 Harsh Sheth
Harsh Sheth Jhanvi Shah
Jhanvi Shah Aadhira Nair1
Aadhira Nair1- 1FRIGE’s Institute of Human Genetics, Ahmedabad, India
- 2Rainbow Superspeciality Hospital and Children’s Orthopedic Centre, Ahmedabad, India
Biallelic mutations in the CCN6 gene are known to cause a rare genetic disorder—progressive pseudorheumatoid dysplasia (PPD). PPD is characterized by distinct joint deformities of interphalangeal joints, stiffness, gait disturbance, abnormal posture, and absence of inflammation, resulting in significant morbidity. The largest case series of PPD from India suggests c.233G>A and c.1010G>A to be the most common mutations in the CCN6 gene, although the distribution of these variants among endogamous communities in India has not been carried out. We here report three cases of PPD from three independent families belonging to the Patni community of Gujarat, a community known to practice endogamy. All three cases had short stature, gait disturbance, scoliosis, and interphalangeal joint deformities. Analysis by whole-exome sequencing in the first case showed the presence of a previously known, homozygous, missense variant c.298T>A (p.Cys100Ser) in exon 3 of the CCN6 gene in all cases. Due to all three families belonging to the same community, analysis by Sanger sequencing in the remaining two cases for the variant mentioned earlier showed both cases to be of homozygous mutant genotype. Unaffected family members, i.e., parents and siblings, were either heterozygous carriers or wildtype for the said variant. The present case series is the first report of a recurrent variant occurring across multiple PPD-affected individuals from unrelated families belonging to the same community from India.
Introduction
Progressive pseudorheumatoid dysplasia (PPD; OMIM#208230), also known as spondyloepiphyseal tarda with progressive arthropathy or progressive pseudorheumatoid arthropathy of childhood, is a rare autosomal recessive disorder (Dalal et al., 2012). PPD patients are asymptomatic at birth but progressively develop joint deformities, specifically interphalangeal joints, stiffness, joint contractures, gait imbalance, scoliosis, and/or kyphosis, leading to abnormal posture and swelling in hips, knees, wrists, and fingers (Dalal et al., 2012; Bhavani et al., 2015). Short stature (<third centile), absence of inflammation, and pain are considerable phenotypes in these patients. The condition is part of a group of 42 genetic and clinically stratified groups of skeletal disorders (Chen et al., 2016).
PPD occurs due to missense and/or nonsense variants in the CCN6 (Wnt1 lnducible signaling pathway 3) gene located on chromosome 6q22 (Hurvitz et al., 1999). CCN6 protein is a member of the connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family of the extracellular matrix of proteins involved in the regulation of cell migration, adhesion, proliferation, differentiation, and survival (Hurvitz et al., 1999). CCN6 protein function has been reported to be in maintaining cartilage integrity, and abnormal protein function could lead to degradation and loss in articular cartilage (Wang et al., 2013).
The majority of the variants in the CCN6 gene have been reported from the United States, China, and Middle-East countries. More recently, variant data from the largest series of PPD patients from 79 families from India were published, showcasing a series of novel mutations and reclassification of one variant (Dalal et al., 2012; Bhavani et al., 2015). Interestingly, five previously reported variants—c.156C>A, c.233G>A, c.383C>A, c.739_740delTG, and c.1010G>A—were observed in several patients across several families, with c.233G>A and c.1010G>A suggested to be specific to the Indian population (Bhavani et al., 2015).
Herein, we report three PPD cases from three self-reportedly unrelated families belonging to the Patni community of Gujarat. Furthermore, we present a novel observation of a previously known pathogenic variant c.298T>A to be present in all cases from a given community.
Case Description
Clinical Findings
Three probands from three self-reportedly unrelated families belonging to the Patni community of Gujarat in India came for genetic counseling.
Case 1
A 6-year-old male proband, born to a phenotypically normal and endogamous couple (Figure 1A), presented with prominent distal and proximal bilateral metaphysis of fingers, gait disturbance, mild scoliosis, anterior beaking of the chest, square vertebrae, and osteopenia (Figures 2A–F). He had a normal complete blood count and rheumatoid factor of 6.20 IU/ml (normal range: 0–20 IU/ml). Phenotypic analysis of the mother showed a normal clinical picture (Figures 2G–I). However, the father showed mild enlargement of interphalangeal joints of the hands (Figure 2J–L).
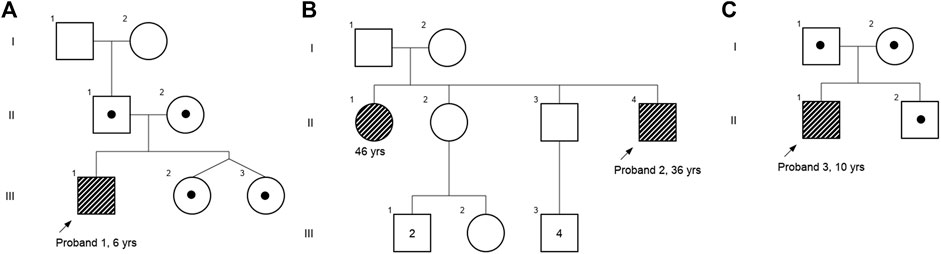
FIGURE 1. Pedigree chart of (A) case 1, (B) case 2, and (C) case 3. Affected individuals are shaded in gray strips, and carriers are depicted with a dot.
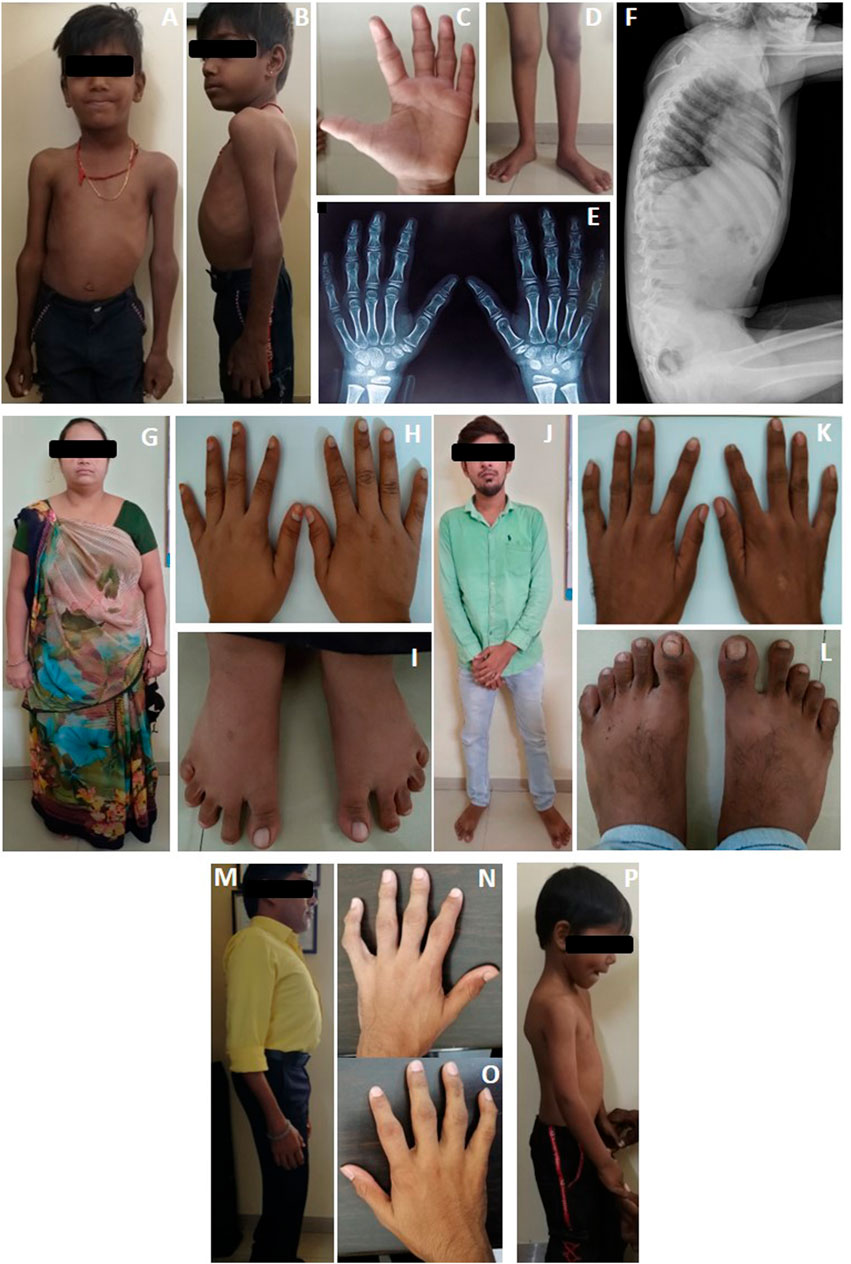
FIGURE 2. Clinical photographs and radiographs of proband's (A–F) case 1, (G–H) case 1’s mother, (J–L) case 1’s father, (M–O) case 2, and (P) Case 3. Case 1 is observed with (A) chest deformity and enlarged elbow joints; (B) scoliosis; (C) deformities of interphalangeal joints; (D) knocked knees; (E) radiograph anterior-posterior view, showing osteopenia; (F) anterior beaking of vertebrae. (G–I) Clinical picture of mother (carrier) of case 1; (J–L) clinical picture of father (carrier) of case 1. Case 2 is observed with (M) scoliosis and (N,O)clinodactyly with deformities of interphalangeal joints. Case 3 is observed with (P) mild scoliosis.
Case 2
A 36-year-old male proband, born to a phenotypically normal and endogamous couple (Figure 1B), presented with short stature, bilateral clinodactyly, gait disturbance, scoliosis, square vertebrae, enlarged chest, and inward knocked knees (Figures 2M–O). His 46-year-old female sibling was reported to have a similar set of phenotypes. Other family members were reported to be phenotypically normal.
Case 3
A 10-year-old male proband, born to a phenotypically normal and endogamous couple (Figure 1C), presented with short stature, mild scoliosis, reduced flexion of fingers with incomplete fist formation, and bilateral elbow terminal flexion restriction (Figure 2P).
Molecular Findings
Genomic DNA was extracted from ethylenediaminetetraacetic acid-treated peripheral whole blood using the desalting method and quantified using QIAxpert (Qiagen, Germany). Whole-exome sequencing was carried out in case 1 only. Library preparation was carried out using the Agilent SureSelect Clinical Research Exome V2 capture kit (Agilent, United States) and sequenced on the Illumina HiSeq platform (Illumina, United States). Fastq files were aligned against the GRCh37/hg19 reference genome using BWA v0.7.17, and an average coverage of 118.48 × post-alignment was observed (Li and Durbin, 2009). Variants were called using GATK’s HaplotypeCaller v4.1 by following GATK’s best practices framework (Poplin et al., 2018). Variants were filtered and prioritized using human phenotype ontology-coded phenotype-driven Exomiser v12.1.0 tool (Smedley et al., 2015). A known pathogenic homozygous variant CCN6(NM_003880.4):c.298T>A (p.Cys100Ser) was observed in exon 3 of the CCN6 gene at a depth of 80×.
Because probands from all three families belonged to the same ethnic community, we prepared an a priori hypothesis that the variant detected in case 1 would likely be observed in cases 2 and 3 due to the prevalence of endogamous practices within the community. Therefore, Sanger sequencing of variant c.298T>A was carried out in cases 2 and 3 and their parents. Interestingly, we observed both cases 2 and 3 to be homozygous for the said variant, and their first-degree relatives (i.e., unaffected parents and/or siblings) were found to be heterozygous carriers (Figure 3).
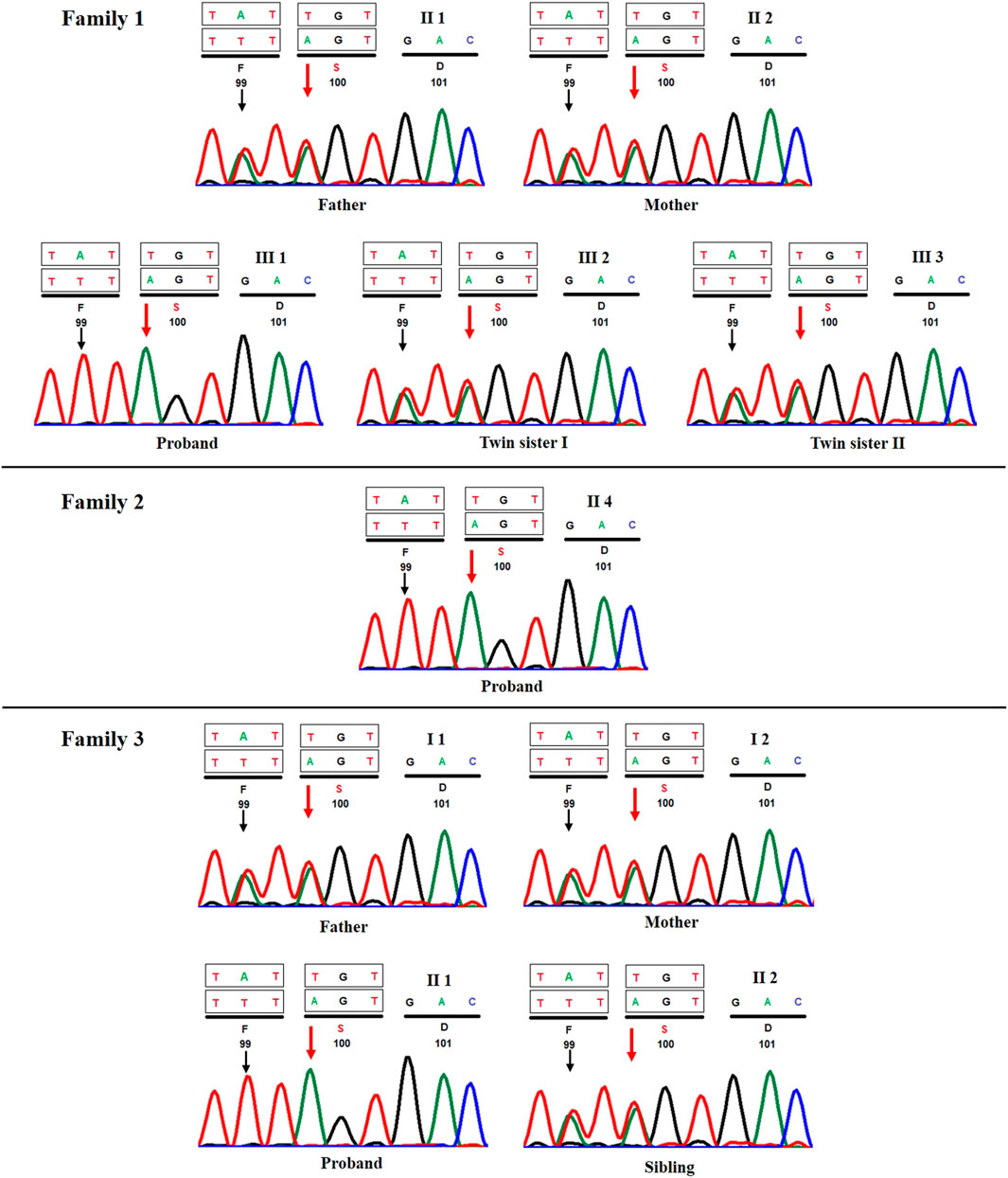
FIGURE 3. Sanger sequencing chromatograms of all three proband's and their respective first-degree relatives (A) case 1, (B) case 2, and (C) case 3. Red and black arrows show the position of c.298T>A and c.296A>T variants, respectively.
Discussion
PPD is a rare skeletal dysplasia with disruption of cartilage homeostasis and progressive joint stiffness in the absence of inflammation (Wynne-Davies et al., 1982). The disease is caused due to either dysfunction or loss of the CCN6 protein. Its prevalence has been estimated to be 1 per 1 million in the United Kingdom population. However, it is likely that the disease may be underdiagnosed due to overlapping clinical presentations with juvenile idiopathic arthritis (Hurvitz et al., 1999).
To date, approximately 210 families have been reported with an underlying genetic cause of PPD, with the most common occurrences in communities practicing endogamy/consanguinity (Progressive Pseudorheumat, 2021). The largest set of case series comprising 79 families has been published from India (Dalal et al., 2012; Bhavani et al., 2015). In total, Bhavani et al. reported 9 novel mutations and 17 mutations that have so far been observed only in the Indian population (Bhavani et al., 2015). Interestingly, the authors observed c.156C>A (p.Cys52Ter) and c.233G>A (p.Cys78Thr) in exon 2 and c.1010G>A (p.Cys337Thr) in exon 5 to account for 70.7% of the total number of mutations and 18.9, 21.5, and 30.3%, respectively, across 79 families from India (Bhavani et al., 2015). Although c.156C>A (p.Cys52Ter) is the most common recurrent variant worldwide, c.233G>A (p.Cys78Thr) and c.1010G>A (p.Cys337Thr) were observed to be the most common variants in the Indian population (Bhavani et al., 2015; Bhavani et al., 2020).
Interestingly, c.298T>A (p.Cys100Ser) variant, which was reported as a novel by Bhavani et al., was observed in three separate families in our study. Furthermore, Bhavani et al. had also observed c.296A>T (p.Tyr99Phe) in cis with c.298T>A variant in their patient. In concordance with this observation, we also observed c.296A>T variant across all three patients (Figure 3) in our study, although in contrast, we could not find evidence to classify it as benign/tolerated based on in silico predictions from DANN (Quang et al., 2015), FATHMM-MKL (Shihab et al., 2015), PROVEAN (Choi and Chan, 2015), and MetaDome (Wiel et al., 2019). Determination of the functional effect of these variants would require functional studies, which is beyond the scope of the current study.
Across all three cases, we observed interphalangeal joint involvement, large joint involvement, spine involvement, and short stature (<third centile) as key clinical features (Table 1). Enlarged chest and square vertebrae were observed in cases 1 and 2 but not in case 3. Although families of cases 2 and 3 were lost to follow-up, the family of case 1 were subsequently invited for genetic counseling and treatment of phenotype manifestation. Following counseling, case 1 is treated by physical therapy to manage small joint arthropathy, although surgical interventions such as bracing for scoliosis were not carried out. Phenotypic assessment of parents of case 1 suggests no obvious clinical PPD phenotype, although interestingly, the father had telltale signs of enlargement of interphalangeal joints of hands (Figure 2K), which is in contrast to the asymptomatic obligate carriers observed in the literature. Although this is a noteworthy observation, the search for a cause of this phenotype in the father was beyond the scope of the study.
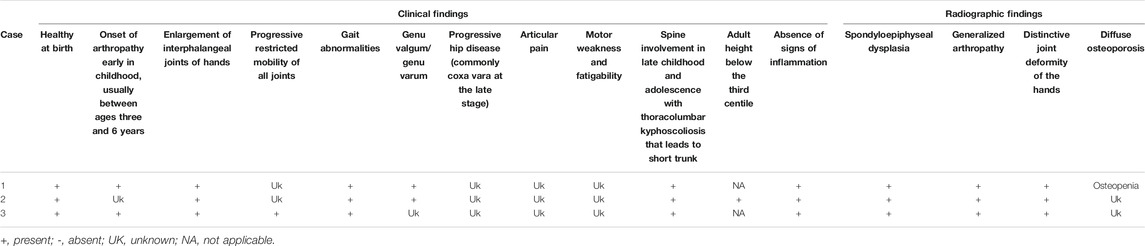
TABLE 1. Summary of the clinical and radiographic findings of all three cases.
In concordance with the literature, we observe a higher than anticipated number of people affected with PPD in the Patni community of Gujarat. This community lives across the western part of India and Sindh province in Pakistan and is known to practice endogamy frequently. This could explain the observation of the same causative variant across three independent families from this community. Therefore, this suggests the utility of testing c.298T>A variant first in a case suspected with PPD belonging to the Patni community.
Conclusion
We reported three PPD cases of individuals from three independent families belonging to the Patni community of Gujarat who were homozygous for a pathogenic variant c.298T>A in the CCN6 gene. The literature review suggests this to be the first instance from India with a recurrent variant being detected in the CCN6 gene in patients belonging to an endogamous community. In summary, our study suggests c.298T>A in CCN6 gene to be a recurrent variant in patients clinically suspected with PPD belonging to the Patni community.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional ethics committee at FRIGE’s Institute of Human Genetics. Written informed consent to participate in this study was provided by the participant’s legal guardian/next of kin.
Author Contributions
HS, PN, and JaS: conception and collection. HS, JhS, AN, and JaS: analysis and interpretation of data. HS, JhS, and AN: drafting the manuscript. All the authors reviewed and approved the final version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to the family of the patients and their families for kind cooperation and permission.
References
Bhavani, G. S., Shah, H., Dalal, A. B., Shukla, A., Danda, S., Aggarwal, S., et al. (2015). Novel and Recurrent Mutations in WISP3 and an Atypical Phenotype. Am. J. Med. Genet. 167, 2481–2484. doi:10.1002/ajmg.a.37164
Bhavani, G. S., Shah, H., Shukla, A., Dalal, A., and Girisha, K. M. (2020). Progressive Pseudorheumatoid Dysplasia, GeneReviews. Seattle, WA: The University of Washington.
Chen, C., Jiang, Y., Xu, C., Liu, X., Hu, L., Xiang, Y., et al. (2016). Skeleton Genetics: A Comprehensive Database for Genes and Mutations Related to Genetic Skeletal Disorders, 2016. Wenzhou, China: Database Oxford. doi:10.1093/database/baw127
Choi, Y., and Chan, A. P. (2015). PROVEAN Web Server: a Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 31, 2745–2747. doi:10.1093/bioinformatics/btv195
Dalal, A., Bhavani G, S. L., Togarrati, P. P., Bierhals, T., Nandineni, M. R., Danda, S., et al. (2012). Analysis of theWISP3gene in Indian Families with Progressive Pseudorheumatoid Dysplasia. Am. J. Med. Genet. 158a, 2820–2828. doi:10.1002/ajmg.a.35620
Hurvitz, J. R., Suwairi, W. M., Van Hul, W., El-Shanti, H., Superti-Furga, A., Roudier, J., et al. (1999). Mutations in the CCN Gene Family Member WISP3 Cause Progressive Pseudorheumatoid Dysplasia. Nat. Genet. 23, 94–98. doi:10.1038/12699
Li, H., and Durbin, R. (2009). Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760. doi:10.1093/bioinformatics/btp324
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples. bioRxiv.
Progressive Pseudorheumatoid Dysplasia (2021). GeneReviews. Seattle, WA: The University of Washington.
Quang, D., Chen, Y., and Xie, X. (2015). DANN: a Deep Learning Approach for Annotating the Pathogenicity of Genetic Variants. Bioinformatics (Oxford, England) 31, 761–763. doi:10.1093/bioinformatics/btu703
Shihab, H. A., Rogers, M. F., Gough, J., Mort, M., Cooper, D. N., Day, I. N. M., et al. (2015). An Integrative Approach to Predicting the Functional Effects of Non-coding and Coding Sequence Variation. Bioinformatics 31, 1536–1543. doi:10.1093/bioinformatics/btv009
Smedley, D., Jacobsen, J. O. B., Jäger, M., Köhler, S., Holtgrewe, M., Schubach, M., et al. (2015). Next-generation Diagnostics and Disease-Gene Discovery with the Exomiser. Nat. Protoc. 10, 2004–2015. doi:10.1038/nprot.2015.124
Wang, M., Man, X. F., Liu, Y. Q., Liao, E. Y., Shen, Z. F., Luo, X. H., et al. (2013). Dysfunction of Collagen Synthesis and Secretion in Chondrocytes Induced by Wisp3 Mutation. Int. J. Endocrinol. 2013.679763 doi:10.1155/2013/679763
Wiel, L., Baakman, C., Gilissen, D., Veltman, J. A., Vriend, G., and Gilissen, C. (2019). MetaDome: Pathogenicity Analysis of Genetic Variants through Aggregation of Homologous Human Protein Domains. Hum. Mutat. 40, 1030–1038. doi:10.1002/humu.23798
Keywords: progressive pseudorheumatoid dysplasia, recurrent variant, Patni, Gujarat, CCN6
Citation: Sheth H, Shah J, Nair A, Naik P and Sheth J (2021) Case Report: Recurrent Variant c.298 TA in CCN6 Gene Found in Progressive Pseudorheumatoid Dysplasia Patients From Patni Community of Gujarat: A Report of Three Cases. Front. Genet. 12:724824. doi: 10.3389/fgene.2021.724824
Received: 14 June 2021; Accepted: 01 September 2021;
Published: 28 September 2021.
Edited by:
Aleksander Jamsheer, Poznan University of Medical Sciences, PolandReviewed by:
Jernej Kovac, University Children’s Hospital, SloveniaEmanuele Micaglio, IRCCS Policlinico San Donato, Italy
Copyright © 2021 Sheth, Shah, Nair, Naik and Sheth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harsh Sheth, aGFyc2guc2hldGhAZnJpZ2UuY28uaW4=