1G183R/+ mouse model shows spontaneous thoracic aortic rupture resulting in a 50% mortality rate at 24 weeks of age which was lower in female (25%) compared to male (60%) mice (Fontaine et al., 2015). The observed arterial fragility was not preceded by dilatation of the aorta, but reduced abdominal aortic stiffening was observed (Fontaine et al., 2015; Goudot et al., 2018). TEM analysis of the aorta of Col3a1G183R/+ mice showed a lower density of collagen fibrils with heterogeneous diameters and dilated endoplasmic reticulum in adventitial fibroblasts (Fontaine et al., 2015). Recently, two additional murine Col3a1 knock-in models were created, Col3a1G209S/+ and Col3a1G938D/+, harboring helical glycine substitutions previously identified in vEDS patients (Bowen et al., 2019). Both models suffer from sudden premature death due to spontaneous aortic rupture occasionally accompanied by dissection of the proximal descending thoracic aorta but without aneurysms. As expected from the location of the substituted glycine and the assembly of type III procollagen from the C- to N-terminus (Mizuno et al., 2013), Col3a1G938D/+ mice showed a more severe phenotype, including smaller body size, smaller aortas with decreased collagen content and a median survival of 45 days compared to 400 days for Col3a1G209S/+ mice. Histology of the aortic wall revealed occasional elastic fiber breaks and TEM of the proximal descending thoracic aorta showed disrupted elastic lamellar units with thickened elastic fibers with a moth-eaten appearance, disarray of vascular smooth muscle cells (VSMCs) and paucity of collagen fibrils in the space between VSMCs and elastic fibers in both models. Collagen fibrils in the aortic media of Col3a1G938D/+ mice had a wide variation in diameter and were generally smaller whereas adventitial fibroblasts showed a dilated endoplasmic reticulum. Pharmacological blood pressure reduction using losartan, propranolol, atenolol and amlodipine had no impact on survival rate of the severe Col3a1G938D/+ model. Surprisingly, and in contrast to the ameliorated biomechanical findings in Col3a1m1Lsmi/+ aortas (Dubacher et al., 2019) and decreased arterial rupture incidence in vEDS patients (Ong et al., 2010), celiprolol accelerated death from aortic dissection in both Col3a1G209S/+ and Col3a1G938D/+ mice. Transcriptome profiling of the descending thoracic aorta revealed that the vascular rupture risk is mediated by excessive PLC/IP3/PKC/ERK signaling and pharmacological attenuation of this pathway by inhibition of IP3 (hydralazine), PKCβ (ruboxistaurin), or MEK/ERK (cobimetinib) increased survival in the severe Col3a1G938D/+ model. Furthermore, the increased risk for life-threatening vascular events associated with pregnancy and puberty, also seen in vEDS patients (Murray et al., 2014; Byers et al., 2017), was shown to be rescued by inhibiting oxytocin signaling (by removal of pups or oxytocin receptor antagonist), MEK (trametinib) or IP3 (hydralazine) in mild Col3a1G209S/+ female mice and androgen signaling (with androgen receptor antagonists bicalutamide or spironolactone) in Col3a1G938D/+ mice, respectively.
In summary, vEDS is currently the only EDS subtype for which mice are available with Col3a1 haploinsufficiency, a genomic multi-exon deletion or a glycine substitution (knock-in). Strikingly, sexual dimorphism is seen in almost all of the described vEDS models, with greater phenotypic severity in male mice compared to female mice, reflecting what is seen in vEDS patients (Pepin et al., 2014). Furthermore, vEDS mouse models are the only preclinical models in which pharmacological studies have been performed (summarized in Supplementary Table 3).
Although the majority of heterozygous mutations in COL1A1 or COL1A2, encoding the pro-α1- and pro-α2-chain of type I collagen, respectively, result in the brittle bone disorder osteogenesis imperfecta (OI), specific type I collagen defects can also give rise to rare EDS subtypes.
Biallelic COL1A2 mutations resulting in complete loss of pro-α2(I)-chains are associated with the cardiac-valvular EDS subtype (cvEDS). The clinical hallmarks of cvEDS include severe and progressive cardiac-valvular problems, combined with variable skin hyperextensibility, atrophic scarring, and joint hypermobility (Brady et al., 2017). A recent study from our team focusing on type I collagen defects in zebrafish, described a col1a2 knockout (col1a2–/–) zebrafish lacking pro-α2(I)-chains as the first zebrafish mutant for EDS (Figure 6A; Gistelinck et al., 2018). The skin of col1a2–/– zebrafish showed a disturbance of the typical stripe pattern, increased fragility and 50% reduction in dermal thickness. Consistent with these findings, biomechanical testing revealed decreased strength of the soft connective tissues, which is in line with the human phenotype. col1a2–/– zebrafish also displayed marked kyphosis, most likely as a consequence of local distortion and dislocation of the intervertebral ligament in parts of the vertebral column. Additionally, a mild reduction of bone thickness and mineralization in the vertebral column was observed in col1a2–/– zebrafish. These findings are consistent with the presence of joint dislocations and the lack of a severe skeletal phenotype in cvEDS patients (Malfait et al., 2006). Despite the prominent propensity for cardiac-valvular disease in human patients, initial histological analysis of the adult heart from col1a2–/– mutants did not show overt morphological abnormalities of the cardiac valves and cardiac function and blood flow was normal in col1a2–/– larvae. More in-depth studies are necessary to study the effect of absent pro-α2(I)-chains on the zebrafish cardiac morphology and functioning.
Dermatosparaxis was first described in cattle showing severe fragility of the skin and was the first collagen disorder that was characterized at the ultrastructural and biochemical level in the animal world (Lapière et al., 1971; Hanset and Lapiere, 1974). Human dermatosparaxis EDS (dEDS) is an autosomal recessive condition clinically hallmarked by extreme skin fragility with excessive skin folds at the wrists and ankles, severe bruisability and growth retardation. Patients have a typical facial gestalt with prominent and protuberant eyes with puffy, edematous eyelids, excessive periorbital skin, large fontanels and/or wide cranial sutures, a hypoplastic chin and bluish or grayish sclerae. Animal dermatosparaxis and human dEDS are caused by biallelic mutations in ADAMTS2, encoding a disintegrin and metalloproteinase with thrombospondin-motifs Type 1 Motif-2 (ADAMTS-2) (Malfait et al., 2017). ADAMTS-2 is the most prominent type I procollagen N-proteinase but can also cleave the N-propeptide of types II, III, and V procollagen chains and possibly also other substrates (Colige et al., 2005).
A murine homozygous knockout model of Adamts2 (Adamts2–/–) was generated. At birth, Adamts2–/– mice were indistinguishable from their wild-type littermates. Around the age of two months, Adamts2–/– mice developed triangular facies with a shorter snout, had less dense hair with thinner hair follicles, but lacked craniofacial abnormalities or growth retardation, unlike dEDS patients (Li et al., 2001; Goff et al., 2006). Additionally, the skin of Adamts2–/– mice felt thinner and softer and was extremely fragile resulting in easy tearing upon handling. While TEM analysis of the skin of 2-day-old Adamts2–/– mice was unremarkable, 2-month-old Adamts2–/– mice showed collagen fibrils with an unusually curled morphology, a finding consistent with the “hieroglyphic” appearance of cross-sectional collagen fibrils in human dEDS patients (Colige et al., 2004). Although the cutaneous features of Adamts2–/– mice largely mimic findings in human dEDS skin, age-dependent changes have not been reported in dEDS patients or animal dermatosparaxis. Histological and/or ultrastructural evaluation of cartilage, skeleton and aorta of Adamts2–/– mice were unremarkable (Li et al., 2001; Goff et al., 2006). Only mild dental changes were noted in Adamts2–/– mice, with normal incisors but subtle loss of the surface contour of the molar teeth, which contrasts the rather severe secondary dentition abnormalities (e.g., micro- and hypodontia) seen in dEDS patients (Goff et al., 2006; Brady et al., 2017). Adamts2–/– mice had abnormal lungs, characterized by decreased parenchymal density, with a pseudo-emphysematous appearance, but without inflammatory exudate or obvious fibrosis. Collagen fibrils in the lung appeared unaffected on TEM (Goff et al., 2006). Female Adamts2–/– mice were fertile, but male mice were sterile, pointing to an unexpected role for ADAMTS-2 in maturation of spermatogonia (Li et al., 2001). To date, no pregnancies have been reported in affected individuals and no information on reproduction is available (Brady et al., 2017).
Notably, not all type I collagen-rich tissues of Adamts2–/– mice (and human dEDS) are affected to the same degree. Although pro-α(I)-chains with a retained N-propeptide were seen in skin extracts of Adamts2–/– mice as expected, fully processed mature α-chains were also observed. Similar findings were obtained for type II (pro)collagen in cartilage (Li et al., 2001), and types I and III (pro)collagen in aorta and lung (Goff et al., 2006) of Adamts2–/– mice, thereby indicating that other enzymes (e.g., ADAMTS-3 and ADAMTS-14) can, at least partially, compensate for the loss of ADAMTS-2, in a tissue-specific way (Goff et al., 2006).
Of interest, two additional murine Adamts2 knockout models, Adamts2Δ28 and Adamts2Δ245, harboring a 28 and 245 bp genomic deletion, respectively, were recently established to investigate the proteolytic inactivation of the ECM glycoprotein, reelin, by Adamts-2 in the adult brain and did not focus on the associated EDS phenotype, but both models showed fragile skin and dull fur (Yamakage et al., 2019). This is the first report highlighting a neuronal function for ADAMTS-2 in addition to its role in collagen biosynthesis and connective tissue integrity.
Individuals with kyphoscoliotic EDS (kEDS) mainly suffer from congenital muscle hypotonia, progressive kyphoscoliosis and generalized joint hypermobility. Patients also present with abnormal scarring and easy bruising and have an increased risk of fatal arterial ruptures and, in some patients, ocular fragility. The majority of kEDS patients harbor biallelic mutations in the PLOD1 gene, encoding lysyl hydroxylase 1 (LH1), which catalyzes hydroxylation of specific helical lysine residues within the pro-α collagen chains (Brady et al., 2017). As a result of LH1 deficiency, lysyl residues are underhydroxylated and hydroxylysyl residues underglycosylated, leading to impaired cross-link formation and mechanical instability of affected tissues (Rohrbach et al., 2011). kEDS-PLOD1 was the first EDS type to be characterized at the biochemical level (Krane et al., 1972).
A homozygous Plod1 knockout (Plod1–/–) murine model for kEDS-PLOD1 was generated and the resulting Plod1–/– mice were viable and fertile but showed gait abnormalities and tired quickly (Figure 4C). They were passive when being handled and felt soft and floppy and their movements were powerless, consistent with muscle hypotonia, which is also observed in kEDS-PLOD1 patients (Takaluoma et al., 2007). Although the majority of Plod1–/– mice survived, 15% of these mice (17% of males and 9% of females) died before the age of 1 year (mostly at 1–4 months of age) due to aortic rupture, a life-threatening complication that can also occur in kEDS-PLOD1 patients. Although the thickness of the aortic wall was normal in surviving Plod1–/– mice, VSMCs appeared to be less regularly ordered and presented with degenerative ultrastructural changes, including vacuolization and mitochondrial swelling. Ultrastructural analysis showed more variation with overall increased collagen fibril diameters in the aorta and some fibrils had irregular contours. Similar ultrastructural alterations were seen in the skin but skin hyperextensibility or fragility were lacking in Plod1–/– mice. In contrast to the human phenotype, no kyphoscoliosis was observed in these mice. In line with defective LH1 function, tissue-specific decreases in total hydroxylysine residues were apparent in Plod1–/– mice, ranging from 22% in skin to 86% in lung. These findings suggest partial compensation by the two other isoenzymes, LH2 and LH3 (Takaluoma et al., 2007).
Overall, Plod1–/– mice seem to only partially capture the human kEDS-PLOD1 phenotype with the presence of muscle hypotonia and aortic ruptures but without kyphoscoliosis or a clear skin phenotype. The latter might reflect differences in remaining hydroxylysine content between mice (22%) and in human (5%) (Steinmann et al., 2003; Takaluoma et al., 2007).
Classical-like EDS (clEDS) is a rare autosomal recessive condition, characterized by generalized joint hypermobility and skin hyperextensibility with easy bruising but without atrophic scarring. Patients harbor biallelic mutations in the TNXB gene, resulting in complete absence of the corresponding tenascin-X (TNX) protein (Brady et al., 2017; Green et al., 2020). TNX is a glycoprotein with an architectural function in the ECM and has been shown to localize with collagen fibrils in tendon and dermis (Valcourt et al., 2015).
Tenascin-X was the first structural protein beyond fibrillar collagens or their modifying enzymes associated with EDS and to investigate its hitherto unknown functions, two groups independently created homozygous Tnxb knockout (Tnxb–/–) mice (Matsumoto et al., 2001; Mao et al., 2002). Although Tnxb–/– mice appeared morphologically normal at birth they showed progressive skin hyperextensibility, similar to clEDS patients, accompanied by a decreased ultrastructural density of fibrils with normal size and shape, resulting in a reduced dermal collagen content. Despite near-normal collagen synthesis in vitro, Tnxb–/– skin fibroblasts showed defective deposition of type I collagen. Together with the ability of TNX to regulate the expression of several ECM molecules, these findings pointed to a regulatory role in collagen fibril deposition (Mao et al., 2002; Minamitani et al., 2004a). Nevertheless, conflicting results have been reported for Tnxb–/– skin with normal dermal collagen content (Egging et al., 2007) and increased collagen fibril diameters with normal fibril density (Minamitani et al., 2004b), which might be influenced by murine age, sample site and genetic background. Of note, opposed to the elastin fragmentation in clEDS patients, Tnxb–/– mice showed increased dermal elastin density, which is probably less stable (Egging et al., 2006).
In contrast to cEDS, clEDS patients do not present with atrophic scarring or delayed wound healing. Tnxb–/– mice mimicked the macroscopically normal wound closure of the dorsal skin in vivo, associated with reduced breaking strength (Egging et al., 2007). Subsequent in vitro experiments using Tnxb–/– mouse embryonic fibroblasts suggested that the near-normal wound healing may be due to accelerated local matrix contraction and tissue remodeling caused by increased activation of MMPs, upregulation of TGF-β1 expression and upregulated collagen synthesis followed by the promotion of cell proliferation and migration (Hashimoto et al., 2018). Together, these findings pointed to a role for TNX during the later phase of wound healing when remodeling and maturation of the ECM occurs (Egging et al., 2007). Additionally, subcutaneous adipose tissue of Tnxb–/– mice was thicker and contains an increased amount of triglycerides and an altered fatty acid composition, suggesting a role during lipogenesis (Matsumoto et al., 2004).
Unlike clEDS patients, Tnxb–/– mice did not show overt signs of joint hypermobility or ligamentous laxity (Egging et al., 2006), although tail and Achilles tendon displayed ultrastructural changes in collagen fibril density similar to observations in the skin (Mao et al., 2002). Tnxb–/– mice did present with mild muscle weakness, histological evidence of myopathy and increased turnover of the ECM in quadriceps muscle (Voermans et al., 2011) as well as altered myofascial force transmission (Huijing et al., 2010), consistent with the mild to moderate muscle weakness observed in clEDS patients (Brady et al., 2017). Histology and TEM studies of the sciatic nerve revealed mildly reduced diameters of myelinated fibers and reduced collagen fibril density in the endoneurium in older, but not young, Tnxb–/– mice, which may correspond to axonal polyneuropathy seen in clEDS patients (Matsumoto et al., 2002; Voermans et al., 2009, 2011; Sakai et al., 2017). Additionally, blood vessel formation (but not morphology) was altered in the peripheral nervous system of Tnxb–/– mice, with a decreased density of blood vessels with increased diameters (Sakai et al., 2017). Interestingly, Tnxb–/– mice were reported to have mechanical allodynia but not thermal hypersensitivity. A chemical pain stimulus with formalin injection in the hind paw evoked an increased pain response during the first (acute) phase (0–5 min) and early during the second (inflammatory) phase (16–30 min) in Tnxb–/– mice. Transcutaneous sine wave stimuli elicited a hypersensitive response of myelinated Aδ and Aβ fibers, but not of unmyelinated C fibers. Additionally, Tnxb–/– mice showed molecular alterations in the dorsal horn of the spinal cord suggestive of spinal central sensitization (Okuda-Ashitaka et al., 2020).
Additional studies in Tnxb–/– mice revealed the presence of reduced femoral bone mass with enhanced osteoclast differentiation and bone-resorbing ability (Kajitani et al., 2019), very mild genito-urinary complications (rectal prolapse in <1%) in female mice compared to uterine and vaginal prolapse seen in clEDS patients (Egging et al., 2008), attenuated stromal neovascularization following cauterization (Sumioka et al., 2018) and gastric dysfunction associated with abnormal gastric sensory function (Aktar et al., 2019). Additionally, Tnxb–/– mice showed increased anxiety-like behavior as well as superior sensorimotor coordination and emotional learning and memory, without differences in locomotor or home cage activity (Kawakami and Matsumoto, 2011; Voermans et al., 2011). Cognitive development was not studied in clEDS patients (Brady et al., 2017).
In summary, Tnxb–/– mice recapitulate the dermatological, mild muscular findings, and some neurological (including pain) findings of human clEDS but lack the articular aspects. These mice have been instrumental in demonstrating a regulatory role for TNX in a broad range of tissues.
Myopathic EDS (mEDS) is a rare EDS subtype clinically characterized by congenital muscle hypotonia and weakness with delayed motor development, proximal joint contractures in combination with distal joint hypermobility, scoliosis or kyphosis and abnormal scarring (Malfait et al., 2017; Delbaere et al., 2019b). mEDS is caused by mutations in COL12A1 encoding the pro-α1-chain of type XII collagen, a homotrimer that is the largest member of the FACIT family. Type XII collagen interacts with several ECM molecules (e.g., decorin and tenascin-X) and regulates the organization and mechanical properties of collagen fibrils (Chiquet et al., 2014). The majority of defects identified in COL12A1 give rise to heterozygous substitutions or in-frame exon skips, while a single biallelic loss-of-function COL12A1 mutation was identified that results in a more severe phenotype (Hicks et al., 2014; Zou et al., 2014).
Before disease-causing mutations in COL12A1 were identified, type XII collagen-deficient (Col12a1–/–) mice were generated. Col12a1–/– mice were smaller and exhibited skeletal abnormalities with shorter, more slender and fragile long bones as well as aberrant vertebrae structures (Izu et al., 2011). In depth analyses revealed that the bone phenotype is associated with defective differentiation, organization, polarization, morphology and interactions of Col12a1–/– osteoblasts (Izu et al., 2011), thereby affecting cell–cell communication (Izu et al., 2016) and resulting in decreased bone matrix formation with altered quality. Although these skeletal manifestations were not observed in human mEDS, patients have kyphosis and/or scoliosis, comparable to the kyphoscoliosis seen in Col12a1–/– mice (Izu et al., 2011; Zou et al., 2014). Together with the identification of COL12A1 defects in human patients, the phenotype of the Col12a1–/– mouse model was revisited. Col12a1–/– mice showed evidence of (mild) muscle weakness, delayed transition in muscle fiber-type composition and altered force transmission in the muscle-tendon-bone unit. Ultrastructural studies showed more diffusely dispersed collagen fibrils throughout the endomysium of Col12a1–/– muscle (Zou et al., 2014). Given the combination of distal joint hypermobility and proximal contractures in mEDS, tendon development was evaluated in Col12a1–/– mice, which also showed decreased collagen fibril packing. Tenocytes from Col12a1–/– mice had an altered shape and function with lower type I collagen secretion (Izu et al., 2020). Together, these data indicated the crucial role of type XII collagen in the development and maturation of tendon.
Although all animal studies were performed on homozygous Col12a1–/– mice, only two siblings with autosomal recessive mEDS due to complete loss of type XII collagen were identified to date (Zou et al., 2014). Nevertheless, muscle weakness and skeletal abnormalities were observed in both Col12a1–/– mice and mEDS patients, but the muscular phenotype seems to be milder in mice compared to patients. To date, phenotypic characteristics of heterozygous Col12a1+/– mice have not been reported.
Proteoglycans represent major ECM components and play essential roles as structural macromolecules, modulators of cell adhesion and motility, ECM and collagen fibril assembly and signal transduction during development, tissue repair, and angiogenesis. Proteoglycans are composed of a specific core protein substituted with one or more glycosaminoglycan (GAG)-chains (Prydz and Dalen, 2000; Bishop et al., 2007; Couchman and Pataki, 2012). GAG biosynthesis is initiated by the stepwise addition of a tetrasaccharide linker to the core protein followed by the addition of repeating disaccharide units that define the GAG-chain as heparan sulfate (HS), chondroitin sulfate (CS), and/or dermatan sulfate (DS) (Figure 1B).
The first hint for the involvement of proteoglycans in the pathophysiology of EDS came from a mouse model deficient for decorin, the prototypic member of a specific class of proteoglycans, the SLRPs. The resulting decorin knockout (Dcn–/–) mice had thin and fragile skin with reduced tensile strength and an abnormal ultrastructural morphology of collagen fibrils in skin and tendon with larger and more irregular fibril outlines, thereby highlighting the important regulatory role of decorin during collagen fibrillogenesis (Danielson et al., 1997). Subsequently, knockout mice for other SLRPs, lumican (Chakravarti et al., 1998), dermatopontin (Takeda et al., 2002), or mimecan (Tasheva et al., 2002) also displayed signs of skin laxity and fragility and similar ultrastructural collagen fibril abnormalities as seen in Dcn–/– mice (Supplementary Table 4). Given the phenotypic resemblance of these animal models to human EDS, the genes encoding SLRPs were considered good candidates for molecularly unresolved EDS patients, but defects in SLRP core proteins could never be linked with EDS (Malfait et al., 2005). Nevertheless, the subsequent identification of genetic defects in enzymes involved in the synthesis and modification of GAG-chains unequivocally confirmed a role for aberrant proteoglycan biosynthesis in human EDS pathogenesis, but the causal defects compromise proper GAG-formation rather than affecting the core protein.
Biallelic mutations in the B4GALT7 and B3GALT6 genes, encoding the linker enzymes galactosyltransferase-I (β4GalT7) and -II (β3GalT6), respectively, that catalyze subsequent steps of the GAG tetrasaccharide linker region biosynthesis (Figure 1B), give rise to two subtypes of spondylodysplastic EDS (spEDS), spEDS-B4GALT7 and spEDS-B3GALT6. Both subtypes are severe, pleiotropic conditions that are generally characterized by short stature, muscle hypotonia, skeletal abnormalities, skin hyperextensibility and translucency, delayed motor development, and osteopenia. Additionally, subtype specific clinical features can be observed (Table 1; Malfait et al., 2017).
To date, no mouse models with complete β4GalT7- or β3GalT6-deficiency have been reported. The lack of relevant in vivo models to investigate the pathogenic mechanisms in these EDS subtypes prompted our group to generate zebrafish models for these conditions (Delbaere et al., 2019a, 2020).
Since reported B4GALT7 mutations result in a variable reduction, but not complete loss, of β4GalT7 activity (Salter et al., 2016), hypomorphic zebrafish with partial loss of β4GalT7 function were generated to model spEDS-B4GALT7, including two different morpholino-based b4galt7 knockdown (morphant) as well as two mosaic F0 b4galt7 knockout (crispant) models (Delbaere et al., 2019a). In both models, defective β4GalT7 activity led to hampered GAG biosynthesis with diminished amounts of sulfated GAGs and strongly reduced HS and CS proteoglycan levels at 4 days post fertilization (dpf). Comparable to the short stature seen in spEDS-B4GALT7 patients, morphant and crispant embryos were smaller than wild-type embryos, mainly attributed to their smaller head. During early development, craniofacial features (e.g., triangular, flat face, and narrow mouth) of human spEDS-B4GALT7 are mimicked in morphant and crispant b4galt7 zebrafish, which show craniofacial abnormalities with underdeveloped, misshapen and partly absent cartilage structures (Figure 6B). Cranial neural crest cells are also less abundant and more disorganized in the developing jaw. Additionally, severely reduced to absent mineralized bone structures were observed, with delayed ossification, reminiscent of osteopenia seen in some spEDS-B4GALT7 patients. Furthermore, the bowing of the pectoral fins in the morphants and crispants corresponds to the bowing of the limbs observed in spEDS-B4GALT7 patients. Only in the b4galt7 morphant created with a translation-blocking morpholino compromised muscle function and structure were suggested by deficient hatching from the chorion, a delayed touch-evoked escape response and disturbed filamentous actin patterning in trunk muscle. Nevertheless, despite its presence in only one of the b4galt7 models, muscle hypotonia and delayed motor development are prominent clinical features in spEDS-B4GALT7 patients.
Homozygous b4galt7 knockout (b4galt7–/–) zebrafish were created for validation purposes and displayed similar craniofacial features as seen in the morphant and crispant models. However, as expected from complete loss of β4GalT7, b4galt7–/– embryos and larvae showed a total lack of HS and CS GAGs leading to a more severe bone phenotype with delay or absence of ossification of several bone structures, complete loss of cartilage staining and disorganized chondrocyte stacking in the head. These animals died before the age of 10dpf, confirming that complete loss of β4GalT7 activity is not compatible with life.
Taken together, larval b4galt7 zebrafish models largely phenocopy the human spEDS-B4GALT7 phenotype and highlight a key role for β4GalT7 during early development, affecting cartilage, bone and possibly muscle formation. The generation of stable knock-in models harboring (reported) missense variants could overcome the phenotypic variability of b4galt7 crispants and lethality of the b4galt7–/– knockout zebrafish and would allow to study the adult phenotype in more depth (Delbaere et al., 2019a).
In contrast to b4galt7–/– zebrafish, homozygous b3galt6 knockout (b3galt6–/–) zebrafish did survive to adulthood (Delbaere et al., 2020). b3galt6–/– zebrafish showed progressive skeletal malformations with variable severity, including reduced body length, misshapen cranial bones, smaller teeth, and general skeletal dysplasia with kyphosis and scoliosis (Figure 6C), which are reminiscent of the skeletal dysplasia phenotype observed in spEDS-B3GALT6 patients. Additionally, extra intramembranous bone and bony elements were found on the vertebra as well as a generalized reduction in bone volume and thickness, and relatively increased tissue mineral density. Ultrastructural analysis showed less organized type I collagen fibrils in the intervertebral ligament and the inner layer of the vertebral bone. Additionally, electron dense spots were observed in the vertebral bone, which might be remnants of extrafibrillar mineral aggregates, and could contribute to the increased mineralization and brittleness of the skeleton. This might be related to bone fragility with spontaneous fractures observed in spEDS-B3GALT6. Adult b3galt6–/– zebrafish displayed interruptions of the horizontal stripes. Ultrastructural analysis of the skin showed a thicker epidermis covering the scales and loosely packed dermal collagen fibrils with increased interfibrillar spaces. The latter findings mimic ultrastructural features of spEDS-B3GALT6 patients and could be attributed to alterations in the GAG-chains of SLRPs which regulate collagen fibrillogenesis. Consistent with the muscle hypotonia and delayed motor development observed in patients, functional and structural muscle abnormalities were detected in b3galt6–/– zebrafish, with lower endurance during a swim-tunnel experiment, thicker endomysium surrounding the muscle fibers and increased sarcomere length on ultrastructure. These phenotypic findings are in accordance with the strongly reduced amounts of CS, DS, and HS disaccharides observed in bone, skin, and muscle of b3galt6–/– zebrafish. Surprisingly, low amounts of GAGs were still produced in absence of compensatory upregulation of other galactosyltransferases or linker enzymes, and residual proteoglycans contained an immature linker region consisting of only three instead of four sugars (lacking a galactose residue), thereby presenting a rescue mechanism for a deficiency of one of the linker enzymes. The presence of this non-canonical trilinker region could be confirmed in urine of human spEDS-B3GALT6 samples (unpublished). As such, b3galt6–/– zebrafish largely phenocopy the human spEDS-B3GALT6 phenotype, including craniofacial dysmorphism, generalized skeletal dysplasia, skin involvement and indications for muscle hypotonia.
Collectively, these studies provided proof-of-concept that zebrafish are good models to study the phenotypic and biomolecular consequences of impaired GAG linker region synthesis and further underscore their usefulness as animal models for EDS research.
Musculocontractural EDS (mcEDS) is clinically characterized by multiple congenital malformations including characteristic craniofacial features and contractures as well as progressive multisystemic complications such as skin hyperextensibility and fragility, recurrent dislocations, progressive talipes or spinal deformities, and large subcutaneous hematomas (Malfait et al., 2017). mcEDS is caused by biallelic mutations in either CHST14 or DSE, encoding dermatan 4-O-sulfotransferase-1 (D4ST1) and dermatan sulfate epimerase-1 (DS-epi1), respectively. Both enzymes are specifically involved in DS GAG biosynthesis (Figure 1B). In contrast to defects in B3GALT6 and B4GALT7 that compromise linker formation and affect the synthesis of both CS/DS and HS proteoglycans, D4ST1- or DS-epi1-deficiency only interferes with formation of DS proteoglycans.
The first murine homozygous Chst14 knockout (Chst14–/–) model was created to study the roles of DS GAGs during neural development and nerve regeneration (Bian et al., 2011). Chst14–/– mice showed reduced viability (8% of offspring). Surviving Chst14–/– mice had a normal life span but were smaller and had reduced fertility, kinked tail, tooth malformations and increased skin fragility (Akyüz et al., 2013). Chst14–/– mice provided evidence for an overall negative impact of DS on peripheral nerve regeneration (Akyüz et al., 2013) but beneficial activity in central nervous system regeneration (Rost et al., 2016), which could be attributed to differences in levels of DS proteoglycans and their interaction partners between the peripheral nervous system and the central nervous system (Rost et al., 2016). Furthermore, Chst14–/– mice showed impaired spatial learning and memory and demonstrated that DS is indispensable for synaptic plasticity in the hippocampus, which might be attributed to impaired Akt/mTOR-signaling (Li et al., 2019).
Subsequently, another homozygous Chst14 knockout (Chst14–/–) mouse model was studied that was initially created as part of a largescale effort to create a knockout library for secreted and transmembrane proteins (Chst14tm1Lex) (Tang et al., 2010). This Chst14–/– model also suffered from perinatal lethality, with a markedly decreased survival of 14.8% at embryonic day 18.5 to 1.3% at adulthood. Placentas from Chst14–/– fetuses showed a reduced weight, with smaller vascular diameters in the placental villi (the part of the fetal placenta), and hypoxia- and/or necrotic-like changes in a small subset (6%). TEM analysis demonstrated decreased thickness and fragmentation of basement membranes in the capillaries of the villus (Yoshizawa et al., 2018). These placental vascular abnormalities possibly relate to the high perinatal lethality in Chst14–/– mice as well as the development of large subcutaneous hematomas in mcEDS-CHST14 patients. Changing the genetic background (from mixed C57BL/6-129/SvJ to BALB/c), improved the birth rate for Chst14–/– mice to 6.12–18.64% (Shimada et al., 2020). Subsequent analysis of the skin, which is affected in mcEDS-CHST14 patients, show reduced tensile strength Chst14–/– mice and TEM analysis revealed increased intrafibrillar spaces with a decreased density and disorganization of collagen fibrils. Additionally, rod-shaped, linear GAG-chains protruded from collagen fibrils in Chst14–/– mice compared to the curved GAG-chains wrapped around the collagen fibrils in wild-type (Hirose et al., 2020), similar to TEM findings for mcEDS-CHST14 patients (Hirose et al., 2019b). Glycobiological analyses of the skin revealed very low levels of DS disaccharides and an excess of CS disaccharides (Hirose et al., 2020).
The characteristic alterations in GAG-structure and disorganization of the collagen network in de skin are similar to ultrastructural observations in mcEDS-CHST14 patients, thereby making Chst14–/– mice a reasonable model for this EDS subtype although the reduced viability compromises further studies.
Before DSE defects were identified in mcEDS patients, homozygous Dse knockout (Dse–/–) mice were already generated. Dse–/– mice were smaller, presented with a kinked tail that resolved before 4 weeks of age and had reduced fertility (Figure 4D). The skin of Dse–/– mice had reduced tensile strength and sparser loose hypodermal connective tissue on light microscopy despite normal collagen content. TEM analysis revealed dermal collagen fibrils with larger diameters but normal fibril density while Dse–/– tail tendons showed only a minor shift towards thicker fibrils with mostly fibrils with normal diameter and no differences in diameter in Achilles tendon (Maccarana et al., 2009). This contrasts the normal ultrastructure of dermal collagen fibrils in mcEDS-DSE patients, although morphometric analysis was not performed (Syx et al., 2015). Glycobiological examination revealed a decreased, but not totally absent, DS content in the skin, similar to findings in a mcEDS-DSE patient (Müller et al., 2013). The residual DS moieties may be attributed to partial compensation by DS-epi2 activity. Changing the genetic background (from mixed 129SvJ-C57BL/6 to NFR), resulted in Dse–/– mice with normal body weights, a kinked tail and some developmental malformations, including an abdominal wall defect with herniated intestines in 16% of Dse–/– embryos and neural tube defects (e.g., exencephaly and spina bifida) in 5%. Additionally, Dse–/– embryos and newborns had thicker epidermal layers with increased amounts of epidermal basal and spinous layer markers (Gustafsson et al., 2014).
The relatively milder phenotype in Dse–/– mice is consistent with the findings in mcEDS-DSE patients, which also present an apparently milder phenotype compared to mcEDS due to CHST14 defects, but only a limited number of patients have been reported so far (Lautrup et al., 2020).
Biallelic mutations in the SLC39A13 gene, encoding the metal transporter Zrt/irt-like protein 13 (ZIP13), result in a phenotype which clinically overlaps with spEDS due to B4GALT7 and B3GALT6 mutations and is therefore grouped within the same clinical entity (Malfait et al., 2017). spEDS-SLC39A13 patients present with moderate short stature, combined with specific facial characteristics, hypo- or oligodontia, distal joint hypermobility, thin and fragile skin, wrinkled palms, and characteristic radiographic abnormalities (Brady et al., 2017; Malfait et al., 2017; Kumps et al., 2020). spEDS-SLC39A13 patients display collagen underhydroxylation, despite normal activity of lysyl and prolyl hydroxylases (Fukada et al., 2008; Giunta et al., 2008). ZIP13 is a homodimeric transmembrane protein and putative zinc (Zn2+) transporter located on the endoplasmic reticulum/Golgi apparatus which regulates the influx of Zn2+ into the cytosol (Bin et al., 2011). Interestingly, studies in Drosophila melanogaster revealed that the fruit fly homolog of human ZIP13 exports iron (Fe2+) and its depletion might attenuate the action of iron-dependent enzymes such as lysyl hydroxylases, possibly explaining the observed underhydroxylation (Xiao et al., 2014).
Homozygous Slc39a13 knockout (Slc39a13-KO) mice showed growth retardation, progressive kyphosis after 3–4 weeks of age, osteopenia, and abnormal cartilage development (Fukada et al., 2008). Additionally, altered craniofacial features, including sunken and downslanting eyes were apparent as well as dental abnormalities, such as incisor deformities and aberrant dentin formation (Figure 4E). The skin of Slc39a13-KO mice was more fragile and showed a thinner dermis with smaller and less densely packed collagen fibrils on ultrastructure, the latter possibly attributed to a decreased dermal DS content (Fukada et al., 2008; Hirose et al., 2015). Subcutaneous adipose tissue was thinner (Fukada et al., 2008; Fukunaka et al., 2017). Corneal stroma showed reduced thickness in Slc39a13-KO mice with smaller and more widely spaced collagen fibrils which were more randomly oriented (Fukada et al., 2008; Hirose et al., 2018). Except for severe varicosity in the lower extremities, spEDS-SLC39A13 patients do not present with cardiovascular symptoms. Similarly, Slc39a13-KO mice did not show aneurysms or arterial ruptures, however, abnormalities were observed in the tunica media of the thoracic aorta which could potentially result in increased fragility (Hirose et al., 2019a). Slc39a13-KO mice showed evidence for reduced osteoblast activity, impaired chondrocyte and adipocyte differentiation as well as irregular cellular morphology of odontoblasts in the molar tooth, dermal fibroblasts, corneal keratocytes, and aortic VSMCs (Fukada et al., 2008; Fukunaka et al., 2017; Hirose et al., 2018, 2019a). Furthermore, murine studies illustrated dysregulation of the BMP- and TGFβ-signaling pathways in several of these connective tissue forming cells, which might contribute to the observed multisystemic phenotype (Fukada et al., 2008).
Taken together, Slc39a13-KO mice display craniofacial, dental, skeletal, dermal, and ocular features that are reminiscent of the human spEDS-SLC39A13 phenotype and highlight a role for altered BMP- and TGFβ-signaling in spEDS-SLC39A13 pathogenesis.
Brittle cornea syndrome (BCS) is clinically hallmarked by a severe ocular phenotype with thin and fragile cornea leading to increased risk of spontaneous rupture and often vision loss. Additionally, patients also present with hearing loss and signs of generalized connective tissue fragility such as distal joint hypermobility and mild cutaneous and craniofacial abnormalities. BCS is caused by biallelic defects in either ZNF469 or PRDM5. ZNF469 encodes the zinc finger protein ZNF469, which is believed to be a (transcriptional) regulator of the synthesis and/or organization of collagen fibrils (Abu et al., 2008) but its function remains elusive. PRDM5 encodes PR domain zinc finger protein 5 (PRDM5), a transcription factor that was identified as a putative tumor suppressor gene in several cancers but has also been shown to regulate transcription of collagen and other ECM genes (Galli et al., 2012).
Although no animal models have been generated to study BCS pathogenesis, two independent studies investigated the role of PRDM5 in zebrafish larvae as a putative tumor suppressor and during craniofacial development (Meani et al., 2009; Ding et al., 2013). Interestingly, morpholino-based prdm5 knockdown affects early stages of zebrafish development with craniofacial malformation including cyclopia or smaller eyes and axial mesendodermal defects (jaw, heart, and blood) (Figure 6D; Meani et al., 2009). Craniofacial malformations with neurocranial defects were subsequently confirmed in prdm5 knockout (prdm5hi61Tg) zebrafish larvae (Amsterdam et al., 2004; Ding et al., 2013). These findings hint to the ocular phenotype and mild craniofacial abnormalities seen in BCS patients. Furthermore, prdm5 knockdown results in impaired morphogenetic movement during gastrulation as a consequence of increased canonical Wnt-signaling (Meani et al., 2009; Ding et al., 2013).
To date, no stable animal models of BCS due to ZNF469 defects have been described.
The most recent defects associated with human EDS reside in adipocyte enhancer-binding protein-1 (AEBP1), also known as aortic carboxypeptidase-like protein (ACLP). AEBP1 is expressed in tissues with a high collagen content and is a secreted ECM-associated protein that preferentially binds to fibrillar types I, III, and V collagen and acts as a positive regulator of collagen polymerization in vitro and collagen fibrillogenesis in vivo (Layne et al., 2001; Blackburn et al., 2018). Biallelic mutations in the AEBP1 gene, encoding AEBP1, cause an EDS phenotype hallmarked by skin hyperextensibility with atrophic scarring, generalized joint hypermobility, foot deformities and early-onset osteopenia, provisionally referred to as classical-like EDS type 2 (clEDS2) (Blackburn et al., 2018; Syx et al., 2019).
Before AEBP1 defects were linked to EDS, two groups independently created homozygous Aebp1 knockout (Aebp1–/–) mice, which presented some phenotypic discrepancies. While the majority of the first Aebp1–/– mouse model died perinatally due to gastroschisis, surviving Aebp1–/– mice developed spontaneous skin lesions and present with delayed wound healing, which correlated with reduced dermal fibroblast proliferation (Layne et al., 2001). These wound healing defects are reminiscent of the phenotype of clEDS2 patients, indicating a role for AEBP1 in wound repair. Additional studies in mice suggested role(s) of AEBP1 in fibroblast-to-myofibroblast transition in lung through TGFβ-signaling (Schissel et al., 2009; Tumelty et al., 2014), and activation of the canonical Wnt-signaling pathway through frizzled-8 and LRP6 in hepatic stellate cells (Teratani et al., 2018). Deregulation of these pathways could potentially contribute to the defective wound healing and bone development and homeostasis seen in patients. For the second model, about half of Aebp1–/– mice survived and they were smaller, with markedly reduced white adipose tissue and defective proliferation and lower MAPK activity in Aebp1–/– pre-adipocytes. Additionally, lactation defects were observed in females and infertility in males (Ro et al., 2007; Zhang et al., 2011).
As such, studies in Aebp1–/– mice suggest the involvement of AEBP1 in the development and homeostasis of adipose tissue, mammary gland and connective tissue, with recapitulation of the dermal manifestations of clEDS2 in one model. Nevertheless, the exact mechanisms leading to the observed EDS phenotype remain currently unknown.
Before the first genetically engineered EDS mouse models were generated, naturally occurring EDS(-like) phenotypes were already described for decades in domesticated animals. There are several reports of cats, dogs, rabbits, mink, cattle, sheep, and horses displaying a disease reminiscent of EDS (Fjølstad and Helle, 1974; Hegreberg, 1975; Counts et al., 1977; Holbrook et al., 1980; Brown et al., 1993; Hansen et al., 2015). The phenotypes of these animals were hallmarked by thin and hyperextensible skin, which was fragile leading to hemorrhagic wounds and atrophic scars; and commonly described as cutis hyperelastica, hyperelastosis cutis, dermatosparaxis, dermal/collagen dysplasia, dermal/cutaneous asthenia, or Ehlers–Danlos-like syndromes (Halper, 2013).
While the genetic defects causing EDS-like pathologies in domestic animals were less well studied in early reports, the advent of next generation sequencing technologies such as whole exome sequencing and whole genome sequencing, provided molecular proof of the underlying disorder in more recent reports. Mutations have been reported in Col5a1 in cats (Spycher et al., 2018) and dogs (Bauer et al., 2019a), Col5a2 in cattle (Jacinto et al., 2020), Adamts2 in cattle (Colige et al., 1999), sheep (Zhou et al., 2011; Monteagudo et al., 2015; Joller et al., 2017), and dogs (Jaffey et al., 2019), Plod1 in horse (Monthoux et al., 2015), Tnxb in dogs (Bauer et al., 2019b) and B4galt7 in Finish horse (Leegwater et al., 2016). These animals display phenotypic features reminiscent of the corresponding human EDS counterpart. Additionally, horses presenting with an EDS-like condition with loose skin have been described and coined Hereditary Equine Regional Dermal Asthenia (HERDA). This condition is caused by defects in the Ppib gene, encoding cyclophilin B. This gene is not associated with EDS in humans but results in OI (van Dijk et al., 2009; Rashmir-Raven, 2013). Functional studies in these animals are restricted but sometimes accompanied by biochemical, histopathological and/or ultrastructural data. A summary of the findings of spontaneous models with confirmed genetic defects is provided in Table 2.
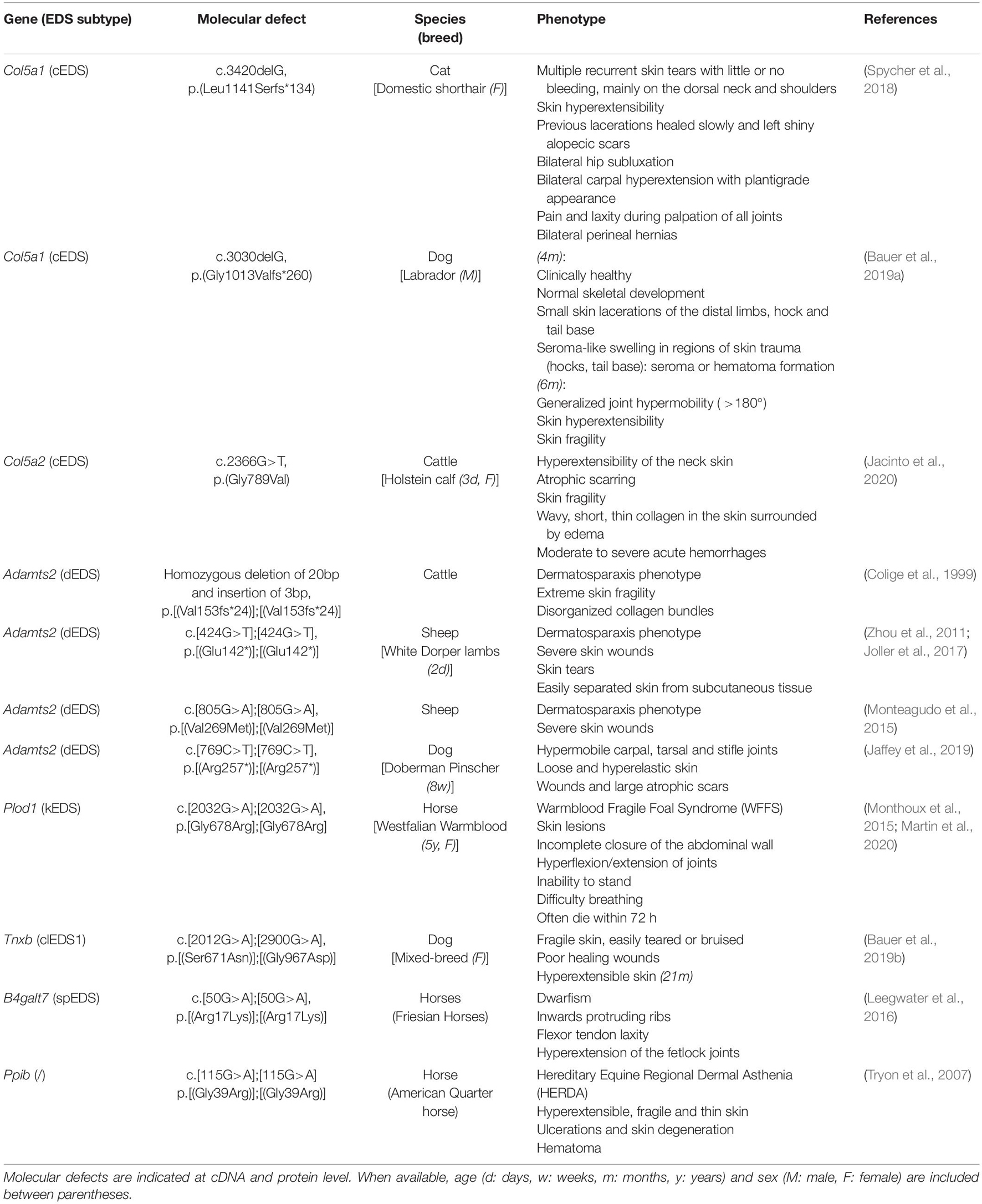
Table 2. Overview of naturally occurring animals with a molecularly proven EDS subtype.
Taken together, it is clear that EDS also occurs naturally in mammals and can be prominent in specific (in)breeding schemes. However, due to limited resources, especially in sequencing capacity for animals, many cases are not molecularly confirmed, and in-depth functional analyses are often lacking.
For 14 of the 20 hitherto known EDS-associated genes, animal models, either mouse or zebrafish, have been created. While for the more common cEDS and vEDS subtypes different engineered mouse models are available and often extensively characterized, animal models are lacking for some of the rarer EDS subtypes (e.g., periodontal EDS, kEDS-FKBP14, arthrochalasia EDS and BCS-ZNF469). Recently, zebrafish models were introduced into the EDS research field [col1a2–/– (cvEDS) and b4galt7 and b3galt6–/– (spEDS)]. Additionally, numerous domestic animals displaying an EDS(-like) phenotype have been reported, with the genetic defect identified for some. Although these models are valuable tools to learn about the EDS phenotype in animals other than rodents or zebrafish, they are less amendable for large-scale research purposes.
Especially engineered models have been instrumental in discerning the functions of these particular proteins during development, maturation and repair and in portraying their roles during collagen biosynthesis and fibrillogenesis, for some even before their contribution to an EDS phenotype was elucidated. Extensive phenotypical characterization of these models has shown that they largely phenocopy their human counterparts, with recapitulation of several key clinical features of the corresponding EDS subtype, including dermal, cardiovascular, musculoskeletal and ocular alterations. Moreover, these models provide valuable tools to perform in-depth studies of several disease-related aspects (e.g., wound healing); and allow to investigate organs and tissues that are not easily accessible from humans (e.g., cardiovascular or nervous tissue) to assess physicochemical, ultrastructural and biomechanical properties during early development and adult stages. This can be particularly helpful to elucidate disease mechanisms in rarer EDS subtypes for which only a limited number of patients are reported. Since for most EDS types, the exact link between a single gene defect and the observed phenotype remains elusive, more studies are needed to investigate the underlying pathways and understand the pathophysiological consequences.
As such, investigation of these models hinted toward the involvement of several signaling pathways, such as Wnt-signaling in prdm5 knockdown zebrafish and Aebp1–/– mice, BMP1- and/or TGFβ-signaling in Slc39a13-KO and Aebp1–/– mice; and PLC/IP3/PKC/ERK-signaling in Col3a1 knock-in mice and highlighted potential compensatory mechanisms such as the presence of a trilinker region in b3galt6–/– zebrafish. Further validation of these findings in patient samples is warranted and additional investigations are required to further pinpoint common or distinct pathways in several EDS subtypes, thereby explaining phenotypic overlap and differences.
Nevertheless, direct comparison between different EDS models is limited and complicated by several variables, such as: (1) differences in genetic background influencing the phenotypic outcome and survival of animals (e.g., Chst14–/– mice) (Montagutelli, 2000); (2) age at which the animals were examined; and (3) anatomical location of investigated tissues. Furthermore, sex differences have been described in most murine models of vEDS, with a more severe skin and vascular phenotype in males compared to females. Although the sex of the EDS animals was not always clearly indicated in literature, male-female divergences are increasingly being recognized in several organ systems, including, skin thickness, cardiovascular function, musculoskeletal integrity as well as in physiological processes, such as pain, and need to be taken into account in future preclinical EDS research (Huxley, 2007; Lang, 2011; Rosen et al., 2017; Sorge and Totsch, 2017; Rahrovan et al., 2018).
The majority of the models generated to date harbor heterozygous or homozygous null mutations, which are indispensable to study the consequences of complete protein deficiency, but which usually do not capture the complete mutation spectrum associated with a particular EDS subtype (e.g., structural variants like glycine substitutions or exon skips in COL5A1, COL5A2, or COL12A1). With exception of Col3a1 models of vEDS, no knock-in EDS models have been developed. Advances in genome editing technologies such as CRISPR/Cas9 and base editing can assist in creating these models (Adli, 2018; Porto et al., 2020).
Currently, treatment and management options for EDS patients are limited and for none of the EDS subtypes a targeted therapy is available. So far, preclinical therapeutic interventions have only been explored in mouse models of vEDS and contradicting results were obtained depending on the model used. When the elucidation of the pathogenic mechanisms and deregulated pathways progresses, novel druggable targets will likely be identified. Additionally, the introduction of zebrafish into the EDS research field holds promising potential for therapeutic discovery, since these animals are highly amenable for drug/compound screening.
Despite the availability of animal models for several EDS subtypes, the study of these models for understanding the pathophysiology of EDS is still in its infancy but they can supply a tremendous bulk of information to the scientific community. The generation of additional models (both murine and zebrafish), harboring different genetic defects and different types of genetic variants, will further boost our knowledge and might lead us into a new era of disease research and drug discovery. In summary, these EDS animal models proved to be valid models that are and will continue to be valuable resources that, when explored to full extend, can uncover novel insights in pathogenic mechanisms, identify reliable biomarkers and pinpoint relevant cellular processes or signaling pathways that can serve as targets for the development of disease-specific (and/or personalized) therapies.
RV and DS wrote the manuscript. RM, A-MM, and FM read and edited the draft of the manuscript. All authors approved the final version of the manuscript.
This work was supported by the Research Foundation Flanders (FWO), Belgium [1842318N to FM, 3G041519 to FM, and 12Q5920N to DS]; Association française des syndromes d’Ehlers-Danlos (AFSED) to FM; The Ehlers-Danlos Society to FM; the National Institutes of Health [National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS)] [grant numbers K01AR070328 to RM, R01AR077019-01A1 to RM, R01AR060364 and R01AR064251 to A-MM]. A-MM was supported by the George W. Stuppy, MD, Chair of Arthritis at Rush University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.726474/full#supplementary-material
Abu, A., Frydman, M., Marek, D., Pras, E., Nir, U., Reznik-Wolf, H., et al. (2008). Deleterious mutations in the zinc-finger 469 gene cause brittle cornea syndrome. Am. J. Hum. Genet. 82, 1217–1222. doi: 10.1016/j.ajhg.2008.04.001
Adli, M. (2018). The CRISPR tool kit for genome editing and beyond. Nat. Commun. 9:1911. doi: 10.1038/s41467-018-04252-2
Aktar, R., Peiris, M., Fikree, A., Eaton, S., Kritas, S., Kentish, S. J., et al. (2019). A novel role for the extracellular matrix glycoprotein-Tenascin-X in gastric function. J. Physiol. 597, 1503–1515. doi: 10.1113/jp277195
Akyüz, N., Rost, S., Mehanna, A., Bian, S., Loers, G., Oezen, I., et al. (2013). Dermatan 4-O-sulfotransferase1 ablation accelerates peripheral nerve regeneration. Exp. Neurol. 247, 517–530. doi: 10.1016/j.expneurol.2013.01.025
Amsterdam, A., Nissen, R. M., Sun, Z., Swindell, E. C., Farrington, S., and Hopkins, N. (2004). Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. U.S.A. 101, 12792–12797. doi: 10.1073/pnas.0403929101
Andrikopoulos, K., Liu, X., Keene, D. R., Jaenisch, R., and Ramirez, F. (1995). Targeted mutation in the col5a2 gene reveals a regulatory role for type V collagen during matrix assembly. Nat. Genet. 9, 31–36. doi: 10.1038/ng0195-31
Angwin, C., Brady, A. F., Pope, F. M., Vandersteen, A., Baker, D., Cheema, H., et al. (2020). Arterial complications in classical Ehlers-Danlos syndrome: a case series. J. Med. Genet. 57, 769–776. doi: 10.1136/jmedgenet-2019-106689
Backer, J. D., and Backer, T. D. (2019). Vascular Ehlers-Danlos syndrome management the paris way, a step forward on a long road ∗. J. Am. Coll. Cardiol. 73, 1958–1960. doi: 10.1016/j.jacc.2019.02.025
Bauer, A., Bateman, J. F., Lamandé, S. R., Hanssen, E., Kirejczyk, S. G. M., Yee, M., et al. (2019a). Identification of two independent COL5A1 variants in dogs with Ehlers–Danlos syndrome. Genes Basel 10:731. doi: 10.3390/genes10100731
Bauer, A., de Lucia, M., Leuthard, F., Jagannathan, V., and Leeb, T. (2019b). Compound heterozygosity for TNXB genetic variants in a mixed-breed dog with Ehlers-Danlos syndrome. Anim. Genet. 50, 546–549. doi: 10.1111/age.12830
Beighton, P., Paepe, A. D., Steinmann, B., Tsipouras, P., and Wenstrup, R. J. (1998). Ehlers-Danlos syndromes: revised nosology, villefranche, 1997. Am. J. Med. Genet. 77, 31–37. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o
Bian, S., Akyüz, N., Bernreuther, C., Loers, G., Laczynska, E., Jakovcevski, I., et al. (2011). Dermatan sulfotransferase Chst14/D4st1, but not chondroitin sulfotransferase Chst11/C4st1, regulates proliferation and neurogenesis of neural progenitor cells. J. Cell Sci. 124, 4051–4063. doi: 10.1242/jcs.088120
Bin, B.-H., Fukada, T., Hosaka, T., Yamasaki, S., Ohashi, W., Hojyo, S., et al. (2011). Biochemical characterization of human ZIP13 protein: a homo-dimerized zinc transporter involved in the spondylocheiro dysplastic Ehlers-Danlos syndrome. J. Biol. Chem. 286, 40255–40265. doi: 10.1074/jbc.m111.256784
Birk, D. E. (2001). Type V collagen: heterotypic type I/V collagen interactions in the regulation of fibril assembly. Micron 32, 223–237. doi: 10.1016/s0968-4328(00)00043-3
Birk, D. E., and Brückner, P. (2010). The Extracellular Matrix: an Overview. Berlin: Springer, 77–115. doi: 10.1007/978-3-642-16555-9_3
Bishop, J. R., Schuksz, M., and Esko, J. D. (2007). Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037. doi: 10.1038/nature05817
Blackburn, P. R., Xu, Z., Tumelty, K. E., Zhao, R. W., Monis, W. J., Harris, K. G., et al. (2018). Bi-allelic alterations in AEBP1 lead to defective collagen assembly and connective tissue structure resulting in a variant of Ehlers-Danlos syndrome. Am. J. Hum. Genet. 102, 696–705. doi: 10.1016/j.ajhg.2018.02.018
Bowen, C. J., Giadrosic, J. F. C., Burger, Z., Rykiel, G., Davis, E. C., Helmers, M. R., et al. (2019). Targetable cellular signaling events mediate vascular pathology in vascular Ehlers-Danlos syndrome. J. Clin. Invest. 130, 686–698. doi: 10.1172/jci130730
Bowen, J. M., Sobey, G. J., Burrows, N. P., Colombi, M., Lavallee, M. E., Malfait, F., et al. (2017). Ehlers–Danlos syndrome, classical type. Am. J. Med. Genet. Part C Semin. Med. Genet. 175, 27–39. doi: 10.1002/ajmg.c.31548
Brady, A. F., Demirdas, S., Fournel-Gigleux, S., Ghali, N., Giunta, C., Kapferer-Seebacher, I., et al. (2017). The Ehlers–Danlos syndromes, rare types. Am. J. Med. Genet. Part C Semin. Med. Genet. 175, 70–115. doi: 10.1002/ajmg.c.31550
Briest, W., and Talan, M. I. (2011). Genes and Cardiovascular Function. New York, NY: Springer, 241–251. doi: 10.1007/978-1-4419-7207-1_23
Briest, W., Cooper, T. K., Tae, H.-J., Krawczyk, M., McDonnell, N. B., and Talan, M. I. (2011). Doxycycline ameliorates the susceptibility to aortic lesions in a mouse model for the vascular type of Ehlers-Danlos syndrome. J. Pharmacol. Exp. Ther. 337, 621–627. doi: 10.1124/jpet.110.177782
Brown, P. J., Young, R. D., and Cripps, P. J. (1993). Abnormalities of collagen fibrils in a rabbit with a connective tissue defect similar to Ehlers-Danlos syndrome. Res. Vet. Sci. 55, 346–350. doi: 10.1016/0034-5288(93)90105-o
Byers, P. H., Belmont, J., Black, J., Backer, J. D., Frank, M., Jeunemaitre, X., et al. (2017). Diagnosis, natural history, and management in vascular Ehlers–Danlos syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 175, 40–47. doi: 10.1002/ajmg.c.31553
Canty, E. G., and Kadler, K. E. (2005). Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 118, 1341–1353. doi: 10.1242/jcs.01731
Chakravarti, S., Magnuson, T., Lass, J. H., Jepsen, K. J., LaMantia, C., and Carroll, H. (1998). Lumican regulates collagen fibril assembly: skin fragility and corneal opacity in the absence of lumican. J. Cell Biol. 141, 1277–1286. doi: 10.1083/jcb.141.5.1277
Chanut-Delalande, H., Bonod-Bidaud, C., Cogne, S., Malbouyres, M., Ramirez, F., Fichard, A., et al. (2004). Development of a functional skin matrix requires deposition of collagen V heterotrimers. Mol. Cell Biol. 24, 6049–6057. doi: 10.1128/mcb.24.13.6049-6057.2004
Chiquet, M., Birk, D. E., Bönnemann, C. G., and Koch, M. (2014). Collagen XII: protecting bone and muscle integrity by organizing collagen fibrils. Int. J. Biochem. Cell Biol. 53, 51–54. doi: 10.1016/j.biocel.2014.04.020
Colige, A., Nuytinck, L., Hausser, I., van Essen, A. J., Thiry, M., Herens, C., et al. (2004). Novel types of mutation responsible for the dermatosparactic type of Ehlers–Danlos syndrome (Type VIIC) and common polymorphisms in the ADAMTS2 gene. J. Invest. Dermatol. 123, 656–663. doi: 10.1111/j.0022-202x.2004.23406.x
Colige, A., Ruggiero, F., Vandenberghe, I., Dubail, J., Kesteloot, F., Beeumen, J. V., et al. (2005). Domains and maturation processes that regulate the activity of ADAMTS-2, a metalloproteinase cleaving the aminopropeptide of fibrillar procollagens types I–III and V∗. J. Biol. Chem. 280, 34397–34408. doi: 10.1074/jbc.m506458200
Colige, A., Sieron, A. L., Li, S.-W., Schwarze, U., Petty, E., Wertelecki, W., et al. (1999). Human Ehlers-Danlos syndrome type Vii C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am. J. Hum. Genet. 65, 308–317. doi: 10.1086/302504
Colman, M., Syx, D., Wandele, I. D., Dhooge, T., Symoens, S., and Malfait, F. (2021). Clinical and molecular characteristics of 168 probands and 65 relatives with a clinical presentation of classical Ehlers-Danlos syndrome. Matrix Biol. 42, 1294–1306. doi: 10.1002/humu.24258
Cooper, T. K., Zhong, Q., Krawczyk, M., Tae, H.-J., Müller, G. A., Schubert, R., et al. (2010). The haploinsufficient Col3a1 mouse as a model for vascular Ehlers-Danlos syndrome. Vet. Pathol. 47, 1028–1039. doi: 10.1177/0300985810374842
Couchman, J. R., and Pataki, C. A. (2012). An introduction to proteoglycans and their localization. J. Histochem. Cytochem. 60, 885–897. doi: 10.1369/0022155412464638
Counts, D. F., Knighten, P., and Hegreberg, G. (1977). Biochemical changes in the skin of mink with Ehlers-Danlos syndrome: increased collagen biosynthesis in the dermis of affected mink. J. Invest. Dermatol. 69, 521–526. doi: 10.1111/1523-1747.ep12687965
D’hondt, S., Guillemyn, B., Syx, D., Symoens, S., Rycke, R. D., Vanhoutte, L., et al. (2018). Type III collagen affects dermal and vascular collagen fibrillogenesis and tissue integrity in a mutant Col3a1 transgenic mouse model. Matrix Biol. 70, 72–83. doi: 10.1016/j.matbio.2018.03.008
Dalgleish, R. (1998). The human collagen mutation database 1998. Nucleic Acids Res. 26, 253–255. doi: 10.1093/nar/26.1.253
Danielson, K. G., Baribault, H., Holmes, D. F., Graham, H., Kadler, K. E., and Iozzo, R. V. (1997). Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J. Cell Biol. 136, 729–743. doi: 10.1083/jcb.136.3.729
Delbaere, S., Clercq, A. D., Mizumoto, S., Noborn, F., Bek, J. W., Alluyn, L., et al. (2020). b3galt6 knock-out zebrafish recapitulate β3GalT6-deficiency disorders in human and reveal a trisaccharide proteoglycan linkage region. Front. Cell Dev. Biol. 8:597857. doi: 10.3389/fcell.2020.597857
Delbaere, S., Damme, T. V., Syx, D., Symoens, S., Coucke, P., Willaert, A., et al. (2019a). Hypomorphic zebrafish models mimic the musculoskeletal phenotype of β4GalT7-deficient Ehlers-Danlos syndrome. Matrix Biol. 89, 59–75. doi: 10.1016/j.matbio.2019.12.002
Delbaere, S., Dhooge, T., Syx, D., Petit, F., Goemans, N., Destrée, A., et al. (2019b). Novel defects in collagen XII and VI expand the mixed myopathy/Ehlers-Danlos syndrome spectrum and lead to variant-specific alterations in the extracellular matrix. Genet. Med. Official J. Am. Coll. Med. Genet. 22, 112–123. doi: 10.1038/s41436-019-0599-6
DeNigris, J., Yao, Q., Birk, E. K., and Birk, D. E. (2015). Altered dermal fibroblast behavior in a collagen V haploinsufficient murine model of classic Ehlers–Danlos syndrome. Connect. Tissue Res. 57, 1–9. doi: 10.3109/03008207.2015.1081901
Ding, H., Clouthier, D. E., and Artinger, K. B. (2013). Redundant roles of PRDM family members in zebrafish craniofacial development. Dev. Dynam. 242, 67–79. doi: 10.1002/dvdy.23895
Dubacher, N., Münger, J., Gorosabel, M. C., Crabb, J., Ksiazek, A. A., Caspar, S. M., et al. (2019). Celiprolol but not losartan improves the biomechanical integrity of the aorta in a mouse model of vascular Ehlers–Danlos syndrome. Cardiovasc. Res. 116, 457–465. doi: 10.1093/cvr/cvz095
Egging, D. F., van Vlijmen, I., Starcher, B., Gijsen, Y., Zweers, M. C., Blankevoort, L., et al. (2006). Dermal connective tissue development in mice: an essential role for tenascin-X. Cell Tissue Res. 323, 465–474. doi: 10.1007/s00441-005-0100-5
Egging, D. F., van Vlijmen-Willems, I., Choi, J., Peeters, A. C. T. M., Rens, D., van, et al. (2008). Analysis of obstetric complications and uterine connective tissue in tenascin-X-deficient humans and mice. Cell Tissue Res. 332, 523–532. doi: 10.1007/s00441-008-0591-y
Egging, D., van Vlijmen-Willems, I., Tongeren, T., van, Schalkwijk, J., and Peeters, A. (2007). Wound healing in tenascin-X deficient mice suggests that tenascin-X is involved in matrix maturation rather than matrix deposition. Connect. Tissue Res. 48, 93–98. doi: 10.1080/03008200601166160
Epstein, E. H., and Munderloh, N. H. (1975). Isolation and characterization of CNBr peptides of human (alpha 1 (III))3 collagen and tissue distribution of (alpha 1 (I))2 alpha 2 and (alpha 1 (III))3 collagens. J. Biol. Chem. 250, 9304–9312. doi: 10.1016/s0021-9258(19)40644-3
Faugeroux, J., Nematalla, H., Li, W., Clement, M., Robidel, E., Frank, M., et al. (2013). Angiotensin II promotes thoracic aortic dissections and ruptures in Col3a1 haploinsufficient mice. Hypertens. Dallas Tex 1979, 203–208. doi: 10.1161/hypertensionaha.111.00974
Fjølstad, M., and Helle, O. (1974). A hereditary dysplasia of collagen tissues in sheep. J. Pathol. 112, 183–188. doi: 10.1002/path.1711120309
Fontaine, E., Faugeroux, J., Beugnon, C., Verpont, M.-C., Nematalla, H., Bruneval, P., et al. (2015). Caractérisation d’un modèle murin du Syndrome d’Ehlers-Danlos vasculaire. J. Mal. Vascul. 40:119. doi: 10.1016/j.jmv.2014.12.024
Fukada, T., Civic, N., Furuichi, T., Shimoda, S., Mishima, K., Higashiyama, H., et al. (2008). The Zinc Transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-β signaling pathways. PLoS One 3:e3642. doi: 10.1371/journal.pone.0003642
Fukunaka, A., Fukada, T., Bhin, J., Suzuki, L., Tsuzuki, T., Takamine, Y., et al. (2017). Zinc transporter ZIP13 suppresses beige adipocyte biogenesis and energy expenditure by regulating C/EBP-β expression. PLoS Genet. 13:e1006950. doi: 10.1371/journal.pgen.1006950
Galli, G. G., de Lichtenberg, K. H., Carrara, M., Hans, W., Wuelling, M., Mentz, B., et al. (2012). Prdm5 regulates collagen gene transcription by association with RNA polymerase II in developing bone. PLoS Genet. 8:e1002711. doi: 10.1371/journal.pgen.1002711
Gistelinck, C., Kwon, R. Y., Malfait, F., Symoens, S., Harris, M. P., Henke, K., et al. (2018). Zebrafish type I collagen mutants faithfully recapitulate human type I collagenopathies. Proc. Natl. Acad. Sci. U.S.A. 115, 201722200. doi: 10.1073/pnas.1722200115
Giunta, C., Elçioglu, N. H., Albrecht, B., Eich, G., Chambaz, C., Janecke, A. R., et al. (2008). Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome—an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am. J. Hum. Genet. 82, 1290–1305. doi: 10.1016/j.ajhg.2008.05.001
Goff, C. L., Somerville, R. P. T., Kesteloot, F., Powell, K., Birk, D. E., Colige, A. C., et al. (2006). Regulation of procollagen amino-propeptide processing during mouse embryogenesis by specialization of homologous ADAMTS proteases: insights on collagen biosynthesis and dermatosparaxis. Development 133, 1587–1596. doi: 10.1242/dev.02308
Gorosabel, M. C., Dubacher, N., Meienberg, J., and Matyas, G. (2019). Vascular Ehlers-Danlos syndrome: Can the beneficial effect of celiprolol be extrapolated to bisoprolol? Eur. Hear J. Cardiovasc. Pharmacother. 6, 199–200. doi: 10.1093/ehjcvp/pvz067
Goudot, G., Papadacci, C., Dizier, B., Baudrie, V., Ferreira, I., Boisson-Vidal, C., et al. (2018). Arterial stiffening with ultrafast ultrasound imaging gives new insight into arterial phenotype of vascular Ehlers-Danlos mouse models. Ultraschall. Med. Eur. J. Ultrasound 40, 734–742. doi: 10.1055/a-0599-0841
Green, C., Ghali, N., Akilapa, R., Angwin, C., Baker, D., Bartlett, M., et al. (2020). Classical-like Ehlers–Danlos syndrome: a clinical description of 20 newly identified individuals with evidence of tissue fragility. Genet. Med. 22, 1576–1582. doi: 10.1038/s41436-020-0850-1
Greenspan, D. S. (2005). Collagen, Primer in Structure, Processing and Assembly. Berlin: Springer, 149–183. doi: 10.1007/b103822
Gustafsson, R., Stachtea, X., Maccarana, M., Grottling, E., Eklund, E., Malmström, A., et al. (2014). Dermatan sulfate epimerase 1 deficient mice as a model for human abdominal wall defects. Birth Defects Res. Part Clin. Mol. Teratol. 100, 712–720. doi: 10.1002/bdra.23300
Halper, J. (2013). Progress in Heritable Soft Connective Tissue Diseases. Dordrecht: Springer, 231–240. doi: 10.1007/978-94-007-7893-1_14
Hansen, N., Foster, S. F., Burrows, A. K., Mackie, J., and Malik, R. (2015). Cutaneous asthenia (Ehlers–Danlos-like syndrome) of burmese cats. J. Feline Med. Surg. 17, 954–963. doi: 10.1177/1098612x15610683
Hanset, R., and Lapiere, C. M. (1974). Inheritance of dermatosparaxis in the Calf. J. Hered. 65, 356–358. doi: 10.1093/oxfordjournals.jhered.a108549
Hashimoto, K., Kajitani, N., Miyamoto, Y., and Matsumoto, K. (2018). Wound healing-related properties detected in an experimental model with a collagen gel contraction assay are affected in the absence of tenascin-X. Exp. Cell Res. 363, 102–113. doi: 10.1016/j.yexcr.2017.12.025
Hausser, I., and Anton-Lamprecht, I. (1994). Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I–IV as a means of diagnostics and classification. Hum. Genet. 93, 394–407. doi: 10.1007/bf00201664
Hegreberg, G. A. (1975). Animal model of human disease: Ehlers-Danlos syndrome. Am. J. Pathol. 79, 383–386.
Hicks, D., Farsani, G. T., Laval, S., Collins, J., Sarkozy, A., Martoni, E., et al. (2014). Mutations in the collagen XII gene define a new form of extracellular matrix-related myopathy. Hum. Mol. Genet. 23, 2353–2363. doi: 10.1093/hmg/ddt637
Hirose, T., Mizumoto, S., Hashimoto, A., Takahashi, Y., Yoshizawa, T., Nitahara-Kasahara, Y., et al. (2020). Systematic investigation of the skin in Chst14-/- mice: a model for skin fragility in musculocontractural Ehlers–Danlos syndrome caused by CHST14 variants (mcEDS-CHST14). Glycobiology 31, 137–150. doi: 10.1093/glycob/cwaa058
Hirose, T., Ogura, T., Tanaka, K., Minaguchi, J., Yamauchi, T., Fukada, T., et al. (2015). Comparative study of dermal components and plasma TGF-β1 levels in Slc39a13/Zip13-KO mice. J. Vet. Med. Sci. 77, 1385–1389. doi: 10.1292/jvms.15-0015
Hirose, T., Shimazaki, T., Takahashi, N., Fukada, T., Watanabe, T., Tangkawattana, P., et al. (2019a). Morphometric analysis of thoracic aorta in Slc39a13/Zip13-KO mice. Cell Tissue Res. 376, 137–141. doi: 10.1007/s00441-018-2977-9
Hirose, T., Suzuki, I., Takahashi, N., Fukada, T., Tangkawattana, P., and Takehana, K. (2018). Morphometric analysis of cornea in the Slc39a13/Zip13-knockout mice. J. Vet. Med. Sci. 80, 18–0019. doi: 10.1292/jvms.18-0019
Hirose, T., Takahashi, N., Tangkawattana, P., Minaguchi, J., Mizumoto, S., Yamada, S., et al. (2019b). Structural alteration of glycosaminoglycan side chains and spatial disorganization of collagen networks in the skin of patients with mcEDS-CHST14. Biochim. Biophys. Acta BBA Gen. Subj. 1863, 623–631. doi: 10.1016/j.bbagen.2018.12.006
Holbrook, K. A., Byers, P. H., Counts, D. F., and Hegreberg, G. A. (1980). Dermatosparaxis in a himalayan cat: II. ultrastructural studies of dermal collagen. J. Invest. Dermatol. 74, 100–104. doi: 10.1111/1523-1747.ep12520000
Horn, D., Siebert, E., Seidel, U., Rost, I., Mayer, K., Jamra, R. A., et al. (2017). Biallelic COL3A1 mutations result in a clinical spectrum of specific structural brain anomalies and connective tissue abnormalities. Am. J. Med. Genet. A 173, 2534–2538. doi: 10.1002/ajmg.a.38345
Huijing, P. A., Voermans, N. C., Baan, G. C., Busé, T. E., van Engelen, B. G. M., Haan, A., et al. (2010). Muscle characteristics and altered myofascial force transmission in tenascin-X-deficient mice, a mouse model of Ehlers-Danlos syndrome. J. Appl. Physiol. 109, 986–995. doi: 10.1152/japplphysiol.00723.2009
Huxley, V. H. (2007). Sex and the cardiovascular system: the intriguing tale of how women and men regulate cardiovascular function differently. Adv. Physiol. Educ. 31, 17–22. doi: 10.1152/advan.00099.2006
Izu, Y., Adams, S. M., Connizzo, B. K., Beason, D. P., Soslowsky, L. J., Koch, M., et al. (2020). Collagen XII mediated cellular and extracellular mechanisms regulate establishment of tendon structure and function. Matrix Biol. 95, 52–67. doi: 10.1016/j.matbio.2020.10.004
Izu, Y., Ezura, Y., Koch, M., Birk, D. E., and Noda, M. (2016). Collagens VI and XII form complexes mediating osteoblast interactions during osteogenesis. Cell Tissue Res. 364, 623–635. doi: 10.1007/s00441-015-2345-y
Izu, Y., Sun, M., Zwolanek, D., Veit, G., Williams, V., Cha, B., et al. (2011). Type XII collagen regulates osteoblast polarity and communication during bone formation. J. Cell Biol. 193, 1115–1130. doi: 10.1083/jcb.201010010
Jacinto, J. G. P., Häfliger, I. M., Veiga, I. M. B., Letko, A., Benazzi, C., Bolcato, M., et al. (2020). A heterozygous missense variant in the COL5A2 in holstein cattle resembling the classical Ehlers–Danlos syndrome. Animals 10:2002. doi: 10.3390/ani10112002
Jaffey, J. A., Bullock, G., Teplin, E., Guo, J., Villani, N. A., Mhlanga-Mutangadura, T., et al. (2019). A homozygous ADAMTS2 nonsense mutation in a Doberman Pinscher dog with Ehlers Danlos syndrome and extreme skin fragility. Anim. Genet. 50, 543–545. doi: 10.1111/age.12825
Jansen, L. H. (1955). The structure of the connective tissue, an explanation of the symptoms of the Ehlers-Danlos syndrome. Dermatology 110, 108–120. doi: 10.1159/000256442
Jeong, S.-J., Li, S., Luo, R., Strokes, N., and Piao, X. (2012). Loss of Col3a1, the gene for Ehlers-Danlos syndrome type IV, results in neocortical dyslamination. PLoS One 7:e29767. doi: 10.1371/journal.pone.0029767
Johnston, J. M., Connizzo, B. K., Shetye, S. S., Robinson, K. A., Huegel, J., Rodriguez, A. B., et al. (2017). Collagen V haploinsufficiency in a murine model of classic Ehlers–Danlos syndrome is associated with deficient structural and mechanical healing in tendons. J. Orthopaed. Res. 35, 2707–2715. doi: 10.1002/jor.23571
Joller, S., Veiga, I. B., and Drögemüller, C. (2017). Dermatosparaxis in white dorper sheep: confirmation of a causative nonsense mutation in ADAMTS2. Anim. Genet. 48, 729–730. doi: 10.1111/age.12591
Jørgensen, A., Fagerheim, T., Rand-Hendriksen, S., Lunde, P. I., Vorren, T. O., Pepin, M. G., et al. (2014). Vascular Ehlers-Danlos syndrome in siblings with biallelic COL3A1 sequence variants and marked clinical variability in the extended family. Eur. J. Hum. Genet. EJHG 23, 796–802. doi: 10.1038/ejhg.2014.181
Kadler, K. E., Hill, A., and Canty-Laird, E. G. (2008). Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell. Biol. 20, 495–501. doi: 10.1016/j.ceb.2008.06.008
Kajitani, N., Yamada, T., Kawakami, K., and Matsumoto, K. (2019). TNX deficiency results in bone loss due to an increase in multinucleated osteoclasts. Biochem. Bioph. Res. Commun. 512, 659–664. doi: 10.1016/j.bbrc.2019.03.134
Kawakami, K., and Matsumoto, K. (2011). Behavioral alterations in mice lacking the gene for tenascin-X. Biol. Pharm. Bull. 34, 590–593. doi: 10.1248/bpb.34.590
Kimmel, C. B. (1989). Genetics and early development of zebrafish. Trends Genet. 5, 283–288. doi: 10.1016/0168-9525(89)90103-0
Krane, S. M., Pinnell, S. R., and Erbe, R. W. (1972). Lysyl-protocollagen hydroxylase deficiency in fibroblasts from siblings with hydroxylysine-deficient collagen. Proc. Natl. Acad. Sci. U.S.A. 69, 2899–2903. doi: 10.1073/pnas.69.10.2899
Kumps, C., Campos-Xavier, B., Hilhorst-Hofstee, Y., Marcelis, C., Kraenzlin, M., Fleischer, N., et al. (2020). The connective tissue disorder associated with recessive variants in the SLC39A13 zinc transporter gene (spondylo-dysplastic Ehlers–Danlos syndrome type 3): insights from four novel patients and follow-up on two original cases. Genes Basel 11:420. doi: 10.3390/genes11040420
Lang, T. F. (2011). The bone-muscle relationship in men and women. J. Osteoporos. 2011:702735. doi: 10.4061/2011/702735
Lapière, C. M., Lenaers, A., and Kohn, L. D. (1971). procollagen peptidase: an enzyme excising the coordination peptides of procollagen. Proc. Natl. Acad. Sci. U.S.A. 68, 3054–3058. doi: 10.1073/pnas.68.12.3054
Lautrup, C. K., Teik, K. W., Unzaki, A., Mizumoto, S., Syx, D., Sin, H. H., et al. (2020). Delineation of musculocontractural Ehlers–Danlos Syndrome caused by dermatan sulfate epimerase deficiency. Mol. Genet. Genom. Med. 8:e1197. doi: 10.1002/mgg3.1197
Layne, M. D., Yet, S.-F., Maemura, K., Hsieh, C.-M., Bernfield, M., Perrella, M. A., et al. (2001). Impaired abdominal wall development and deficient wound healing in mice lacking aortic carboxypeptidase-like protein. Mol. Cell Biol. 21, 5256–5261. doi: 10.1128/mcb.21.15.5256-5261.2001
Leegwater, P. A., Vos-Loohuis, M., Ducro, B. J., Boegheim, I. J., van Steenbeek, F. G., Nijman, I. J., et al. (2016). Dwarfism with joint laxity in Friesian horses is associated with a splice site mutation in B4GALT7. BMC Genomics 17:839. doi: 10.1186/s12864-016-3186-0
Li, Q., Wu, X., Na, X., Ge, B., Wu, Q., Guo, X., et al. (2019). Impaired cognitive function and altered hippocampal synaptic plasticity in mice lacking dermatan sulfotransferase Chst14/D4st1. Front. Mol. Neurosci. 12:26. doi: 10.3389/fnmol.2019.00026
Li, S.-W., Arita, M., Fertala, A., Bao, Y., Kopen, G. C., Långsjö, T. K., et al. (2001). Transgenic mice with inactive alleles for procollagen N-proteinase (ADAMTS-2) develop fragile skin and male sterility. Biochem. J. 355:271. doi: 10.1042/0264-6021:3550271
Lieschke, G. J., and Currie, P. D. (2007). Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 8, 353–367. doi: 10.1038/nrg2091
Lincoln, J., Florer, J. B., Deutsch, G. H., Wenstrup, R. J., and Yutzey, K. E. (2006). ColVa1 and ColXIa1 are required for myocardial morphogenesis and heart valve development. Dev. Dyn. 235, 3295–3305. doi: 10.1002/dvdy.20980
Liu, X., Wu, H., Byrne, M., Krane, S., and Jaenisch, R. (1997). Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc. Natl. Acad. Sci. U.S.A. 94, 1852–1856. doi: 10.1073/pnas.94.5.1852
Maccarana, M., Kalamajski, S., Kongsgaard, M., Magnusson, S. P., Oldberg, A., and Malmström, A. (2009). Dermatan sulfate epimerase 1-deficient mice have reduced content and changed distribution of iduronic acids in dermatan sulfate and an altered collagen structure in skin. Mol. Cell. Biol. 29, 5517–5528. doi: 10.1128/mcb.00430-09
Malfait, F. (2018). Vascular aspects of the Ehlers-Danlos syndromes. Matrix Biol. 71–72, 380–395. doi: 10.1016/j.matbio.2018.04.013
Malfait, F., Castori, M., Francomano, C. A., Giunta, C., Kosho, T., and Byers, P. H. (2020). The Ehlers–Danlos syndromes. Nat. Rev. Dis. Primers 6:64. doi: 10.1038/s41572-020-0194-9
Malfait, F., Coucke, P., Symoens, S., Loeys, B., Nuytinck, L., and Paepe, A. D. (2005). The molecular basis of classic Ehlers-Danlos syndrome: a comprehensive study of biochemical and molecular findings in 48 unrelated patients. Hum. Mutat. 25, 28–37. doi: 10.1002/humu.20107
Malfait, F., Francomano, C., Byers, P., Belmont, J., Berglund, B., Black, J., et al. (2017). The 2017 international classification of the Ehlers–Danlos syndromes. Am. J. Med. Genet. Part C Sem. Med. Genet. 175, 8–26. doi: 10.1002/ajmg.c.31552
Malfait, F., Symoens, S., Coucke, P., Nunes, L., Almeida, S. D., and Paepe, A. D. (2006). Total absence of the α2(I) chain of collagen type I causes a rare form of Ehlers-Danlos syndrome with hypermobility and propensity to cardiac valvular problems. J. Med. Genet. 43, e36. doi: 10.1136/jmg.2005.038224
Mao, J. R., Taylor, G., Dean, W. B., Wagner, D. R., Afzal, V., Lotz, J. C., et al. (2002). Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat. Genet. 30, 421–425. doi: 10.1038/ng850
Martin, K., Brooks, S., Vierra, M., Lafayette, W. T., McClure, S., Carpenter, M., et al. (2020). Fragile foal syndrome (PLOD1 c.2032G>A) occurs across diverse horse populations. Anim. Genet. 52, 137–138. doi: 10.1111/age.13020
Matsumoto, K., Sato, T., Oka, S., Inokuchi, J., and Ariga, H. (2004). Comparison of the compositions of phospholipid-associated fatty acids in wild-type and extracellular matrix tenascin-X-deficient mice. Biol. Pharm. Bull. 27, 1447–1450. doi: 10.1248/bpb.27.1447
Matsumoto, K., Sawa, H., Sato, M., Orba, Y., Nagashima, K., and Ariga, H. (2002). Distribution of extracellular matrix tenascin-X in sciatic nerves. Acta Neuropathol. 104, 448–454. doi: 10.1007/s00401-002-0577-x
Matsumoto, K., Takayama, N., Ohnishi, J., Ohnishi, E., Shirayoshi, Y., Nakatsuji, N., et al. (2001). Tumour invasion and metastasis are promoted in mice deficient in tenascin-X. Genes Cells 6, 1101–1111. doi: 10.1046/j.1365-2443.2001.00482.x
Meani, N., Pezzimenti, F., Deflorian, G., Mione, M., and Alcalay, M. (2009). The tumor suppressor PRDM5 regulates Wnt signaling at early stages of zebrafish development. PLoS One 4:e4273. doi: 10.1371/journal.pone.0004273
Miedel, E. L., Brisson, B. K., Hamilton, T., Gleason, H., Swain, G. P., Lopas, L., et al. (2015). Type III collagen modulates fracture callus bone formation and early remodeling. J. Orthopaed. Res. 33, 675–684. doi: 10.1002/jor.22838
Minamitani, T., Ariga, H., and Matsumoto, K. (2004a). Deficiency of tenascin-X causes a decrease in the level of expression of type VI collagen. Exp. Cell Res. 297, 49–60. doi: 10.1016/j.yexcr.2004.03.002
Minamitani, T., Ikuta, T., Saito, Y., Takebe, G., Sato, M., Sawa, H., et al. (2004b). Modulation of collagen fibrillogenesis by tenascin-X and type VI collagen. Exp. Cell Res. 298, 305–315. doi: 10.1016/j.yexcr.2004.04.030
Mizuno, K., Boudko, S., Engel, J., and Bächinger, H. P. (2013). Vascular Ehlers–Danlos syndrome mutations in type III collagen differently stall the triple helical folding. J. Biol. Chem. 288, 19166–19176. doi: 10.1074/jbc.m113.462002
Montagutelli, X. (2000). Effect of the genetic background on the phenotype of mouse mutations. J. Am. Soc. Nephrol. 11, S101–S105. doi: 10.1681/asn.v11suppl_2s101
Monteagudo, L. V., Ferrer, L. M., Catalan-Insa, E., Savva, D., McGuffin, L. J., and Tejedor, M. T. (2015). In silico identification and three-dimensional modelling of the missense mutation in ADAMTS2 in a sheep flock with dermatosparaxis. Vet. Dermatol. 26, e15–e16. doi: 10.1111/vde.12178
Monthoux, C., de Brot, S., Jackson, M., Bleul, U., and Walter, J. (2015). Skin malformations in a neonatal foal tested homozygous positive for warmblood fragile foal syndrome. BMC Vet. Res. 11:12. doi: 10.1186/s12917-015-0318-8
Müller, T., Mizumoto, S., Suresh, I., Komatsu, Y., Vodopiutz, J., Dundar, M., et al. (2013). Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers–Danlos syndrome. Hum. Mol. Genet. 22, 3761–3772. doi: 10.1093/hmg/ddt227
Murray, M. L., Pepin, M., Peterson, S., and Byers, P. H. (2014). Pregnancy-related deaths and complications in women with vascular Ehlers–Danlos syndrome. Genet. Med. 16, 874–880. doi: 10.1038/gim.2014.53
Myllyharju, J., and Kivirikko, K. I. (2004). Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20, 33–43. doi: 10.1016/j.tig.2003.11.004
Nielsen, R. H., Couppé, C., Jensen, J. K., Olsen, M. R., Heinemeier, K. M., Malfait, F., et al. (2014). Low tendon stiffness and abnormal ultrastructure distinguish classic Ehlers-Danlos syndrome from benign joint hypermobility syndrome in patients. FASEB J. 28, 4668–4676. doi: 10.1096/fj.14-249656
Okechukwu, I. B. (2018). Experimental Animal Models of Human Diseases - An Effective Therapeutic Strategy. London: Intech Open. doi: 10.5772/intechopen.70745
Okuda-Ashitaka, E., Kakuchi, Y., Kakumoto, H., Yamanishi, S., Kamada, H., Yoshidu, T., et al. (2020). Mechanical allodynia in mice with tenascin-X deficiency associated with Ehlers-Danlos syndrome. Sci. Rep.Uk 10:6569. doi: 10.1038/s41598-020-63499-2
Ong, K.-T., Perdu, J., Backer, J. D., Bozec, E., Collignon, P., Emmerich, J., et al. (2010). Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial. Lancet 376, 1476–1484. doi: 10.1016/s0140-6736(10)60960-9
Park, A. C., Phan, N., Massoudi, D., Liu, Z., Kernien, J. F., Adams, S. M., et al. (2017). Deficits in Col5a2 expression result in novel skin and adipose abnormalities and predisposition to aortic aneurysms and dissections. Am. J. Pathol. 187, 2300–2311. doi: 10.1016/j.ajpath.2017.06.006
Park, A. C., Phillips, C. L., Pfeiffer, F. M., Roenneburg, D. A., Kernien, J. F., Adams, S. M., et al. (2015). Homozygosity and heterozygosity for null Col5a2 alleles produce embryonic lethality and a novel classic Ehlers-Danlos syndrome–related phenotype. Am. J. Pathol. 185, 2000–2011. doi: 10.1016/j.ajpath.2015.03.022
Pepin, M. G., Schwarze, U., Rice, K. M., Liu, M., Leistritz, D., and Byers, P. H. (2014). Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet. Med. Official J. Am. Coll. Med. Genet. 16, 881–888. doi: 10.1038/gim.2014.72
Plancke, A., Holder-Espinasse, M., Rigau, V., Manouvrier, S., Claustres, M., and Kien, P. K. V. (2009). Homozygosity for a null allele of COL3A1 results in recessive Ehlers–Danlos syndrome. Eur. J. Hum. Genet. 17, 1411–1416. doi: 10.1038/ejhg.2009.76
Porto, E. M., Komor, A. C., Slaymaker, I. M., and Yeo, G. W. (2020). Base editing: advances and therapeutic opportunities. Nat. Rev. Drug. Discov. 19, 839–859. doi: 10.1038/s41573-020-0084-6
Proske, S., Hartschuh, W., Enk, A., and Hausser, I. (2006). Ehlers-Danlos syndrome – 20 years experience with diagnosis and classification. JDDG J. Der Deutsche. Dermatol. Gesellschaft 4, 308–318. doi: 10.1111/j.1610-0387.2006.05958.x
Prydz, K., and Dalen, K. T. (2000). Synthesis and sorting of proteoglycans. J. Cell Sci. 113(Pt 2), 193–205. doi: 10.1242/jcs.113.2.193
Rahrovan, S., Fanian, F., Mehryan, P., Humbert, P., and Firooz, A. (2018). Male versus female skin: what dermatologists and cosmeticians should know. Int. J. Women Dermatol. 4, 122–130. doi: 10.1016/j.ijwd.2018.03.002
Rashmir-Raven, A. (2013). Heritable equine regional dermal asthenia. Vet. Clin. North Am. Equine Pract. 29, 689–702. doi: 10.1016/j.cveq.2013.09.001
Ro, H., Zhang, L., Majdalawieh, A., Kim, S., Wu, X., Lyons, P. J., et al. (2007). Adipocyte enhancer-binding protein 1 modulates adiposity and energy homeostasis. Obesity 15, 288–302. doi: 10.1038/oby.2007.569
Rohrbach, M., Vandersteen, A., Yiş, U., Serdaroglu, G., Ataman, E., Chopra, M., et al. (2011). Phenotypic variability of the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA): clinical, molecular and biochemical delineation. Orphanet. J. Rare Dis. 6:46. doi: 10.1186/1750-1172-6-46
Rosen, S., Ham, B., and Mogil, J. S. (2017). Sex differences in neuroimmunity and pain. J. Neurosci. Res. 95, 500–508. doi: 10.1002/jnr.23831
Rost, S., Akyüz, N., Martinovic, T., Huckhagel, T., Jakovcevski, I., and Schachner, M. (2016). Germline ablation of dermatan-4O-sulfotransferase1 reduces regeneration after mouse spinal cord injury. Neuroscience 312, 74–85. doi: 10.1016/j.neuroscience.2015.11.013
Sakai, H., Yokota, S., Kajitani, N., Yoneyama, T., Kawakami, K., Yasui, Y., et al. (2017). A potential contribution of tenascin-X to blood vessel formation in peripheral nerves. Neurosci. Res. 124, 1–7. doi: 10.1016/j.neures.2017.06.003
Salter, C. G., Davies, J. H., Moon, R. J., Fairhurst, J., Bunyan, D., Study, D., et al. (2016). Further defining the phenotypic spectrum of B4GALT7 mutations. Am. J. Med. Genet. A 170, 1556–1563. doi: 10.1002/ajmg.a.37604
Schissel, S. L., Dunsmore, S. E., Liu, X., Shine, R. W., Perrella, M. A., and Layne, M. D. (2009). Aortic carboxypeptidase-like protein is expressed in fibrotic human lung and its absence protects against bleomycin-induced lung fibrosis. Am. J. Pathol. 174, 818–828. doi: 10.2353/ajpath.2009.080856
Segev, F., He’, E., Cole, W. G., Wenstrup, R. J., Young, F., Slomovic, A. R., et al. (2006). Structural abnormalities of the Cornea and lid resulting from collagen V mutations. Invest. Opthalmol. Vis. Sci. 47:565. doi: 10.1167/iovs.05-0771
Shimada, S., Yoshizawa, T., Takahashi, Y., Nitahara-Kasahara, Y., Okada, T., Nomura, Y., et al. (2020). Backcrossing to an appropriate genetic background improves the birth rate of carbohydrate sulfotransferase 14 gene-deleted mice. Exp. Anim. Tokyo 69, 407–413. doi: 10.1538/expanim.19-0150
Smith, L. B., Hadoke, P. W. F., Dyer, E., Denvir, M. A., Brownstein, D., Miller, E., et al. (2011). Haploinsufficiency of the murine Col3a1 locus causes aortic dissection: a novel model of the vascular type of Ehlers–Danlos syndrome. Cardiovasc. Res. 90, 182–190. doi: 10.1093/cvr/cvq356
Sorge, R. E., and Totsch, S. K. (2017). Sex differences in pain. J. Neurosci. Res. 95, 1271–1281. doi: 10.1002/jnr.23841
Spycher, M., Bauer, A., Jagannathan, V., Frizzi, M., Lucia, M. D., and Leeb, T. (2018). A frameshift variant in the COL5A1 gene in a cat with Ehlers-Danlos syndrome. Anim. Genet. 49, 641–644. doi: 10.1111/age.12727
Steinmann, B., Royce, P. M., and Superti-Furga, A. (2003). Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects, 2nd Edn. New York, NY: Wiley-Liss, 431–523. doi: 10.1002/0471221929.ch9
Stevenson, K., Kucich, U., Whitbeck, C., Levin, R. M., and Howard, P. S. (2006). Functional changes in bladder tissue from type III collagen-deficient mice. Mol. Cell Biochem. 283, 107–114. doi: 10.1007/s11010-006-2388-1
Sumioka, T., Iwanishi, H., Okada, Y., Nidegawa, Y., Miyajima, M., Matsumoto, K., et al. (2018). Loss of tenascin X gene function impairs injury-induced stromal angiogenesis in mouse corneas. J. Cell Mol. Med. 22, 948–956. doi: 10.1111/jcmm.13397
Symoens, S., Syx, D., Malfait, F., Callewaert, B., Backer, J. D., Vanakker, O., et al. (2012). Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum. Mutat. 33, 1485–1493. doi: 10.1002/humu.22137
Syx, D., Damme, T., Symoens, S., Maiburg, M. C., Laar, I., Morton, J., et al. (2015). Genetic heterogeneity and clinical variability in musculocontractural Ehlers–Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum. Mutat. 36, 535–547. doi: 10.1002/humu.22774
Syx, D., Miller, R. E., Obeidat, A. M., Tran, P. B., Vroman, R., Malfait, Z., et al. (2020). Pain-related behaviors and abnormal cutaneous innervation in a murine model of classical Ehlers-Danlos syndrome. Pain 161, 2274–2283. doi: 10.1097/j.pain.0000000000001935
Syx, D., Wandele, I. D., Symoens, S., Rycke, R. D., Hougrand, O., Voermans, N., et al. (2019). Bi-allelic AEBP1 mutations in two patients with Ehlers-Danlos syndrome. Hum. Mol. Genet. 28, 1853–1864. doi: 10.1093/hmg/ddz024
Tae, H.-J., Marshall, S., Zhang, J., Wang, M., Briest, W., and Talan, M. I. (2012). Chronic treatment with a broad-spectrum metalloproteinase inhibitor, doxycycline, prevents the development of spontaneous aortic lesions in a mouse model of vascular Ehlers-Danlos syndrome. J. Pharmacol. Exp. Ther. 343, 246–251. doi: 10.1124/jpet.112.197020
Takaluoma, K., Hyry, M., Lantto, J., Sormunen, R., Bank, R. A., Kivirikko, K. I., et al. (2007). Tissue-specific Changes in the hydroxylysine content and cross-links of collagens and alterations in fibril morphology in Lysyl hydroxylase 1 knock-out mice. J. Biol. Chem. 282, 6588–6596. doi: 10.1074/jbc.m608830200
Takeda, U., Utani, A., Wu, J., Shinkai, H., Adachi, E., Koseki, H., et al. (2002). Targeted disruption of dermatopontin causes abnormal collagen fibrillogenesis. J. Invest. Dermatol. 119, 678–683. doi: 10.1046/j.1523-1747.2002.01863.x
Tang, T., Li, L., Tang, J., Li, Y., Lin, W. Y., Martin, F., et al. (2010). A mouse knockout library for secreted and transmembrane proteins. Nat. Biotechnol. 28, 749–755. doi: 10.1038/nbt.1644
Tasheva, E. S., Koester, A., Paulsen, A. Q., Garrett, A. S., Boyle, D. L., Davidson, H. J., et al. (2002). Mimecan/osteoglycin-deficient mice have collagen fibril abnormalities. Mol. Vis. 8, 407–415.
Teratani, T., Tomita, K., Suzuki, T., Furuhashi, H., Irie, R., Nishikawa, M., et al. (2018). Aortic carboxypeptidase–like protein, a WNT ligand, exacerbates nonalcoholic steatohepatitis. J. Clin. Invest. 128, 1581–1596. doi: 10.1172/jci92863
Tryon, R. C., White, S. D., and Bannasch, D. L. (2007). Homozygosity mapping approach identifies a missense mutation in equine cyclophilin B (PPIB) associated with HERDA in the American Quarter Horse. Genomics 90, 93–102. doi: 10.1016/j.ygeno.2007.03.009
Tumelty, K. E., Smith, B. D., Nugent, M. A., and Layne, M. D. (2014). Aortic carboxypeptidase-like protein (ACLP) enhances lung myofibroblast differentiation through transforming growth factor β receptor-dependent and -independent pathways. J. Biol. Chem. 289, 2526–2536. doi: 10.1074/jbc.m113.502617
Valcourt, U., Alcaraz, L. B., Exposito, J.-Y., Lethias, C., and Bartholin, L. (2015). Tenascin-X: beyond the architectural function. Cell Adhes. Migr. 9, 154–165. doi: 10.4161/19336918.2014.994893
van Dijk, F. S., Nesbitt, I. M., Zwikstra, E. H., Nikkels, P. G. J., Piersma, S. R., Fratantoni, S. A., et al. (2009). PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 85, 521–527. doi: 10.1016/j.ajhg.2009.09.001
Vandervore, L., Stouffs, K., Tanyalçin, I., Vanderhasselt, T., Roelens, F., Holder-Espinasse, M., et al. (2017). Bi-allelic variants in COL3A1 encoding the ligand to GPR56 are associated with cobblestone-like cortical malformation, white matter changes and cerebellar cysts. J. Med. Genet. 54:432. doi: 10.1136/jmedgenet-2016-104421
Viglio, S., Zoppi, N., Sangalli, A., Gallanti, A., Barlati, S., Mottes, M., et al. (2008). Rescue of migratory defects of Ehlers–Danlos syndrome fibroblasts in vitro by type V collagen but not insulin-like binding protein-1. J. Invest. Dermatol. 128, 1915–1919. doi: 10.1038/jid.2008.33
Voermans, N. C., van Alfen, N., Pillen, S., Lammens, M., Schalkwijk, J., Zwarts, M. J., et al. (2009). Neuromuscular involvement in various types of Ehlers–Danlos syndrome. Ann. Neurol. 65, 687–697. doi: 10.1002/ana.21643
Voermans, N. C., Verrijp, K., Eshuis, L., Balemans, M. C. M., Egging, D., Sterrenburg, E., et al. (2011). Mild muscular features in tenascin-X knockout mice, a model of Ehlers–Danlos syndrome. Connect. Tissue Res. 52, 422–432. doi: 10.3109/03008207.2010.551616
Vogel, A., Holbrook, K. A., Steinmann, B., Gitzelmann, R., and Byers, P. H. (1979). Abnormal collagen fibril structure in the gravis form (type I) of Ehlers-Danlos syndrome. Lab. Invest. J. Tech. Methods Pathol. 40, 201–206.
Volk, S. W., Shah, S. R., Cohen, A. J., Wang, Y., Brisson, B. K., Vogel, L. K., et al. (2014). Type III collagen regulates osteoblastogenesis and the quantity of trabecular bone. Calcified Tissue Int. 94, 621–631. doi: 10.1007/s00223-014-9843-x
Volk, S. W., Wang, Y., Mauldin, E. A., Liechty, K. W., and Adams, S. L. (2011). Diminished type III collagen promotes myofibroblast differentiation and increases scar deposition in cutaneous wound healing. Cells Tissues Organs 194, 25–37. doi: 10.1159/000322399
Wenstrup, R. J., Florer, J. B., Davidson, J. M., Phillips, C. L., Pfeiffer, B. J., Menezes, D. W., et al. (2006). Murine model of the Ehlers-Danlos Syndrome col5a1 haploinsufficiency disrupts collagen fibril assembly at multiple stages. J. Biol. Chem. 281, 12888–12895. doi: 10.1074/jbc.m511528200
Wenstrup, R. J., Florer, J., Brunskill, E., Bell, S., Chervoneva, I., and Birk, D. (2004). Type V collagen controls the initiation of collagen fibril assembly. J. Biol. Chem. 279, 53331–53337. doi: 10.1074/jbc.m409622200
Wenstrup, R. J., Smith, S. M., Florer, J. B., Zhang, G., Beason, D. P., Seegmiller, R. E., et al. (2011). Regulation of collagen fibril nucleation and initial fibril assembly involves coordinate interactions with collagens V and XI in developing tendon. J. Biol. Chem. 286, 20455–20465. doi: 10.1074/jbc.m111.223693
Xiao, G., Wan, Z., Fan, Q., Tang, X., and Zhou, B. (2014). The metal transporter ZIP13 supplies iron into the secretory pathway in drosophila melanogaster. Elife 3:e03191. doi: 10.7554/elife.03191
Yamakage, Y., Kato, M., Hongo, A., Ogino, H., Ishii, K., Ishizuka, T., et al. (2019). A disintegrin and metalloproteinase with thrombospondin motifs 2 cleaves and inactivates Reelin in the postnatal cerebral cortex and hippocampus, but not in the cerebellum. Mol. Cell Neurosci. 100:103401. doi: 10.1016/j.mcn.2019.103401
Yoshizawa, T., Mizumoto, S., Takahashi, Y., Shimada, S., Sugahara, K., Nakayama, J., et al. (2018). Vascular abnormalities in the placenta of Chst14-/- fetuses: implications in the pathophysiology of perinatal lethality of the murine model and vascular lesions in human CHST14/D4ST1 deficiency. Glycobiology 28, 80–89. doi: 10.1093/glycob/cwx099
Zhang, L., Reidy, S. P., Bogachev, O., Hall, B. K., Majdalawieh, A., and Ro, H.-S. (2011). Lactation defect with impaired secretory activation in AEBP1-Null Mice. PLoS One 6:e27795. doi: 10.1371/journal.pone.0027795
Zhou, H., Hickford, J. G. H., and Fang, Q. (2011). A premature stop codon in the ADAMTS2 gene is likely to be responsible for dermatosparaxis in Dorper sheep. Anim. Genet. 43, 471–473. doi: 10.1111/j.1365-2052.2011.02275.x
Keywords: Ehlers–Danlos syndromes, EDS, animal models, mouse, zebrafish
Citation: Vroman R, Malfait A-M, Miller RE, Malfait F and Syx D (2021) Animal Models of Ehlers–Danlos Syndromes: Phenotype, Pathogenesis, and Translational Potential. Front. Genet. 12:726474. doi: 10.3389/fgene.2021.726474
Received: 16 June 2021; Accepted: 10 August 2021;
Published: 12 October 2021.
Edited by:
Gaurav K. Varshney, Oklahoma Medical Research Foundation, United StatesReviewed by:
Magdalena Sandu, Spitalul Clinic de Copii Doctor Victor Gomoiu, RomaniaCopyright © 2021 Vroman, Malfait, Miller, Malfait and Syx. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Delfien Syx, RGVsZmllbi5TeXhAdWdlbnQuYmU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.